1. Introduction
In the last decade, we have seen outbreaks of several pathogenic RNA viruses, like Ebola virus (EBOV), Crimean-Congo hemorrhagic fever virus (CCHFV) and Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). EBOV and CCHFV both cause viral hemorrhagic fever with the average case fatality rate around 50–70% [
1,
2,
3] and 5–30% [
4], respectively. SARS-CoV-2 infection is mild or asymptomatic in the majority of cases; however, up to 10% of cases lead to severe respiratory distress, sepsis and may be lethal. The ongoing Coronavirus disease 2019 (COVID-19) pandemic caused by SARS-CoV-2 has led to 1,200,000 deaths as of November 2020 and unseen social and economic disruption worldwide. Common human coronaviruses such as Coronavirus 229E (CoV 229E) cause mild upper respiratory tract infections and around 15–30% of common cold cases in adults [
5]. While RNA viruses cause a variety of diseases, a common denominator is the lack of antivirals to treat them, highlighting the need for the development of antivirals to tackle these diseases.
Hallmarks of RNA viruses include a heavy reliance on the host cell for viral replication [
6] and high mutation rates [
7,
8] resulting in fast resistance against antiviral drugs targeting virus proteins [
9,
10,
11]. Of note, coronaviruses are an exception to the high mutation rates of RNA viruses, as they have a proofreading mechanism [
12,
13]. Given that several host processes such as translation [
14,
15] and protein folding [
16] are commonly exploited by RNA viruses across virus families, targeting host factors required by the virus is an attractive strategy to battle emerging RNA viruses. Host cells experience higher genetic barriers to evade host-targeting antivirals and drug resistance is much less likely to occur [
17]. However, it is challenging to find druggable host proteins as targets for antiviral therapies with limited understanding of host cell–virus interactions. Unlike target-based drug discovery, phenotypic assays enable discovery of new classes of compounds without prior knowledge on the target or its role in the virus life cycle. Combining phenotypic screenings with target identification methods can lead to a discovery of novel antiviral targets as well as host–virus interactions.
During recent years, advanced mass-spectrometry-based target identification method thermal proteome profiling (TPP) has been developed to monitor drug–target interactions in living cells on a proteome-wide scale [
18]. TPP measures changes of protein thermal stability upon compound binding to cellular targets and can be used to detect drug targets and changes in cellular pathways [
18].
One of the pathways activated by RNA viruses is the oxidative stress pathway in the host cell [
19,
20,
21]. The Helleday Laboratory has previously developed small molecule inhibitors targeting oxidative stress and nucleotide metabolism pathways in the context of cancer and inflammation [
22,
23]. Based on this background, we hypothesized that disturbing oxidative balance of the host cell can influence viral replication.
Here, we established an image-based phenotypic high-throughput screening assay coupled with automated image analysis and tested a set of in-house inhibitors targeting oxidative stress and nucleotide metabolism pathways. We identified novel small molecule compounds with antiviral activity against several pathogenic RNA viruses such as SARS-CoV-2, EBOV and CCHFV, demonstrating broad-activity of the hit compound. Lastly, we studied the pharmacological activity of the new antiviral compound using TPP showing its multifaceted effects on cellular HSP70 pathways.
2. Materials and Methods
2.1. Biosafety
All procedures in presence of infectious viruses were carried out under respective biosafety laboratory conditions according to Swedish Work and Health Authorities. Experiments using Hazara virus (HAZV) and CoV 229E were performed under BSL2, SARS-CoV-2 under BSL3 at Karolinska Institutet. CCHFV and EBOV under BSL4 conditions at the Public Health Agency of Sweden.
2.2. Cells and Viruses
Vero, Vero E6 and U87 cells (from ATCC, Manassas VA, USA) were cultured in complete Dulbecco’s modified Eagle medium (DMEM, Gibco, USA), U-2 OS (from ATCC, Manassas, VA, USA) in DMEM GlutaMAX™ medium (Gibco, USA), SW-13 (ATCC, Manassas, VA, USA) in Leibovitz L-15 (Gibco, USA) medium, HEK293T cells (ATCC, Manassas, VA, USA) in DMEM GlutaMAX™ medium, Huh7 cells (from ATCC, Manassas, VA, USA) in DMEM (Gibco, USA). Culture medium of all cell lines were supplemented with 10% (v/v) FBS (Thermo Fisher Scientific, Waltham, MA, USA), 50 U/mL penicillin and 50 μg/mL streptomycin (PS, Thermo Fisher Scientific, Waltham, MA, USA). All cell lines were maintained at 37 °C under 5% CO2, except SW-13 cells which were maintained under atmospheric CO2. All cell lines were routinely tested for Mycoplasma using a luminescence-based MycoAlert kit (Lonza, Basel, Switzerland).
HAZV (isolate JC280; from European Virus Archive, Marseille, France) stocks were amplified in SW13 cells. Virus titers were determined in Vero cells by focus-forming unit (FFU) assay combined with high-throughput immunofluorescence imaging of viral protein staining. CCHFV (IbAr 10200) and EBOV (Sudan Ebolavirus) were both from the Public Health Agency of Sweden. Stocks were grown on Vero cells and titers were determined by FFU in Vero cells, as previously described in [
24]. CoV 229E (VR-740; from ATCC, Manassas, VA, USA) stocks were amplified in Huh7 cells. SARS-CoV-2 (isolate MT093571.1; from the Public Health Agency of Sweden) [
25] were amplified in Vero E6 cells.
2.3. Virus Infections and Compound Treatments
Cells were infected with a MOI of 0.1, 0.5, 1 or 10 in 50 μL corresponding cell culture medium for 1 h. At least three wells of cells were left uninfected as a mock control. Virus-containing medium was removed and replaced with 100 μL dimethyl sulfoxide (DMSO) (Sigma-Adrich, St. Louis, MO, USA, max. 0.2%) or compound-containing medium in at least technical duplicates. Number of infected cells, cytotoxicity and progeny virus yield in supernatants were determined after 24–48 h post infection (hpi) by immunofluorescence assay.
Infections with CCHFV and EBOV were performed in 24-well plates. Vero cells were seeded at the density of 1 × 105 cells per well. After 24 h cells were infected with the appropriate MOI in 1 mL of plenty medium for 1 h. Then, virus-containing medium was removed and replaced with 1 mL DMSO (max. 0.5%) or compound-containing medium. Each condition was tested in duplicates. At 48 hpi supernatants were collected and viral particles quantified by end-point dilution assay using immunostaining and plaques were counted manually.
2.4. Synthesis of Compounds
2.5. Phenotypic Antiviral Screening
Compounds were dispersed in 384-well plates at final concentration of 10 μM, (max DMSO 0.1% v/v) and stored at 4 °C. SW13 cells were inoculated with HAZV in L15 medium in presence of 10% FBS and 1% PS for 1 h. After inoculation, the virus-containing medium was removed and washed with PBS (Gibco, USA). Cells were trypsinized and 3000 cells were seeded on 384-well plates containing screening compounds. Cells were incubated for 24 h. Infected cells were fixed in 4% paraformaldehyde (Santa Cruz, Dallas, TX, USA) in PBS for 20 min and in 1:1 (v/v) methanol-acetone for 20 min in −20 °C. Number of infected cells were quantified by high-throughput microscopy. Data were normalized to infected DMSO-treated controls.
