
Small-Angle X-ray Scattering Models of APOBEC3B Catalytic Domain in a Complex with a Single-Stranded DNA Inhibitor

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of 2′-Deoxyzebularine (dZ) Containing Oligodeoxynucleotide
2.2. Protein Expression and Purification of A3BCTD* Protein
2.3. Fluorescence-Based Deaminase in-Gel Activity Assay
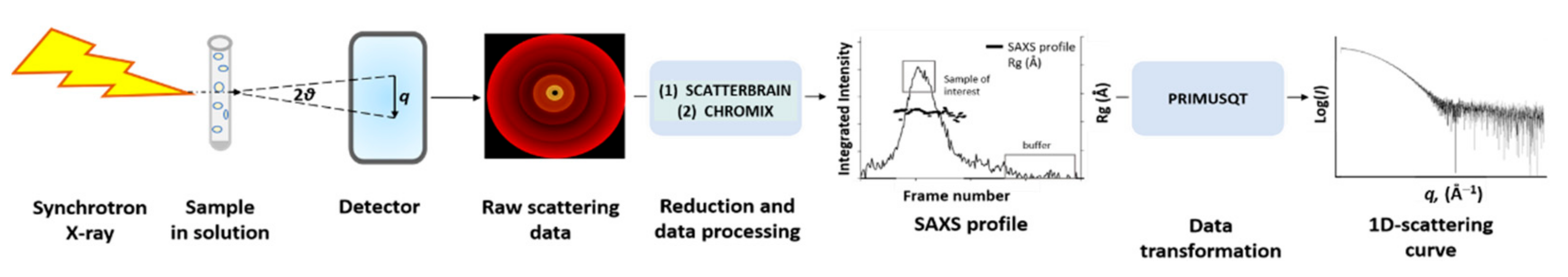
2.4. Small Angle X-ray Scattering (SAXS) Setup
2.5. Analysis of SAXS Data
3. Results
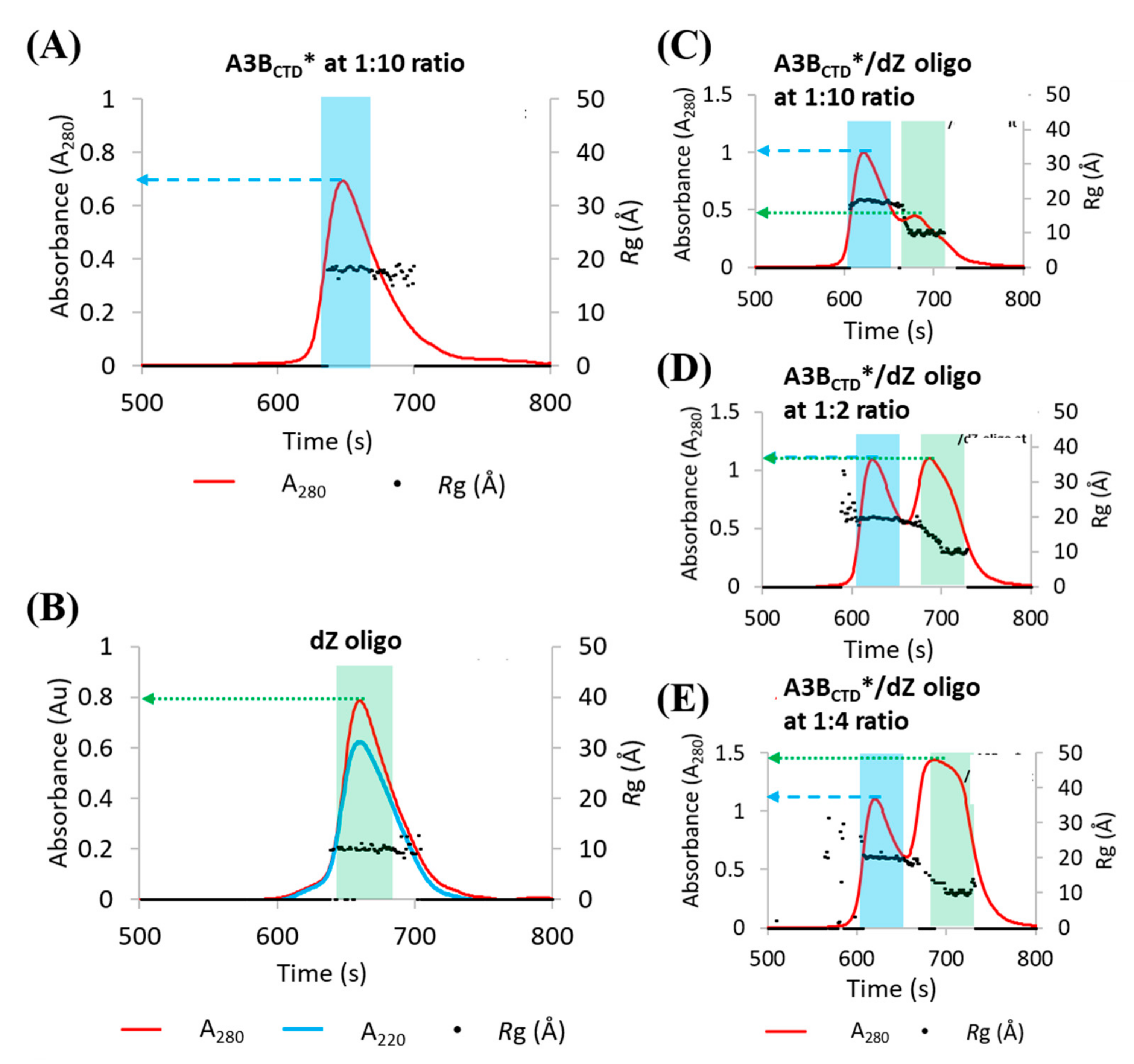
3.1. Methodology of Investigation: Using SAXS to Elucidate the Solution-State Structure of A3BCTD* in Complex with a ssDNA Inhibitor
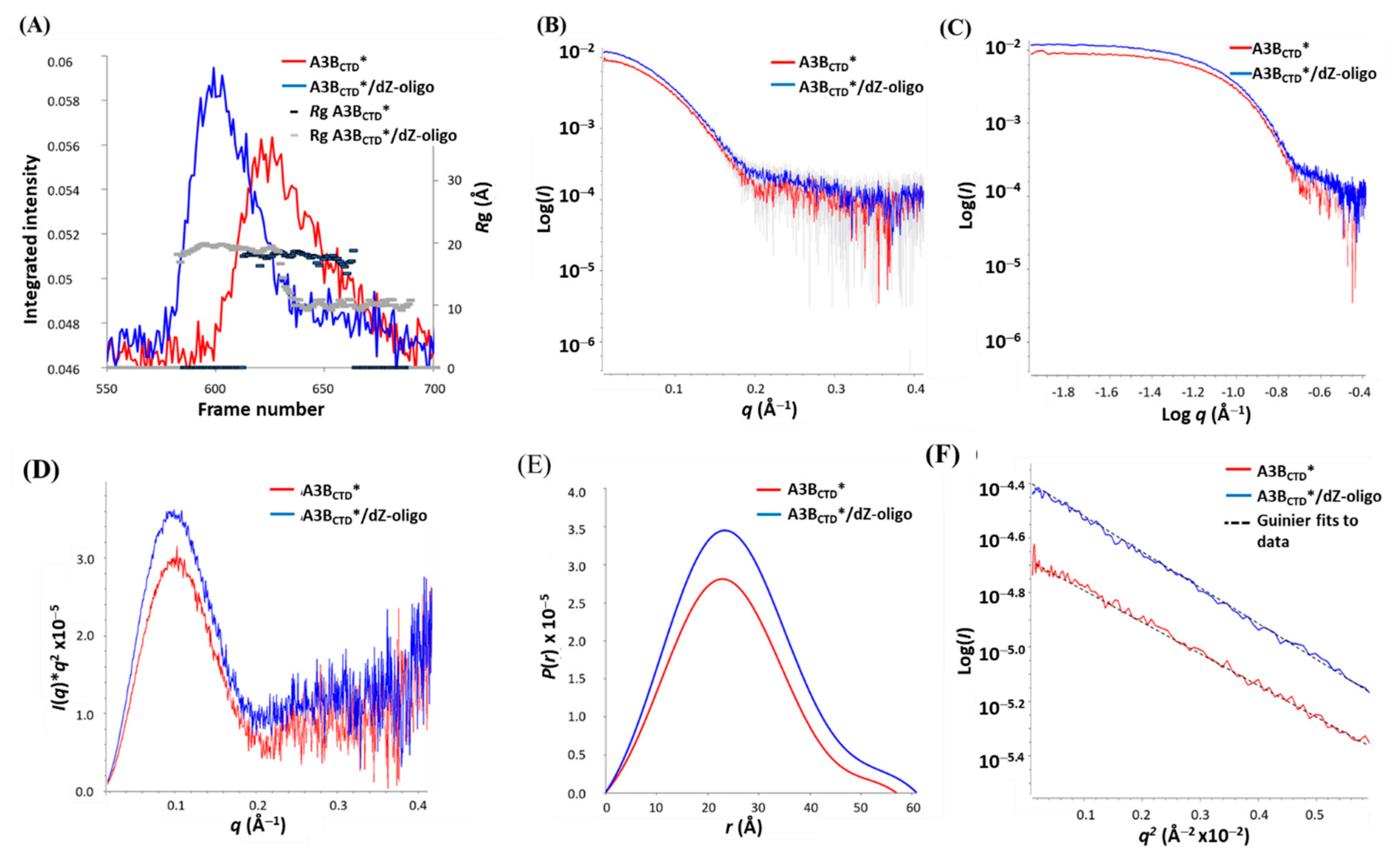
3.2. SAXS Profile Analysis of A3BCTD* Alone and in Complex with dZ-Oligo in Solution
3.3. SAXS Model Structures of A3BCTD* Alone and in Complex with dZ-oligo
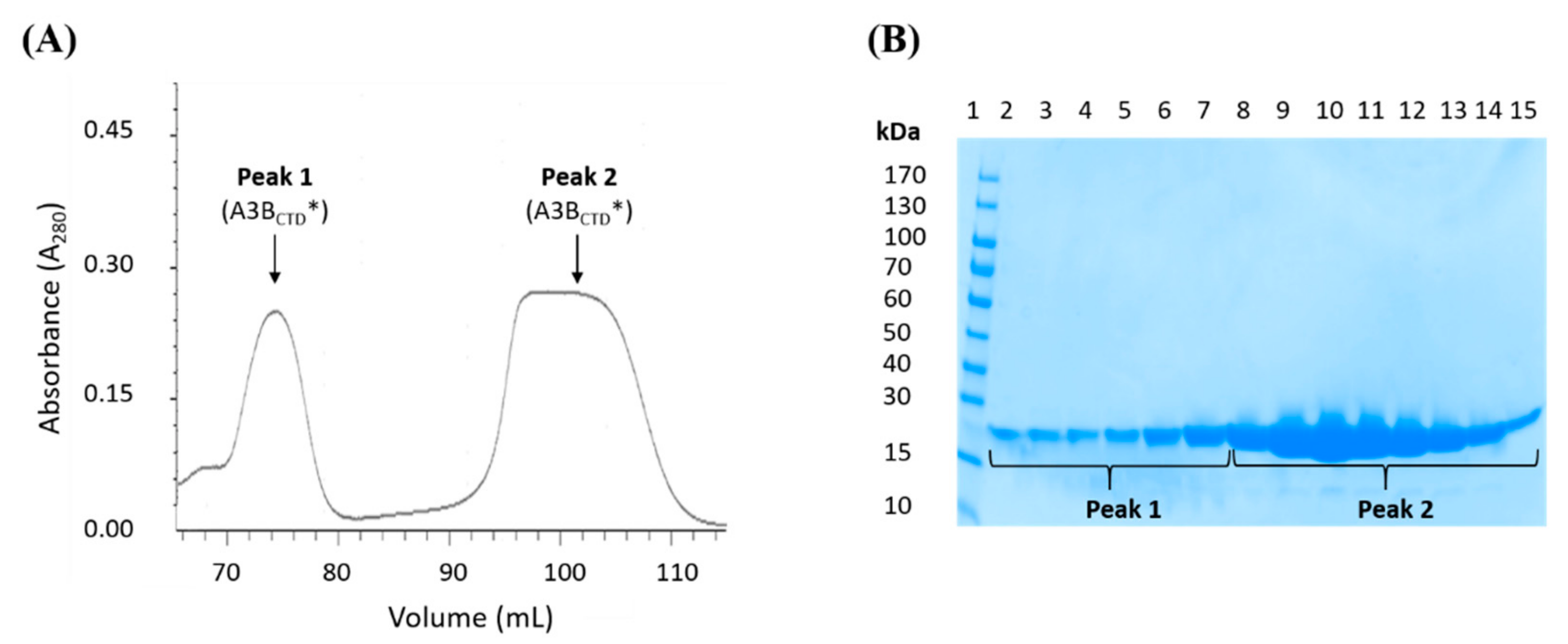
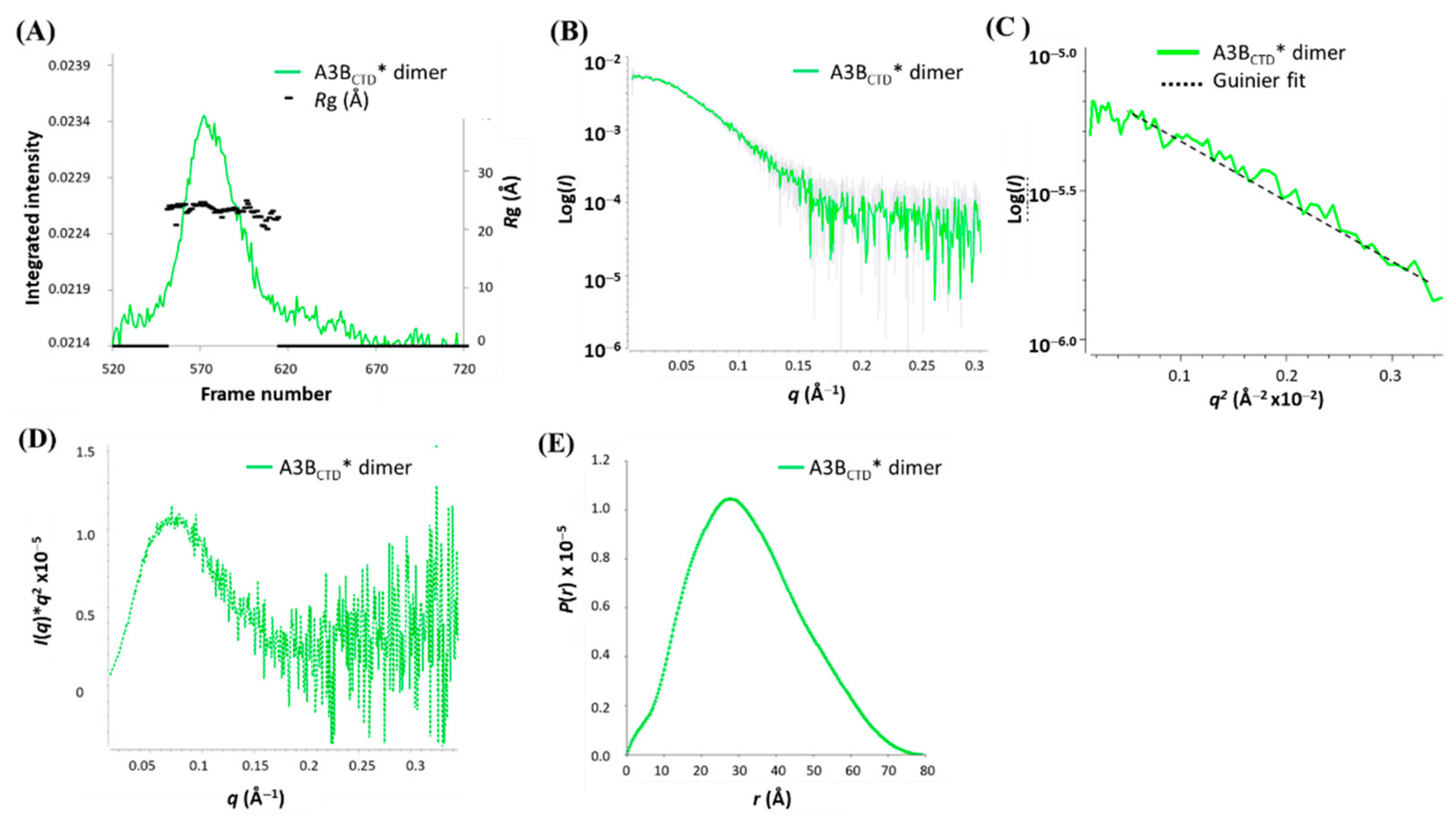
3.4. Multimerization of A3BCTD* in Solution
3.5. Interface Prediction of the A3BCTD* Dimer
3.6. SAXS Model Structure of the A3BCTD* Dimer
3.7. Evaluation of A3BCTD* Dimer with dZ-Oligo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA De-amination Mediates Innate Immunity to Retroviral Infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Refsland, E.W.; Harris, R.S. The APOBEC3 Family of Retroelement Restriction Factors. In Intrinsic Immunity; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–27. [Google Scholar]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.S.; Liddament, M.T. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 2004, 4, 868–877. [Google Scholar] [CrossRef]
- De Maio, N.; Walker, C.R.; Turakhia, Y.; Lanfear, R.; Corbett-Detig, R.; Goldman, N. Mutation rates and selection on synonymous mutations in SARS-CoV-2. bioRxiv 2021. [Google Scholar] [CrossRef]
- Klimczak, L.J.; Randall, T.A.; Saini, N.; Li, J.L.; Gordenin, D.A. Similarity between mutation spectra in hypermutated ge-nomes of rubella virus and in SARS-CoV-2 genomes accumulated during the COVID-19 pandemic. PLoS ONE 2020, 15, e0237689. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Rampant C→U Hypermutation in the Genomes of SARS-CoV-2 and Other Coronaviruses: Causes and Consequences for Their Short- and Long-Term Evolutionary Trajectories. mSphere 2020, 5, e00408-20. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nat. Cell Biol. 2013, 494, 366–370. [Google Scholar] [CrossRef]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, E.K.; Sieuwerts, A.M.; LaPara, K.; Leonard, B.; Starrett, G.J.; Molan, A.M.; Temiz, N.A.; Vogel, R.I.; Gelder, M.E.M.-V.; Sweep, F.C.G.J.; et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Adv. 2016, 2, e1601737. [Google Scholar] [CrossRef] [Green Version]