2.6. Determining Antiviral and Cytotoxicity EC50
Compounds were dispersed in deep-well 96-well plates (Biotix, San Diego, CA, USA #DP-1200-9CUS) using the D300e Digital Dispenser (Tecan, Männedorf, Switzerland) and stored at 4 °C. Compound 1:2 dilution steps between 50 μM and 1.526 μM (final concentration; DMSO 0.5% v/v) were chosen. Compounds were dissolved in culture media. 5000 SW13 cells were seeded into 96-well plates and allowed to attach for 24 h. Cells were inoculated with HAZV in L15 medium in presence of 10% FBS and 1% PS for 1 h. After inoculation, virus-containing medium was removed and replaced with medium containing DMSO or compounds and cells were incubated for 24 h. Cells were fixed in 4% paraformaldehyde in PBS for 20 min and in 1:1 (v/v) methanol-acetone for 20 min in −20 °C and processed for high-throughput microscopy.
2.7. Virus Infectivity Assay
Cells were seeded in 96-well plates (Black 96-well microplate, flat bottom, Falcon). At 24 h after seeding, cells were inoculated with virus in 50 μL corresponding cell culture medium for 1 h. At least three wells of cells were left uninfected as a mock control. After 1 h virus inoculation, the virus-containing medium was removed and replaced with 100 μL DMSO (max. 0.5% v/v) or compound-containing medium in at least technical duplicates. At 24 hpi, virus-containing supernatants were collected and used in end-point dilution assay. Cells were fixed in 4% paraformaldehyde in PBS for 20 min and in 1:1 (v/v) methanol-acetone for 20 min in −20 °C and processed for high-throughput microscopy.
2.8. End-Point Dilution Assay by Immunofluorescence
To quantify viral titer, cells were seeded in 96-well plates (Falcon® 96-well Black/Clear Flat Bottom TC-treated Imaging Microplate, Corning, Glendale, AZ, USA) at a density of 5 × 103 cells per well. 24 h later virus-containing supernatant samples were serially diluted on cells by 10-fold and incubated for 24 h. Supernatants were discarded, cells were fixed in 4% paraformaldehyde in PBS for 20 min and in 1:1 (v/v) methanol-acetone for 20 min in −20 °C and processed for high-throughput microscopy.
2.9. High-Throughput Microscopy
Immunofluorescence staining. Methanol-acetone was removed, plates were washed twice with PBS and incubated with virus-specific primary antibody (see antibody details in
Table S2) in 0.2% BSA 0.1% Triton-X in PBS overnight at 4 °C. Primary antibody was removed and cells were washed 3 × 10 min with PBS and incubated with secondary antibody (donkey-anti-mouse Alexa-488; 1:800) and DAPI (1:1000, see details in
Tables S2 and S3) in 0.2% BSA 0.1% Triton-X in PBS for 1 h. Cells were washed 3 × 10 min with PBS and retained in 100 μL PBS for image acquisition.
Automated image acquisition. Images were automatically captured with ImageXpress Micro XLS Widefield High-Content Analysis System (Molecular Devices, San Jose, CA, USA) typically using two fluorescent channels: DAPI (387/447) and FITC (472/520). Nine images were captured per each well using ×10 Plan Fluor 0.3 NA objective. Images were saved as 16-bit, gray-scale Tiff files, along with metadata in an Oracle database for further analysis.
Quantitative image analysis by CellProfiler. Images were loaded into open-source software CellProfiler (Broad Institute, Cambridge, MA, USA) and analysed by pipeline. Automated analysis had 4 main steps: Identifying primary objects, identifying secondary objects, measuring object intensity and classifying objects. Identifying primary objects were performed on DAPI stained images based on intensity threshold between the foreground and background and referred as “primary objects”. Secondary objects were identified based on the primary objects by expanding the area around each nucleus by 15 pixels and measuring intensity of this area from the FITC channel containing virus-specific staining. Classification based on nuclei expansion is possible given that virus replication is perinuclear. Upon identifying primary and secondary objects, object intensities were measured. Finally, based on secondary object mean intensity, cells were divided into two custom-defined bins: uninfected or infected. The threshold between two bins depends on staining quality, background and foreground intensity and is therefore defined at each CellProfiler analysis based on positive (infected cells) and negative (uninfected cells) controls. Data from object classification, shape, size and intensity measurements were exported to a spreadsheet.
2.10. Z-Factor Calculations
Z-factors were calculated and interpreted as described previously [
26]. Briefly, the main signal of negative controls plus three times the standard deviation (SD) of those valuesx was set as a threshold for negative controls. Then, the mean signal of positive controls minus three times the SD of those values was set as the threshold for positive controls. The difference between the thresholds was calculated and was divided by the absolute value of the difference of thresholds. In infection assays uninfected DMSO-treated cells were used as negative control and infected DMSO-treated cells as positive control. In viability assays, media-only was used as negative control and DMSO treated as positive control.
2.11. Cell Viability Assays
Resazurin assay. Cells were seeded into 96-well plates and allowed to attach for 24 h. Respective media was exchanged for compound-containing with a stable DMSO concentration of maximally 0.5% throughout the plate and cells were incubated for 24, 48 or 72 h. After respective incubation times, 10 μg/mL Resazurin (Sigma-Aldrich, St. Louis, MO, USA, R7017, stock at 1 mg/mL in PBS) was added on top of cells and incubated between 1 to 4 h. Fluorescence was measured at 544/590 (excitation/emission) using the Hidex Sense 425-301 reader (Hidex, Turku, Finland). Quality of the assay was determined by Z-factor using cell-free wells as negative and wells containing DMSO treated cells as positive controls. Data were normalized to DMSO treated controls and curve fitting was performed to determine EC50 values for cell viability using a sigmoidal, 4PL model in GraphPadPrism (San Diego, CA, USA).
MTT assay. Vero cells were seeded into 96-well plates and allowed to attach for 24 h. Culture medium was removed and replaced with medium containing DMSO or compounds at desired concentrations. Cells were incubated for 48 h. Medium was removed and replaced with 100 μL DMEM containing 15 μL MTT ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide)) (Sigma-Aldrich, St. Louis, MO, USA) per well and incubated for 2 h until crystals were present. MTT solution was removed from the cells, 100 μL DMSO was added to dissolve the crystals and optimal density OD was measured at 540 nm after 15 min.
2.12. siRNA-Mediated Depletion of OGG1
Cells were reverse-transfected with individual or with a pool of 4 siRNAs (Dharmacon, Lafayette, CO, USA) specific for OGG1 or with positive control AllStars Hs Cell Death Control siRNA (Qiagen, Hilden, Germany) or negative control ON-TARGETplus Non-targeting Pool siRNAs (Dharmacon, Lafayette, CO, USA). siRNAs were dissolved in 30 μL serum-free DMEM containing 0.6 μL INTERFERin transfection reagent (PolyPlus Transfection, Illkirch-Graffenstaden, France) per well, mixed by pipetting 10 times and incubated at room temperature for 10–15 min for transfection complex formation. U2-OS cells were seeded on top of transfection complexes in 70 μL complete DMEM medium with final density 3000 cells per well, yielding final siRNA concentration 10 nM. Cells were incubated in the presence of siRNA transfection for 48 h. Details of siRNAs used are found in the
Table S1.
2.13. Cellular Thermal Shift Assay (CETSA)
Cetsa-TR. CETSA was performed with intact cells as described previously [
27]. Briefly, U87 or SW13 cells were seeded on 2 × T75 flasks. The following day, cells were incubated with DMSO (0.2%
v/
v) or 20 μM TH6744 for 1.5 or 4 h at 37 °C. Cells were then washed with PBS containing DMSO or compound, harvested by TryplE Express (Gibco, USA) and resuspended in TBS supplemented with cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail (Roche, Basel, Switzerland) at 1.0 × 10
5 cells per 100 μL. Cells from each condition were divided to 10 aliquots and heated to different temperatures (37, 41, 44, 47, 50, 53, 56, 59, 63 or 67 °C) for 3 min using Veriti 96-well Thermal Cycler (Applied Biosystems, Foster City, CA, USA) followed by cooling at 25 °C for 3 min. Thereafter, the cells were lysed by 3 freeze–thaw cycles using dry ice in 100% Ethanol and a water bath set at 25 °C. The lysates were then clarified by centrifugation at 20,000×
g for 40 min at 4 °C and prepared for Western blotting to detect Hsp70 or OGG1. SOD-1 served as the loading control.
Isothermal dose response Cetsa. U87 or SW13 (2 × 105) cells were seeded on a 6-well plate. The following day, cells were incubated with DMSO (0.3% v/v) or different TH6744 concentrations for 1.5 or 4 h at 37 °C. Cells were washed with PBS containing DMSO or compound at respective dose, harvested by TryplE Express and resuspended in 100 μL PBS supplemented with DMSO or compound at respective dose. Cells were heated at 47 °C (for detecting OGG1), 56 °C (Hsp70) or 59 °C (Hsp70) for 3 min using Veriti 96-well Thermal Cycler (Applied Biosystems, Foster City, CA, USA) followed by cooling at 25 °C for 3 min. Thereafter, the cells were lysed by 3 freeze–thaw cycles using dry ice in 100% Ethanol and a water bath set at 25 °C. The lysates were clarified by centrifugation at 20,000× g for 40 min at 4 °C and prepared for Western blotting to detect Hsp70 or OGG1. SOD-1 or β-actin served as the loading control.
2.14. Western Blot
Upon sample preparation and clarification by centrifugation, lysates containing 20–30 μg total protein (measured using Pierce™ BCA Protein Assay Kit, Thermo Fisher Scientific, Waltham, MA, USA) were mixed with 4× Laemmli buffer (Bio-Rad, Hercules, CA, USA) supplemented with mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA) or Dithiothreitol (DTT) (Sigma-Aldrich, St. Louis, MO, USA). Samples were heated at 95 °C for 10 min. Proteins were separated in 4–15% Mini-PROTEAN TGX gels (Bio-Rad, Hercules, CA, USA) and transferred to nitrocellulose membranes using a Trans-Blot Turbo machine (Bio-Rad, Hercules, CA, USA). Membranes were blocked in Odyssey Blocking Buffer (LI-COR, Lincoln, NE, USA) or in 2% non-fat milk (Sigma-Aldrich, St. Louis, MO, USA) in TBS-T, incubated with primary antibodies (details of antibodies is found in
Table S2) against desired protein at 4 °C overnight and with fluorescence-conjugated species-specific secondary antibodies at room-temperature for 1 h. Membranes were washed three times with TBS-T between incubations. Protein bands were directly visualized with an Odyssey Fc Imager (LI-COR, Lincoln, NE, USA) and images were analysed using Image Studio Lite Software (LI-COR, Lincoln, NE, USA). All raw uncropped images are compiled in
Data S7.
2.15. In Vitro OGG1 Activity Assay
Inhibition of human OGG1 enzymatic activity was measured as described previously [
23]. Briefly, compounds were mixed with 0.8 nM OGG1 enzyme, 2 nM AP endonuclease 1, and 10 nM oligonucleotide duplex DNA (5ʹ-FAM-TCTGCCA[8-oxoA]CACTGCGTCGACCTG-3ʹ, annealed to 5ʹ-CAGGTCGACGCAGTGCTGGCAGT[Dabcyl]-3ʹ, where [FAM] indicates fluorescein, and [8-oxoA] indicates 8-oxo-2ʹ-deoxyadenosine (TriLink Biotech, San Diego, CA, USA) in a buffer containing 25 mM Tris-HCl pH 8.0, 15 mM NaCl, 2 mM MgCl2, 0.5 mM DTT, and 0.0025% Tween-20. Fluorescein fluorescence was then recorded in a Hidex Sense plate reader.
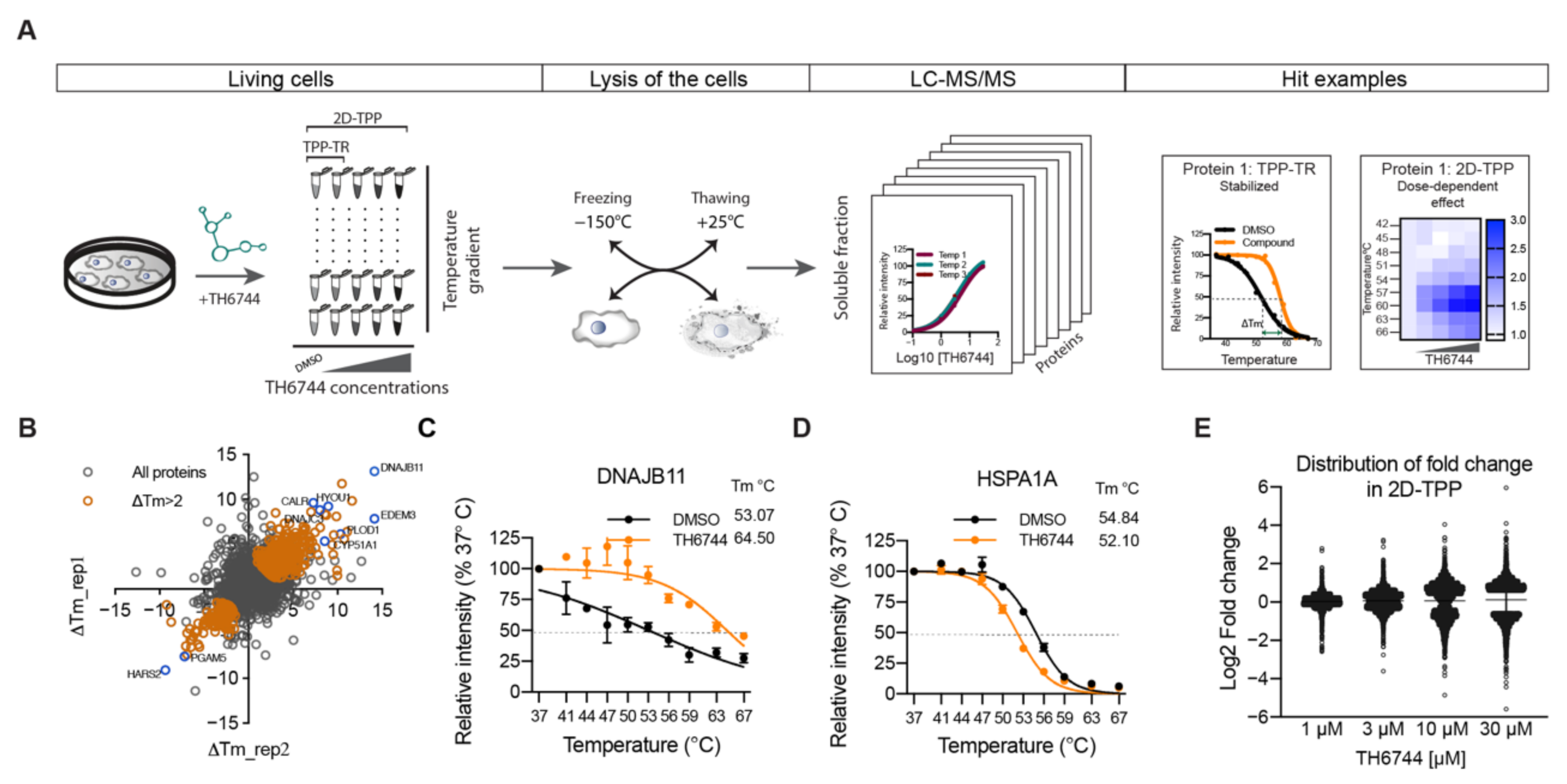
2.16. Thermal Proteome Profiling Sample Preparation
TPP-TR. SW-13 cells were seeded into T175 flasks, after 24 h infected with HAZV (MOI 10) for 16 h and treated with 20 μM TH6744 or DMSO control (0.2% v/v) for 4 h. Cells were washed twice with PBS, harvested by TryplE Express (Gibco, USA) and collected in PBS. Cells were centrifuged at 300× g for 5 min, re-suspended in 1100 μL PBS containing the compound or DMSO, divided in ten 100 μL aliquots and heated individually for 3 min at 37, 41, 44, 47, 50, 53, 56, 59, 63, and 67 °C using Thermocycler (Applied Biosystems, Foster City, CA, USA) followed by cooling at 25 °C for 3 min. Thereafter, the cells were freeze–thawed 3 times by using dry ice in 100% Ethanol and a water bath set at 25 °C. Precipitated proteins were separated from soluble proteins by centrifugation at 20,000× g for 40 min at 4 °C. In total, 70 μL supernatants were transferred into new microtubes and supplemented with SDS to reach final concentration 0.1%. Total protein concentration was measured by PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). TPP-TR experiments were conducted as two biological replicates.