- Sieuwerts, A.M.; Willis, S.; Burns, M.B.; Look, M.P.; Gelder, M.E.M.-V.; Schlicker, A.; Heideman, M.R.; Jacobs, H.; Wessels, L.; Leyland-Jones, B.; et al. Elevated APOBEC3B Correlates with Poor Outcomes for Estrogen-Receptor-Positive Breast Cancers. Horm. Cancer 2014, 5, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Chang, C.-J.; Xie, X.; Xia, W.; Yang, J.-Y.; Wang, S.-C.; Wang, Y.; Xia, J.; Chen, L.; Cai, C.; et al. APOBEC3G promotes liver metastasis in an orthotopic mouse model of colorectal cancer and predicts human hepatic metastasis. J. Clin. Investig. 2011, 121, 4526–4536. [Google Scholar] [CrossRef]
- Swanton, C.; McGranahan, N.; Starrett, G.J.; Harris, R.S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015, 5, 704–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.S. Cancer mutation signatures, DNA damage mechanisms, and potential clinical implications. Genome Med. 2013, 5, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaRue, R.S.; Jónsson, S.R.; Silverstein, K.A.; Lajoie, M.; Bertrand, D.; El-Mabrouk, N.; Hötzel, I.; Andrésdóttir, V.; Smith, T.P.; Harris, R.S. The artiodactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol. Biol. 2008, 9, 104. [Google Scholar] [CrossRef] [Green Version]
- LaRue, R.S.; Andrésdóttir, V.; Blanchard, Y.; Conticello, S.G.; Derse, D.; Emerman, M.; Greene, W.C.; Jónsson, S.R.; Landau, N.R.; Löchelt, M.; et al. Guidelines for Naming Nonprimate APOBEC3 Genes and Proteins. J. Virol. 2008, 83, 494–497. [Google Scholar] [CrossRef] [Green Version]
- Conticello, S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2009, 9, 229. [Google Scholar] [CrossRef] [Green Version]
- Siriwardena, S.U.; Guruge, T.A.; Bhagwat, A.S. Characterization of the Catalytic Domain of Human APOBEC3B and the Critical Structural Role for a Conserved Methionine. J. Mol. Biol. 2015, 427, 3042–3055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Ito, F.; Zhang, G.; Fernandez, B.; Yang, H.; Chen, X.S. DNA cytosine and methylcytosine deamination by APOBEC3B: Enhancing methylcytosine deamination by engineering APOBEC3B. Biochem. J. 2015, 471, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Bonvin, M.; Greeve, J.; Chang, Q.; McLinden, J.H.; Stapleton, J.T.; Sathar, M.A.; Xiang, J. Effects of point mutations in the cytidine deaminase domains of APOBEC3B on replication and hypermutation of hepatitis B virus in vitro. J. Gen. Virol. 2007, 88, 3270–3274. [Google Scholar] [CrossRef]
- Haché, G.; Liddament, M.T.; Harris, R.S. The Retroviral Hypermutation Specificity of APOBEC3F and APOBEC3G Is Governed by the C-terminal DNA Cytosine Deaminase Domain. J. Biol. Chem. 2005, 280, 10920–10924. [Google Scholar] [CrossRef] [Green Version]
- Hakata, Y.; Landau, N.R. Reversed functional organization of mouse and human APOBEC3 cytidine deaminase domains. J. Biol. Chem. 2006, 281, 36624–36631. [Google Scholar] [CrossRef] [Green Version]