2D-TPP. U87 cells were seeded into T175 flasks 72 h prior assay to allow cell growth and obtain approximately 80% confluency. Cells were treated with 30, 10, 3, and 1 μM TH6744 or DMSO for 1.5 h. Cells were washed twice with PBS containing corresponding compound dose, harvested by TryplE Express (Gibco, USA) and collected in PBS containing compound. Cells were centrifuged at 300× g for 3 min, re-suspended in 1000 μL PBS containing the compound or DMSO. Cells from each compound treatment were divided in 9 × 100 μL aliquots and heated individually for 3 min at 42, 45, 48, 51, 54, 57, 60, 63, and 67 °C using Thermocycler (Applied Biosystems, Foster City, CA, USA) followed by cooling at 25 °C for 3 min. Thereafter, the cells were freeze–thawed 3 times by using dry ice in 100% Ethanol and a water bath set at 25 °C. Precipitated proteins were separated from soluble proteins by centrifugation at 20,000× g for 30 min at 4 °C. In total, 70 μL supernatants were transferred into new microtubes. Total protein concentration was measured by PierceTM 660 nm Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA).
2.17. Sample Preparation for LC-MS/MS
Protein digestion and peptide labelling. Total protein concentration of samples was determined by using a DC™ Protein Assay (Bio-Rad, Hercules, CA, USA). Supernatants were supplemented with a stock solution of sodium dodecyl sulfate (SDS) (Sigma-Aldrich, St. Louis, MO, USA), tris(2-carboxyethyl)phosphine (TCEP) (Sigma-Aldrich, St. Louis, MO, USA) and triethylammonium bicarbonate (TEAB) (Sigma-Aldrich, St. Louis, MO, USA) to reach the final concentrations of 0.1% SDS, 5 mM TCEP and 50 mM TEAB. Samples were reduced for 30 min at 65 °C and subsequently alkylated by chloroacetamide at a final concentration of 15 mM for 30 min at RT. At a first digestion step, the protease LysC (Wako, Osaka, Japan) was added to each sample at a protease-to-protein ratio of 1:55 (for TPP-TR) or 1:40 (for 2D-TPP) with regard to the protein amount in the sample with the highest total protein concentration. Digestion was allowed to take place at 37 °C for 16 h. Proteins were digested by adding Pierce™ Trypsin Protease, MS Grade (Thermo Fisher Scientific, Waltham, MA, USA) at a ratio of 1:60 (TPP-TR) or 1:70 (2D-TPP) and incubation for 8 h, followed by adding trypsin once more at a ratio of 1:60 (TPP-TR) or 1:70 (2D-TPP) to complete digestion during a second overnight incubation at 37 °C. Samples were labeled with isobaric TMT tags (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions, but adjusted for three hours. In TPP-TR the full temperature range of each condition (ten samples) was labeled by the ten TMT tags of one TMT10-plex. In 2D-TPP three TMTpro-sets were used, excluding the 134N channel. Each set included samples from five different compound concentrations (0, 1, 3, 10, and 30 μM) and were distributed in a way that the first set comprised the samples that were melted at 42, 45 and 48 °C, the second one at 51, 54 and 57 °C and the third at 60, 63 and 67 °C. Labelling efficiency was determined by LC-MS/MS before pooling of samples into 10- or 15-plexes for TPP-TR and 2D-TPP experiments, respectively. Sample clean-up was performed by solid phase extraction (SPE strata-X-C, Phenomenex, Torrance, CA, USA) and purified samples were dried in a SpeedVac (Thermo Fisher Scientific, Waltham, MA, USA).
Prefractionation by high pH reversed-phase chromatography (HpH RP-HPLC). In the TPP-TR experiment, sample pools were pre fractionated using HpH RP-HPLC [
28] and 200 μg of peptide mixture from each set were fractionated using a Waters XBridge BEH300 C18 3.5 μm 2.1 × 250 mm column (Waters Corporation, Milford, MA, USA) on a Agilent 1200 HPLC system operating at 200 μL per min. Buffer A consisted of 20 mM NH3, and buffer B of 80% acetonitrile, 20 mM NH3. Fractions were eluted using a gradient from 3 to 88% B in 63 min. The column was washed with 88% B for 15 min, followed by a ramp to 100% B in 2.5 min and kept at 100% B for another 13.5 min. Fractions were collected into a polypropylene V-96-well microtiter plates (Microplate, 96-well PP, V-Bottom; Grainer BIOONE, Kremsmünster, Austria). At 97 min, fraction collection was halted, and the gradient was held at 3% B for 20 min. The total number of concatenated fractions was set to 12. Each plate was dried at RT using a SpeedVac (SPD 111V, Thermo Fisher Scientific, Waltham, MA, USA). Plates were stored at −20 °C until LC/MS/MS analysis.
Prefractionation by high-resolution isoelectric focusing (HiRIEF). In the 2D-TPP experiment, HiRIEF prefractionation method was applied as previously described [
29]. Briefly, 300 μg of the dried peptide sample pools were subjected to peptide IEF-IPG (isoelectric focusing by immobilized pH gradient) in the pI range of 3–10. Peptide samples were dissolved in a 250 μL rehydration solution containing 8 M urea and 1% IPG pharmalyte pH 3–10 (GE Healthcare, Chicago, IL, USA) and allowed to adsorb to the gel strip by swelling overnight. The peptides were focused on the gel strip and were thereafter passively eluted into 72 contiguous fractions into a V-bottom 96-well plate (Grainer BIOONE, Kremsmünster, Austria; product #651201) with MilliQ water/35% ACN/35% ACN + 0.1%FA using an in-house built IPG extractor robotics system (GE Healthcare, Chicago, IL, USA, prototype instrument). Samples were dried by using a SpeedVac (Thermo Fisher Scientific, Waltham, MA, USA) at RT and kept at −20 °C.
2.18. Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) Analyses
LC-MS/MS runs of the HpH RP-HPLC fractions (TPP-TR). Each sample was analyzed on a HF Q-Exactive Orbitrap (Thermo Fisher Scientific, Waltham, MA, USA) connected to a Dionex UHPLC system (Thermo Fisher Scientific, Waltham, MA, USA). The UHPLC was equipped with a trap column Acclaim PepMap 100, 75 μm × 2 cm, nanoviper, C18, 3 μm, 100 Å (Thermo Fisher Scientific, Waltham, MA, USA) and an analytical column PepMap RSLC C18, 2 μm, 100 Å, 75 μm × 50 cm (Thermo Fisher Scientific, Waltham, MA, USA). Mobile-phase buffers for nLC separation consisted of 0.1% FA in water (solvent A) and 80% acetonitrile/0.1% formic acid (solvent B). The peptides were eluted during a 2 h gradient and directly sprayed into the mass spectrometer. The flow rate was set at 250 nL per min, and the LC gradient was as follows: 3–6% solvent B within 3 min, 6–35% solvent B within 117 min, 35–47% solvent B within 5 min, 47–100% solvent B within 5 min and 100% B for 8 min and 1% solvent B for 5 min. The mass spectrometer was operated in a data-dependent acquisition mode (top 10 most intense peaks) performing a survey scan from 370 to 1600 m/z (resolution 60,000). Automatic gain control (AGC) target was set to 3e6 and maximum injection time to 250 ms. MS2 scans were acquired on the 10 most-abundant MS1 ions of charge state 2–7 using a Quadrupole isolation window of 1 m/z for higher-energy collision dissociation HCD fragmentation. Collision energy was set to 34%. MS2 spectra were acquired at a resolution of 30,000 using an AGC target of 2e5 and a maximum injection time of 200 ms. Dynamic exclusion was set to 15 s. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [
30] partner repository with the dataset identifier PXD021494.
LC-MS/MS runs of the HiRIEF fractions (2D-TPP). Online LC-MS was performed as previously described [
29] using a Dionex UltiMate™ 3000 RSLCnano System (Thermo Fisher Scientific, Waltham, MA, USA) coupled to a Q-Exactive-HF mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Peptides of each of the 72 plate wells were dissolved in 0.1% formic acid, 3% acetonitrile in water and subjected to LC-MS/MS analysis. Samples were trapped on a C18 guard-desalting column (Acclaim PepMap 100, 75 μm × 2 cm, nanoViper, C18, 5 μm, 100 Å; Thermo Fisher Scientific, Waltham, MA, USA), and separated on a 50 cm long C18 column (Easy spray PepMap RSLC, C18, 2 μm, 100 Å, 75 μm × 50 cm; Thermo Fisher Scientific, Waltham, MA, USA). The nano capillary solvent A consisted of 95% water, 5% DMSO and 0.1% formic acid. Solvent B consisted of 5% water, 5% DMSO, 95% acetonitrile and 0.1% formic acid. At a constant flow of 0.25 μL min−1, the curved gradient went from 10% B up to 40% B for fraction 1–19, 8% B up to 40% B for fraction 20–39 and 6% B up to 40% B for fraction 40–72 in a dynamic range of gradient length, followed by a steep increase to 100% B in 5 min. FTMS master scans with 60,000 resolution (and mass range 300–1500 m/z) were followed by data-dependent MS/MS (30,000 resolution) on the top five ions using higher energy collision dissociation (HCD) at 30% normalized collision energy. Precursors were isolated with a 2 m/z window. Automatic gain control (AGC) targets were 1E6 for MS1 and 1E5 for MS2. Maximum injection times were 100 ms for MS1 and 100 ms for MS2. Dynamic exclusion was set to 30 s duration. Precursors with unassigned charge state or charge state 1 were excluded. An underfill ratio of 1% was used.
2.19. Peptide and Protein Identification and Quantification
The raw data for the TPP-TR experiment were analyzed as previously described [
31]. Mascot was used to search the MS/MS data against the UniProt Homo sapiens database (UniProt Consortium) (containing canonical and isoforms_42144 entries downloaded on 21st of March 2016).
The raw data for the 2D-TPP experiment were analyzed as previously described [
32]. Mascot was used to search the MS/MS data against the UniProt Homo sapiens database (UniProt Consortium) (containing canonical and isoforms_42144 entries downloaded on 21st of March 2016).
2.20. TPP Statistical Analysis and Hit Selection
TPP-TR: TPP-TR results were analyzed as described previously [
31,
33]. First, proteins were sorted by ascending non-parametric analysis of the response curve (NPARC)
p-value “p_adj_NPARC”. The adjusted
p-value from a non-parametric statistical test of the curve-fitting step was used to generate the data in the TPP R package. A 0.0001 threshold was applied, yielding protein melt curves with significant ΔTm (1589 proteins). Next, proteins with a “p_adj_NPARC” below threshold were filtered by the column “meltP_diffs_have_same_sign”, selecting rows that contain a “yes” meaning that the ΔTm between DMSO_1 and TH6744_1 has the same sign (positive or negative) as the ΔTm between DMSO_2 and TH6744_2 (1224 proteins). Next, proteins were filtered on the column “meltP_diffs_T_vs_V_greater_V1_vs_V2”, selecting rows that contain a “yes”, meaning that the ΔTm between treatment and DMSO is larger than between DMSO_1 and DMSO_2 replicates (1154 proteins). Last, a ΔTm > 2 °C threshold was applied (593 proteins).
2D-TPP: 2D-TPP results were analyzed as described previously [
33,
34]. Proteins with changed solubility or expression were excluded and F-statistic threshold of 15 was applied. Half-maximal effective concentration of protein stabilization/destabilization pEC
50 (pEC
50 = −log(EC
50) illustrates binding affinity of TH6744 to proteins.
2.21. Gene Set Enrichment and Interaction Analyses
All gene set enrichment analyses were performed using Metascape (
http://metascape.org/gp/index.html#/main/step1). Gene annotation terms for enrichment analysis were extracted from CORUM, Reactome gene sets, KEGG pathways, gene ontology molecular function and gene ontology biological process databases. Enriched terms with
p < 0.01, 3 or more genes and a ratio between observed and expected count >1.5 were clustered by hierarchical clustering based on term similarities and using kappa scores as a similarity metric. Terms were selected for representation of a cluster when they had the highest statistical significance of the cluster. Protein–protein interaction networks were extracted from the STRING database (STRING Consortium) with a 0.7 confidence threshold and excluding interactions extracted from text mining.