- Navarro, F.; Bollman, B.; Chen, H.; König, R.; Yu, Q.; Chiles, K.; Landau, N.R. Complementary function of the two catalytic domains of APOBEC3G. Virology 2005, 333, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Newman, E.N.; Holmes, R.K.; Craig, H.M.; Klein, K.C.; Lingappa, J.R.; Malim, M.H.; Sheehy, A.M. Antiviral function of APO-BEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 2005, 15, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Bohn, M.-F.; Shandilya, S.M.; Albin, J.S.; Kouno, T.; Anderson, B.D.; McDougle, R.M.; Carpenter, M.A.; Rathore, A.; Evans, L.; Davis, A.N. Crystal structure of the DNA cytosine deaminase APOBEC3F: The catalytically active and HIV-1 Vif-binding do-main. Structure 2013, 21, 1042–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.-M.; Harjes, E.; Gross, P.J.; Fahmy, A.; Lu, Y.; Shindo, K.; Harris, R.S.; Matsuo, H. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 2008, 452, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.-P.; Lin, J.-J.; Hu, C.-Y.; Hsu, Y.-H.; Wang, A.H.-J.; Liaw, S.-H. Crystal structure of yeast cytosine deaminase insights into enzyme mechanism and evolution. J. Biol. Chem. 2003, 278, 19111–19117. [Google Scholar] [CrossRef] [Green Version]
- Conticello, S.G.; Thomas, C.J.; Petersen-Mahrt, S.K.; Neuberger, M.S. Evolution of the AID/APOBEC family of polynucleotide (deoxy) cytidine deaminases. Mol. Biol. Evol. 2005, 22, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Jarmuz, A.; Chester, A.; Bayliss, J.; Gisbourne, J.; Dunham, I.; Scott, J.; Navaratnam, N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 2002, 79, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Carpenter, M.A.; Banerjee, S.; Shaban, N.M.; Kurahashi, K.; Salamango, D.J.; McCann, J.L.; Starrett, G.J.; Duffy, J.V.; Demir, Ö. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017, 24, 131. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, M.A.; Rajagurubandara, E.; Wijesinghe, P.; Bhagwat, A.S. Determinants of sequence-specificity within human AID and APOBEC3G. DNA Repair 2010, 9, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Kohli, R.M.; Abrams, S.R.; Gajula, K.S.; Maul, R.W.; Gearhart, P.J.; Stivers, J.T. A portable hot spot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. J. Biol. Chem. 2009, 284, 22898–22904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, R.M.; Maul, R.W.; Guminski, A.F.; McClure, R.L.; Gajula, K.S.; Saribasak, H.; McMahon, M.A.; Siliciano, R.F.; Gearhart, P.J.; Sti-vers, J.T. Local sequence targeting in the AID/APOBEC family differentially impacts retroviral restriction and anti-body diversification. J. Biol. Chem. 2010, 285, 40956–40964. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Rada, C.; Neuberger, M.S. Altering the spectrum of immunoglobulin V gene somatic hypermutation by modifying the active site of AID. J. Exp. Med. 2010, 207, 141–153. [Google Scholar] [CrossRef] [PubMed]
- McDougall, W.M.; Okany, C.; Smith, H.C. Deaminase Activity on Single-stranded DNA (ssDNA) Occurs in Vitro when APOBEC3G Cytidine Deaminase Forms Homotetramers and Higher-order Complexes*. J. Biol. Chem. 2011, 286, 30655–30661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prohaska, K.M.; Bennett, R.P.; Salter, J.D.; Smith, H.C. The multifaceted roles of RNA binding in APOBEC cytidine de-aminase functions. Wiley Interdiscip. Rev. RNA 2014, 5, 493–508. [Google Scholar] [CrossRef] [Green Version]