2.22. Statistical Analysis
Statistical significance was determined using the GraphPad Prism Software (San Diego, CA, USA, Version 8.4.0). Two-tailed t-tests, one- or two-way ANOVA with appropriate follow up tests were used. For each data panel, the tests used are stated in the according figure legend. If not indicated otherwise, data are presented as mean ± SD from at least n = 2 biological replicates. Threshold for defining significance was p < 0.05 and significance levels were defined as follows: * p < 0.05, ** p < 0.01, *** p < 0.001. For dose-response-testing curve fitting was performed to determine IC50 using a sigmoidal, 4PL model in GraphPad Prism (San Diego, CA, USA) with bottom and top constraints at 0 and 100, respectively. If not indicated otherwise, each image shown is a representative of at least n = 2 biological replicates.
2.23. Data Availability
The data that support the findings of this study are within the article,
Supplementary Materials, supplementary data or available from the corresponding author upon request.
Data S3 is accessible in 10.17044/scilifelab.13089023. The raw mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD021494.
4. Discussion
The increasing incidence of virus emergence and outbreaks in the last decade highlights the need for new, broadly acting antivirals, to diminish the impact of these diseases on health and economy. Phenotypic screening is an attractive approach to discover new host–pathogen interactions as well as new drug targets with potentially broad-spectrum activity [
40]. Previously published antiviral screening cascades are often based on cell viability measured on primary infection [
41,
42] or replicon systems incapable of producing viable virus progeny [
43]. Establishing an image-based phenotypic screening assay using infectious virus in biosafety laboratories allowed us to monitor all steps of the infection cycle. Additionally, follow-up study on virus titer enabled us to distinguish antiviral compounds that poorly inhibited infection in already infected cells, but significantly reduced viral progeny release and thereby virus transmission. Moreover, the developed image-based phenotypic assays provide a single-cell resolution and a detailed overview of the entire heterogeneous cell population instead of pooled population averages when using cell viability-based readouts. Image-based screenings have possibilities to uncover compound mechanistic insights when expanded to multidimensional phenotypic profiling [
44].
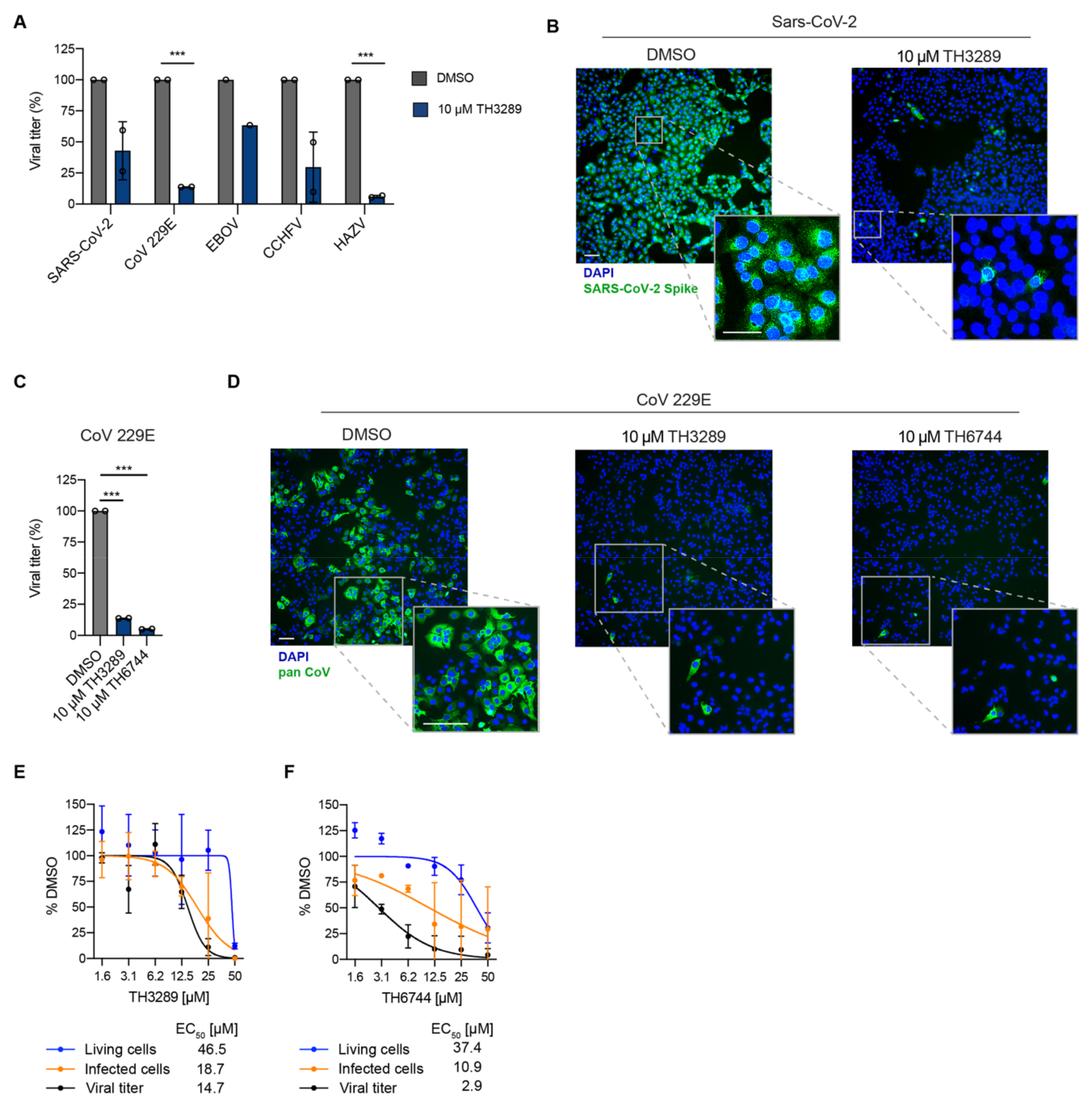
Using the newly developed antiviral assay we first identified a small molecule inhibitor TH3289 as one of the top hit inhibiting HAZV progeny release. Upon the second screening round, a close analogue of TH3289 TH6744 was identified. Both TH3289 and TH6744 reduced HAZV titer to similar effect at 10-fold lower dose as marketed antiviral drug Ribavirin that has been previously shown to have in vitro activity against SARS-CoV-2 [
45] and CCHFV [
46] as well as in a clinical trial setting in combination with other antivirals in COVID-19 patients [
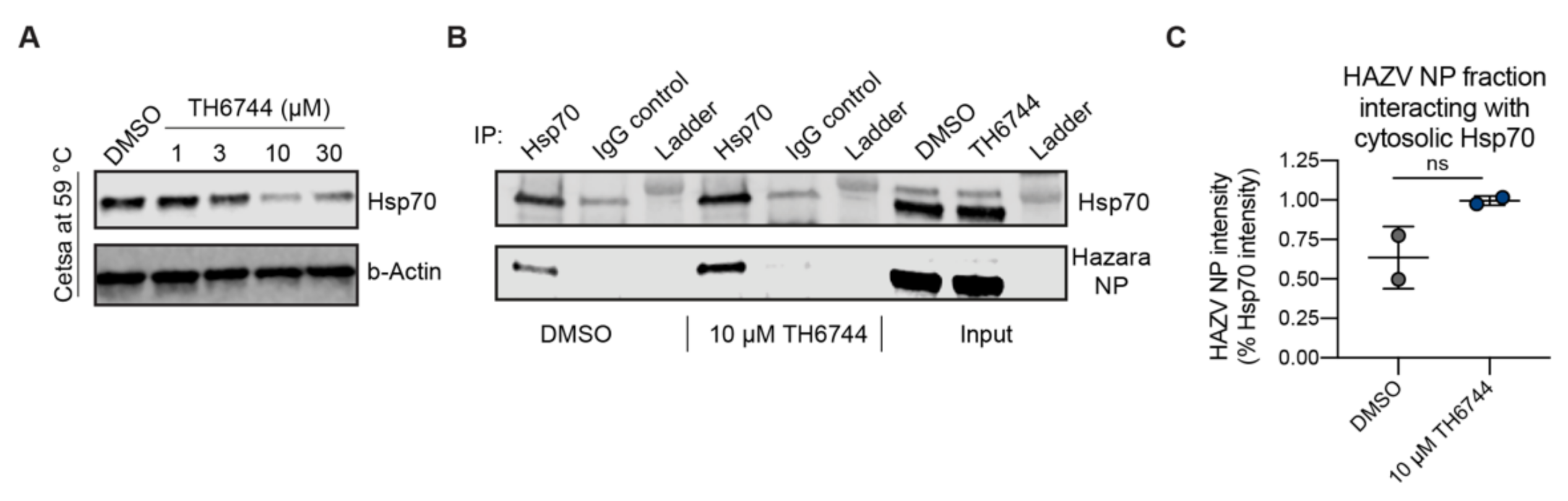
47]. Interestingly, TH3289 had antiviral activity against all tested viruses, SARS-CoV-2, EBOV, CCHFV and CoV 229E, confirming its broad antiviral activity. TH3289 exhibited higher activity against CoV 229E, HAZV and CCHFV than EBOV and SARS-CoV-2, probably due to the differential roles of TH3289 target pathways in virus families. Given that testing compound activity in BSL3 and BSL4 laboratories were limited to the initial screening hit TH3289, the activity of TH6744 was validated in BSL2 setting on CoV 229E and had a significant antiviral activity. Further, high-throughput screenings using single compound doses require a validation of hit compounds. To validate the antiviral effect of our compounds, a dose-dependent activity of TH6744 and TH3289 was evaluated on HAZV primary infection, HAZV titer and cell nuclei. TH6744 exhibited better antiviral potency at non-toxic concentrations compared to TH3289 in a dose-response assay and was therefore chosen for follow-up target identification studies.
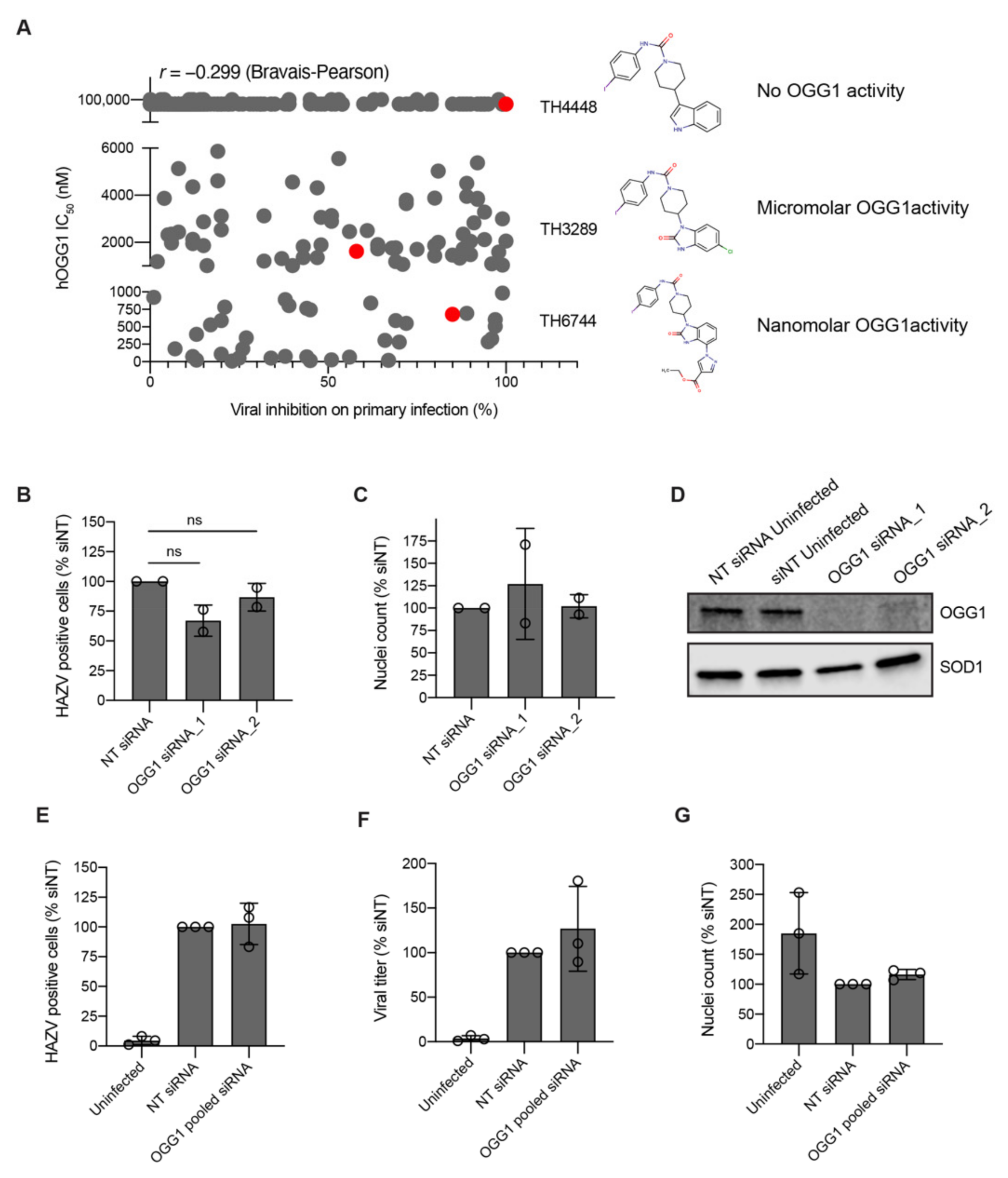
A challenge of phenotypic screening approaches is the lack of information about the target of identified compounds. TH6744 was developed as a part of a compound series targeting OGG1 and considering the link between oxidative stress and virus infection [
19,
20,
21], we investigated the role of OGG1 inhibition on compounds’ antiviral properties. Surprisingly, we could not confirm OGG1 inhibition as the driver for the antiviral effect, pointing towards a multifactorial mechanism of action. It is widely appreciated that small molecule drugs often perform by poly-pharmacological activity [
48]. Advanced proteomic techniques have recently been developed to study the effects of drug responses and to reveal compounds’ direct target and off-targets [
32,
49]. Additionally, TPP method has been used to study changes in cellular pathways in response to infection [
50]. We implemented two distinct TPPs in living cells upon TH6744 treatment, shedding light on compound’s targets, secondary responses and effects on protein pathways. TPP-TR identified a plethora of host proteins responding to TH6744 treatment and therefore we applied 2D-TPP to distinguish proteins that bind to TH6744 in a dose-dependent manner. Overlapping stabilized and destabilized proteins from two TPP analyses show strong enrichment of the host chaperone and co-chaperone network. The most significantly stabilized protein in TPP-TR, co-chaperone DNAJB11, has been described to localize to Dengue virus induced replication complexes in ER promoting vRNA synthesis [
51] and to mediate Simian Virus 40 capsid disassembly from the ER [