- Salter, J.D.; Bennett, R.P.; Smith, H.C. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem. Sci. 2016, 41, 578–594. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.C. RNA binding to APOBEC deaminases; Not simply a substrate for C to U editing. RNA Biol. 2017, 14, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, X.; Tian, C.; Wang, T.; Sarkis, P.T.N.; Fang, Y.; Zheng, S.; Yu, X.-F.; Xu, R. Cytidine deaminase APOBEC3B interacts with heterogeneous nuclear ribonucleoprotein K and suppresses hepatitis B virus expression. Cell. Microbiol. 2007, 10, 112–121. [Google Scholar] [CrossRef]
- Chelico, L.; Pham, P.; Calabrese, P.; Goodman, M.F. APOBEC3G DNA deaminase acts processively 3′→ 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 2006, 13, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Holden, L.G.; Prochnow, C.; Chang, Y.P.; Bransteitter, R.; Chelico, L.; Sen, U.; Stevens, R.C.; Goodman, M.F.; Chen, X.S. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nat. Cell Biol. 2008, 456, 121–124. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.; Li, M.; Carpenter, M.A.; McDougle, R.M.; Luengas, E.M.; Macdonald, P.J.; Harris, R.S.; Mueller, J.D. APOBEC3 Multimerization Correlates with HIV-1 Packaging and Restriction Activity in Living Cells. J. Mol. Biol. 2014, 426, 1296–1307. [Google Scholar] [CrossRef] [Green Version]
- Gallois-Montbrun, S.; Holmes, R.K.; Swanson, C.M.; Fernández-Ocaña, M.; Byers, H.L.; Ward, M.A.; Malim, M.H. Comparison of cellular ribonucleoprotein complexes associated with the APOBEC3F and APOBEC3G antiviral proteins. J. Virol. 2008, 82, 5636–5642. [Google Scholar] [CrossRef] [Green Version]
- Cortez, L.M.; Brown, A.L.; Dennis, M.A.; Collins, C.D.; Brown, A.J.; Mitchell, D.; Mertz, T.M.; Roberts, S.A. APOBEC3A is a prominent cytidine deaminase in breast cancer. PLoS Genet. 2019, 15, e1008545. [Google Scholar] [CrossRef] [Green Version]
- Lau, P.P.; Zhu, H.-J.; Baldini, A.; Charnsangavej, C.; Chan, L. Dimeric structure of a human apolipoprotein B mRNA ed-iting protein and cloning and chromosomal localization of its gene. Proc. Natl. Acad. Sci. USA 1994, 91, 8522–8526. [Google Scholar] [CrossRef] [Green Version]
- Prochnow, C.; Bransteitter, R.; Klein, M.G.; Goodman, M.F.; Chen, X.S. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nat. Cell Biol. 2007, 445, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Wedekind, J.E.; Gillilan, R.; Janda, A.; Krucinska, J.; Salter, J.D.; Bennett, R.P.; Raina, J.; Smith, H.C. Nanostructures of APO-BEC3G support a hierarchical assembly model of high molecular mass ribonucleoprotein particles from dimeric subunits. J. Biol. Chem. 2006, 281, 38122–38126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brar, S.S.; Sacho, E.J.; Tessmer, I.; Croteau, D.L.; Erie, D.A.; Diaz, M. Activation-induced deaminase, AID, is catalytically active as a monomer on single-stranded DNA. DNA Repair 2008, 7, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, A.; Nagata, T.; Matsugami, A.; Habu, Y.; Sugiyama, R.; Hayashi, F.; Kobayashi, N.; Yokoyama, S.; Takaku, H.; Katahira, M. Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 2009, 28, 440–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, S.; Ode, H.; Nakashima, M.; Imahashi, M.; Naganawa, Y.; Kurosawa, T.; Yokomaku, Y.; Yamane, T.; Watanabe, N.; Suzuki, A.; et al. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat. Struct. Mol. Biol. 2012, 19, 1005–1010. [Google Scholar] [CrossRef]
- Shandilya, S.M.D.; Nalam, M.N.L.; Nalivaika, E.A.; Gross, P.J.; Valesano, J.C.; Shindo, K.; Li, M.; Munson, M.; Royer, W.E.; Harjes, E.; et al. Crystal Structure of the APOBEC3G Catalytic Domain Reveals Potential Oligomerization Interfaces. Structure 2010, 18, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Bohn, M.-F.; Shandilya, S.M.; Silvas, T.V.; Nalivaika, E.A.; Kouno, T.; Kelch, B.A.; Ryder, S.P.; Kurt-Yilmaz, N.; Somasundaran, M.; Schiffer, C.A. The ssDNA Mutator APOBEC3A Is Regulated by Cooperative Dimerization. Structure 2015, 23, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouno, T.; Silvas, T.V.; Hilbert, B.J.; Shandilya, S.M.D.; Bohn, M.F.; Kelch, B.A.; Royer, W.E.; Somasundaran, M.; Yilmaz, N.K.; Matsuo, H.; et al. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 2017, 8, 15024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, A.; Myint, W.; Kanai, T.; Delviks-Frankenberry, K.; Rodriguez, C.S.; Pathak, V.K.; Schiffer, C.A.; Matsuo, H. Crystal structure of the catalytic domain of HIV-1 restriction factor APOBEC3G in complex with ssDNA. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Harjes, S.; Jameson, G.B.; Filichev, V.V.; Edwards, P.J.B.; Harjes, E. NMR-based method of small changes reveals how DNA mutator APOBEC3A interacts with its single-stranded DNA substrate. Nucleic Acids Res. 2017, 45, 5602–5613. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Shandilya, S.M.D.; Carpenter, M.A.; Rathore, A.; Brown, W.L.; Perkins, A.L.; Harki, D.A.; Solberg, J.; Hook, D.J.; Pandey, K.K.; et al. First-In-Class Small Molecule Inhibitors of the Single-Strand DNA Cytosine Deaminase APOBEC3G. ACS Chem. Biol. 2012, 7, 506–517. [Google Scholar] [CrossRef] [Green Version]
- Byeon, I.-J.L.; Ahn, J.; Mitra, M.; Byeon, C.-H.; Herc, K.; Hritz, J.; Charlton, L.M.; Levin, J.G.; Gronenborn, A.M. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat. Commun. 2013, 4, 1890. [Google Scholar] [CrossRef] [PubMed]
- Harjes, S.; Solomon, W.C.; Li, M.; Chen, K.-M.; Harjes, E.; Harris, R.S.; Matsuo, H. Impact of H216 on the DNA Binding and Catalytic Activities of the HIV Restriction Factor APOBEC3G. J. Virol. 2013, 87, 7008–7014. [Google Scholar] [CrossRef] [Green Version]
- Kvach, M.V.; Barzak, F.M.; Harjes, S.; Schares, H.A.; Jameson, G.B.; Ayoub, A.M.; Moorthy, R.; Aihara, H.; Harris, R.S.; Filichev, V.V. Inhibiting APOBEC3 activity with single-stranded DNA containing 2′-deoxyzebularine analogs. Biochemistry 2019, 58, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.; Hercík, K.; Byeon, I.-J.L.; Ahn, J.; Hill, S.; Hinchee-Rodriguez, K.; Singer, D.; Byeon, C.-H.; Charlton, L.M.; Nam, G.; et al. Structural determinants of human APOBEC3A enzymatic and nucleic acid binding properties. Nucleic Acids Res. 2014, 42, 1095–1110. [Google Scholar] [CrossRef] [Green Version]
- Barzak, F.M.; Harjes, S.; Kvach, M.V.; Kurup, H.M.; Jameson, G.B.; Filichev, V.V.; Harjes, E. Selective inhibition of APOBEC3 enzymes by single-stranded DNAs containing 2′-deoxyzebularine. Org. Biomol. Chem. 2019, 17, 9435–9441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, K.; Carpenter, M.A.; Kurahashi, K.; Harris, R.S.; Aihara, H. Crystal structure of the DNA deaminase APOBEC3B catalytic domain. J. Biol. Chem. 2015, 290, 28120–28130. [Google Scholar] [CrossRef] [Green Version]
- Summer, H.; Grämer, R.; Dröge, P. Denaturing Urea Polyacrylamide Gel Electrophoresis (Urea PAGE). J. Vis. Exp. 2009, e1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, T.M.; Trewhella, J.; Murphy, J.M.; Keown, J.R.; Casey, L.; Pearce, F.G.; Goldstone, D.C.; Chen, K.; Luo, Z.; Kobe, B. An op-timized SEC-SAXS system enabling high X-ray dose for rapid SAXS assessment with correlated UV measurements for biomolecular structure analysis. J. Appl. Crystallogr. 2018, 51, 97–111. [Google Scholar] [CrossRef]
- Kirby, N.M.; Cowieson, N.P.; Hawley, A.M.; Mudie, S.T.; McGillivray, D.J.; Kusel, M.; Samardzic-Boban, V.; Ryan, T.M. Improved radiation dose efficiency in solution SAXS using a sheath flow sample environment. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Franke, D.; Petoukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, J.B.; Gillilan, R.E.; Skou, S. BioXTAS RAW: Improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 2017, 50, 1545–1553. [Google Scholar] [CrossRef] [Green Version]
- Trewhella, J.; Duff, A.P.; Durand, D.; Gabel, F.; Guss, J.M.; Hendrickson, W.A.; Hura, G.L.; Jacques, D.A.; Kirby, N.M.; Kwan, A.H.; et al. 2017 publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: An update. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 710–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambo, R.P.; Tainer, J.A. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 2011, 95, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Mylonas, E.; Svergun, D.I. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Crystallogr. 2007, 40, s245–s249. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.E.; Bothe, J.R. NMRFAM SAXS Data Processing and Modeling Manual; National Magnetic Resonance Facility at Madison: Madison, WI, USA, 2014. [Google Scholar]