52], illustrating DNAJB11 supportive role for enveloped RNA and non-enveloped DNA viruses. Additionally, we observed uniform stabilization between proteostasis ER residents such as co-chaperones DNAJC13, DNAJC3, EDEM3, chaperones HSP90B1, HYOU1 and CALR and destabilization between cytosolic chaperones such as HSPA1A, HSPA2 and HSPA8. Interestingly, a regulator protein of ERAD, EDEM3 was shown to interact with SARS-Cov2 proteins Orf8 and Orf9b [
53]. Our findings imply that TH6744 affects multiple components of a chaperone network which RNA viruses heavily depend on for propagation [
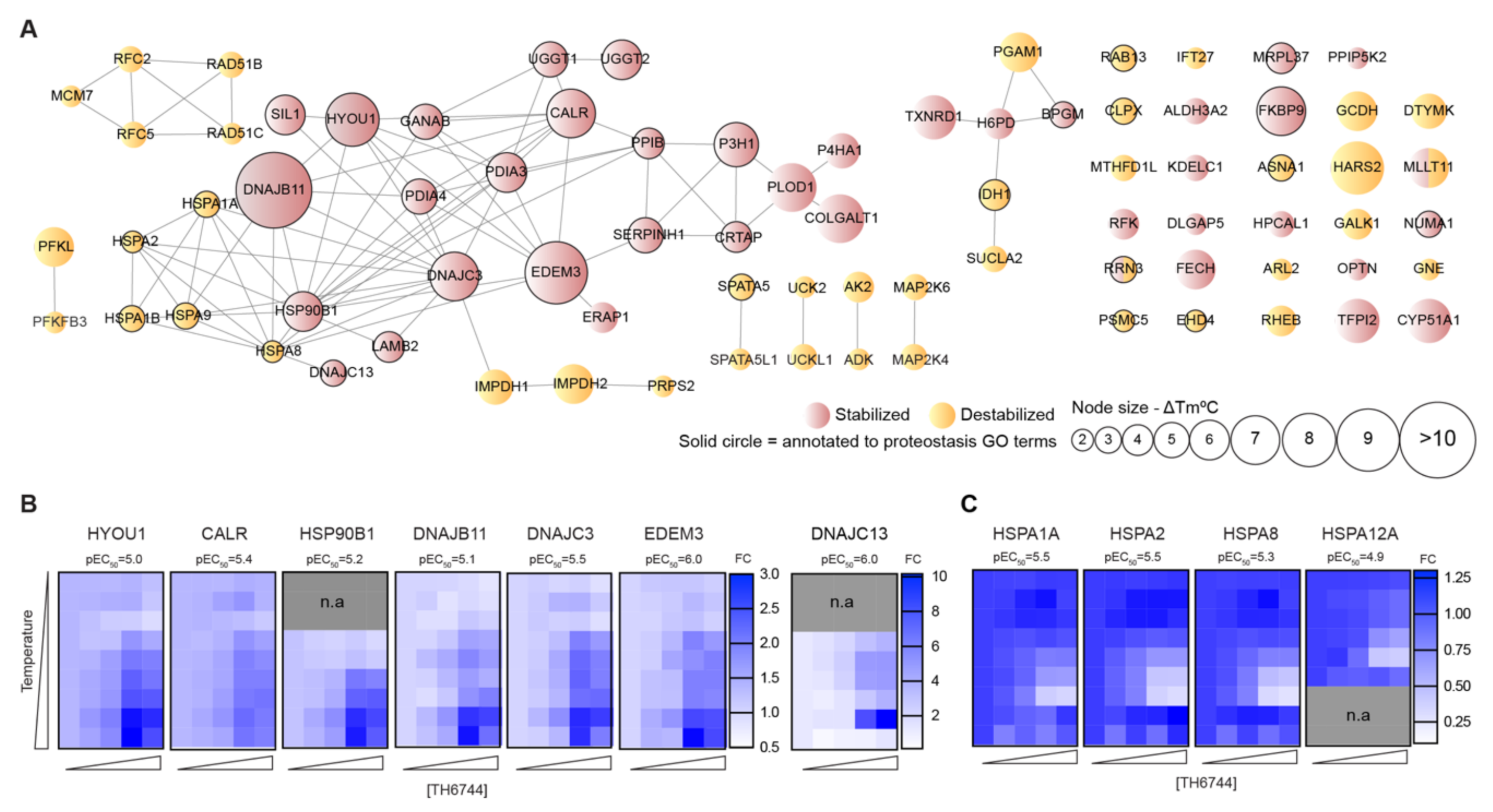
54]. Structural similarities between TH3289 and TH6744 and antiviral phenotypes observed with both compounds suggest that TH3289 targeting same host targets as TH6744. Further target identification studies are needed to confirm TH3289’s target proteins.
Inhibiting the host protein folding machinery has been proposed as a promising broad antiviral strategy [
55] and during recent years, Hsp70 have been validated as a promising antiviral target [
56]. HSP family members possess distinct functions in various steps of RNA and DNA viruses such as supporting CCHFV replication [
39]. We thereby chose to validate Hsp70 as one of TH6744 targets. We confirmed previously reported interaction between HAZV NP and Hsp70 [
39] and show an increase of Hsp70–HAZV NP interaction after TH6744 treatment. Additionally, we validated dose-dependent thermal destabilization of Hsp70 by TH6744 using CETSA, illustrating how TH6744 sensitizes Hsp70 protein to heat-induced denaturation. Based on thermal destabilization of cytosolic Hsp70 and stabilization of numerous co-chaperones, we propose that TH6744 binds to co-chaperones of Hsp70 multiprotein complexes resulting in Hsp70 thermal destabilization and thereby impairing the function of Hsp70. Increased Hsp70–HAZV NP interaction can reflect higher levels of aggregated HAZV NP that cannot be folded due to high viral protein production and compromised Hsp70 folding capacity. We further suggest that potent reduction of HAZV progeny release after TH6744 treatment is due to reduced Hsp70 folding capacity leading to impaired infectivity while being well tolerated by the host cell. Compounds’ antiviral properties or binding targets have not been optimized and thus their antiviral activity is in micromolar range and they bind to many host proteins, as expected from early hit compounds from phenotypic screening. However, these initial hit compounds can be used as tool compounds to study their antiviral activity and to identify their target proteins revealing new host–target interactions. Collectively, numerous target candidates from TPP remain to be further validated and future studies will provide understanding on the direct and indirect TH6744 targets.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