- Kozin, M.B.; Svergun, D.I. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 2001, 34, 33–41. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.A.S.; Andrej, S. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016, 44, W424–W429. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.A.; Sali, A. Accurate SAXS Profile Computation and its Assessment by Contrast Variation Experiments. Biophys. J. 2013, 105, 962–974. [Google Scholar] [CrossRef] [Green Version]
- Betts, L.; Xiang, S.; Short, S.A.; Wolfenden, R.; Carter, C.W. Cytidine Deaminase. The 2·3 Å Crystal Structure of an Enzyme: Transition-state Analog Complex. J. Mol. Biol. 1994, 235, 635–656. [Google Scholar] [CrossRef]
- Xiang, S.; Short, S.A.; Wolfenden, R.; Carter, C.W., Jr. Transition-state selectivity for a single hydroxyl group during catalysis by cytidine deaminase. Biochemistry 1995, 34, 4516–4523. [Google Scholar] [CrossRef]
- Chung, S.J.; Fromme, J.C.; Verdine, G.L. Structure of Human Cytidine Deaminase Bound to a Potent Inhibitor. J. Med. Chem. 2005, 48, 658–660. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 2.1.1.; Schrödinger: New York, NY, USA, 2015. [Google Scholar]
- Bennett, R.P.; Salter, J.D.; Liu, X.; Wedekind, J.E.; Smith, H.C. APOBEC3G subunits self-associate via the C-terminal deaminase domain. J. Biol. Chem. 2008, 283, 33329–33336. [Google Scholar] [CrossRef] [Green Version]
- Kvach, M.V.; Barzak, F.M.; Harjes, S.; Schares, H.A.; Kurup, H.M.; Jones, K.F.; Sutton, L.; Donahue, J.; D’Aquila, R.T.; Jameson, G.B.; et al. Differential inhibition of APOBEC3 DNA-mutator isozymes by fluoro- and non-fluoro-substituted 2′-deoxyzebularine embedded in single-stranded DNA. ChemBioChem 2020, 21, 1028–1035. [Google Scholar] [CrossRef]
- Byeon, I.-J.L.; Byeon, C.-H.; Wu, T.; Mitra, M.; Singer, D.; Levin, J.G.; Gronenborn, A.M. Nuclear Magnetic Resonance Structure of the APOBEC3B Catalytic Domain: Structural Basis for Substrate Binding and DNA Deaminase Activity. Biochemistry 2016, 55, 2944–2959. [Google Scholar] [CrossRef] [Green Version]
- Tuncbag, N.; Gursoy, A.; Nussinov, R.; Keskin, O. Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat. Protoc. 2011, 6, 1341–1354. [Google Scholar] [CrossRef]
- Baspinar, A.; Cukuroglu, E.; Nussinov, R.; Keskin, O.; Gursoy, A. PRISM: A web server and repository for prediction of protein–protein interactions and modeling their 3D complexes. Nucleic Acids Res. 2014, 42, W285–W289. [Google Scholar] [CrossRef]
- Krissinel, E. Crystal contacts as nature’s docking solutions. J. Comput. Chem. 2010, 31, 133–143. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Protein interfaces, surfaces and assemblies service PISA at European Bioinformatics Institute. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Bechtel, T.J.; Weerapana, E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics 2017, 17, 1600391. [Google Scholar] [CrossRef] [Green Version]
- Chelico, L.; Sacho, E.J.; Erie, D.A.; Goodman, M.F. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J. Biol. Chem. 2008, 283, 13780–13791. [Google Scholar] [CrossRef] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Parameters | A3BCTD* | A3BCTD*/dZ-Oligo | A3BCTD* Dimer | Species 1 a (A3BCTD* Dimer) | Species 2 a (A3BCTD*/dZ-Oligo) |
|---|---|---|---|---|---|
| Guinier analysis | |||||
| I(0) (cm−1) | 0.00940 (5) | 0.01031 (5) | 0.00480 (4) | 0.0036 (1) | 0.00110 (5) |
| Rg (Å) | 18.8 (2) | 19.8 ± 0.2 | 24.2 (8) | 24.8 (1.5) | 20.0 (1.5) |
| qmin (Å−1) | 0.0106 | 0.0106 | 0.0165 | 0.01795 | 0.0231 |
| q·Rg max | 1.3 | 1.3 | 1.3 | 1.3 | 1.3 |
| Coeff. correln (R2) | 0.98 | 0.99 | 0.96 | 0.98 | 0.86 |
| P(r) analysis | |||||
| I(0) (cm−1) | 0.00930 (4) | 0.01020 (5) | 0.00480 (1) | 0.00352 (7) | 0.00110 (5) |
| Rg (Å) | 18.7 (1) | 19.7 (1) | 24.5 (6) | 25.1 (5) | 20.1 (8) |
| Dmax (Å) | 57 | 60 | 77.9 | 78.2 | 60.1 |
| q range (Å−1) | 0.011–0.354 | 0.011–0.354 | 0.016–0.345 | 0.0180–0.276 | 0.023- 0.232 |
| Quality estimate | 0.87 | 0.85 | 0.88 | 0.89 | 0.85 |
| Porod vol (Å3) | 35,600 | 36,700 | 72,900 | 74,000 | 38,400 |
| MW (kDa) | 21.4 (0.97) b | 22.0 (0.88) d | 43.7 (1.99) b | 44.4 (2.02) b | 23.0 (0.92) d |
| MW | 23.3 (1.06) c | 25.3 (1.01) e |
| Modelling Parameters | A3BCTD* | A3BCTD*/dZ-Oligo | A3BCTD* Dimer |
|---|---|---|---|
| Ab initio restoration | |||
| DAMMIF a | |||
| q range (Å−1) for fitting | 0.011–0.354 | 0.011–0.354 | 0.0016–0.345 |
| Symmetry | 1 (none) | 1 (none) | P2 |
| NSD (standard deviation) a | 0.559 (0.019) | 0.862 (0.037) | 0.816 (0.110) |
| Resoln (from SASRES) (Å) | 28 (2) | 29 (2) | 41 ± 3 |
| Structure modelling | |||
| q range for modelling (Å−1) | 0.011–0.302 | 0.011–0.302 | |
| PDB structure | 5CQI | 5TD5* b | Interface model 1 |
| FoXS, CRYSOL (Chi2) | 1.16, 1.99 | 1.51, 2.08 | 0.93, 1.49 |
| Interface model 2 | |||
| FoXS, CRYSOL (Chi2) | -- | -- | 0.80, 1.53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barzak, F.M.; Ryan, T.M.; Kvach, M.V.; Kurup, H.M.; Aihara, H.; Harris, R.S.; Filichev, V.V.; Harjes, E.; Jameson, G.B. Small-Angle X-ray Scattering Models of APOBEC3B Catalytic Domain in a Complex with a Single-Stranded DNA Inhibitor. Viruses 2021, 13, 290. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020290
Barzak FM, Ryan TM, Kvach MV, Kurup HM, Aihara H, Harris RS, Filichev VV, Harjes E, Jameson GB. Small-Angle X-ray Scattering Models of APOBEC3B Catalytic Domain in a Complex with a Single-Stranded DNA Inhibitor. Viruses. 2021; 13(2):290. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020290
Chicago/Turabian StyleBarzak, Fareeda M., Timothy M. Ryan, Maksim V. Kvach, Harikrishnan M. Kurup, Hideki Aihara, Reuben S. Harris, Vyacheslav V. Filichev, Elena Harjes, and Geoffrey B. Jameson. 2021. "Small-Angle X-ray Scattering Models of APOBEC3B Catalytic Domain in a Complex with a Single-Stranded DNA Inhibitor" Viruses 13, no. 2: 290. https://0-doi-org.brum.beds.ac.uk/10.3390/v13020290