
Host Components That Modulate the Disease Caused by hMPV

 , , and
, , and
Abstract
:1. Introduction: Human Metapneumovirus
1.1. The Disease Caused by hMPV
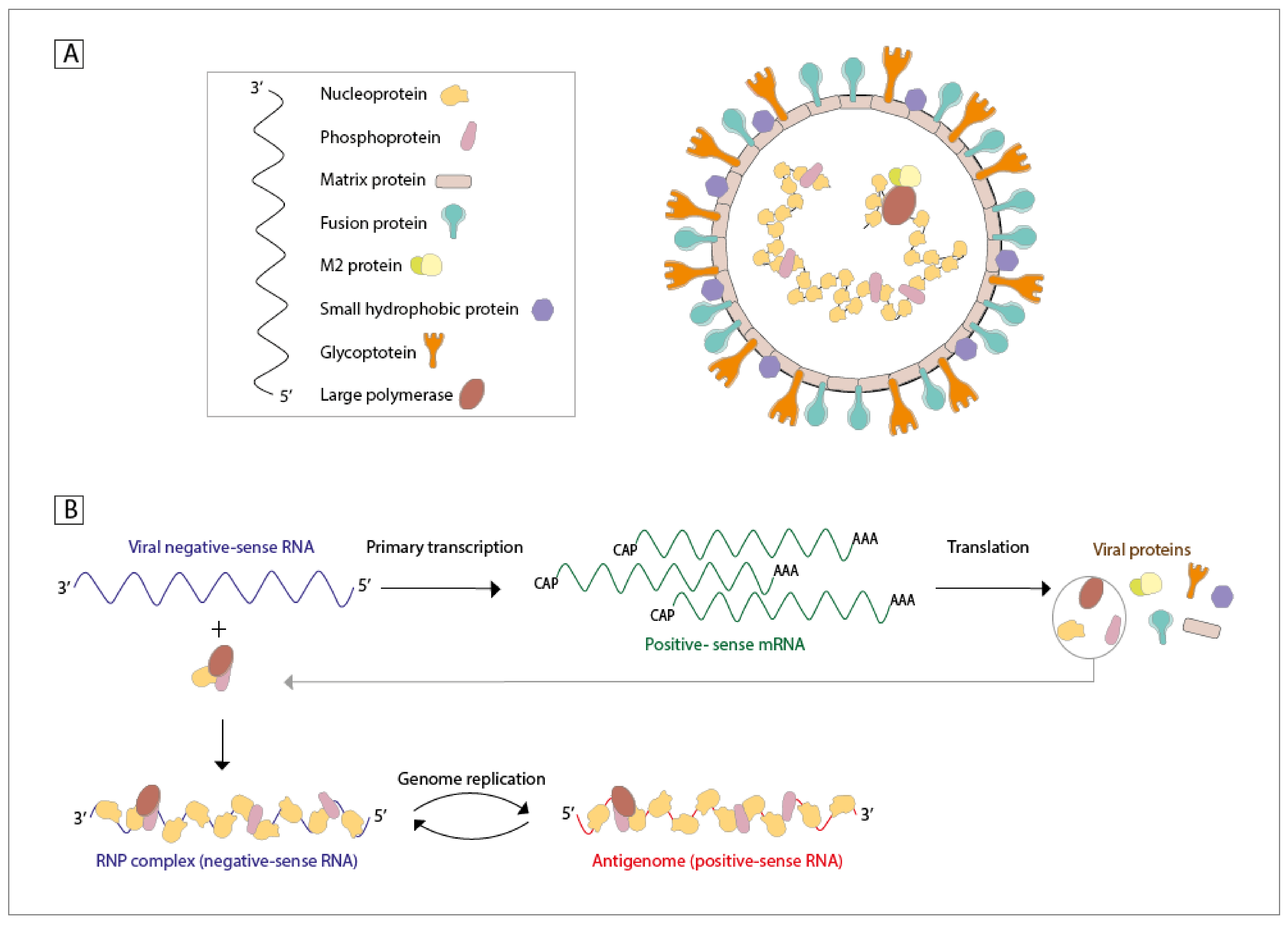
1.2. Proteins, RNA, Viral Structure, and Infective Cycle
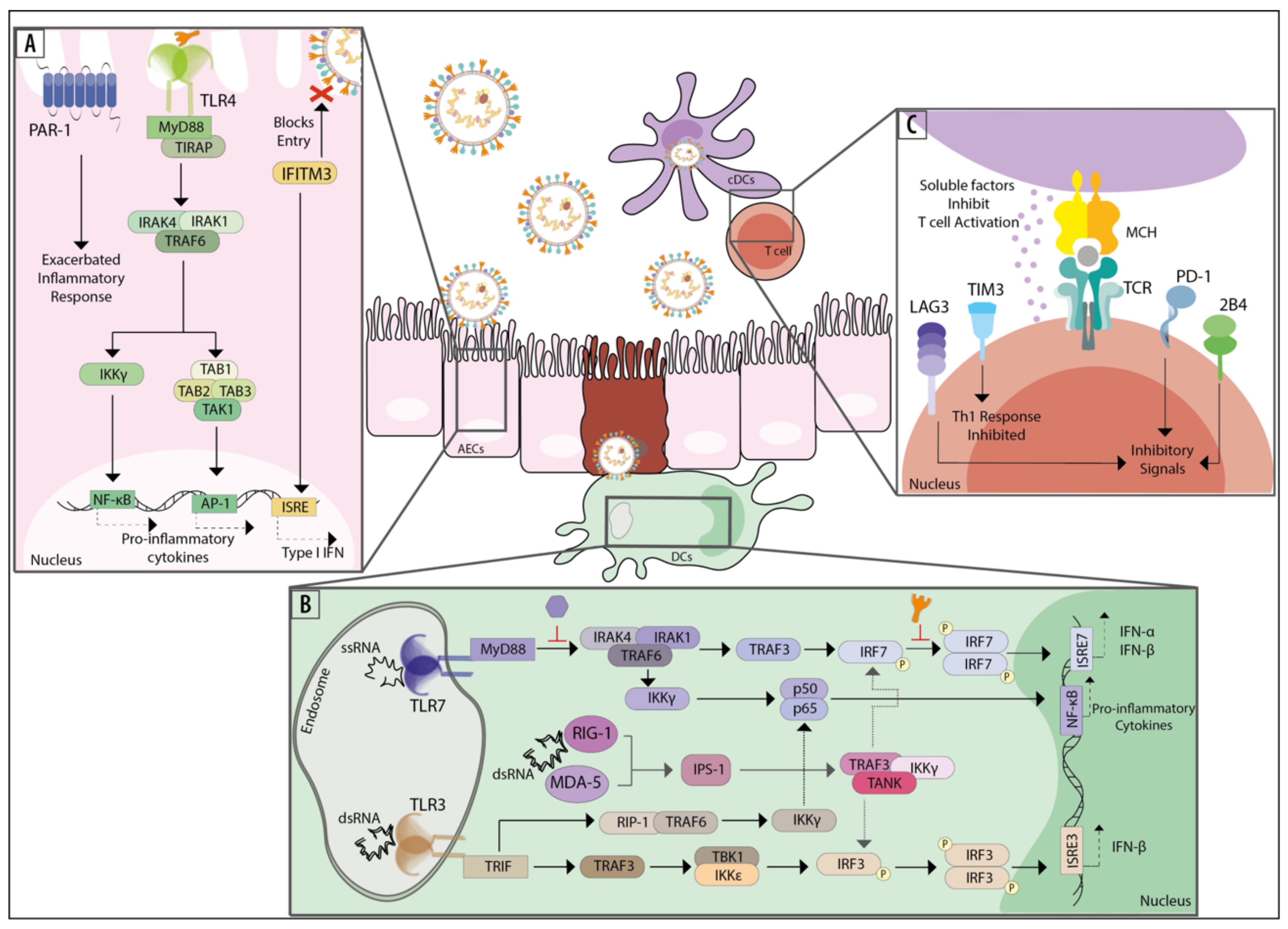
2. Innate Immune Response and Components Recognizing hMPV
2.1. Cells of the Innate Repertoire and the IFN Pathway

{kind=link}
{kind=link}
| hMPV Proteins | Impact on the Host Immune Response | References |
|---|---|---|
| Nucleoprotein | An epitope from this protein promotes a protective CTL response. Along with the P protein, this is the other main component of the inclusion bodies reported during hMPV infections | [20,22,73] |
| Phosphoprotein | Restricts the ability of RIG-I to recognize 5′-triphosphate viral RNA, weakening the expression of IFN-I and ISGs | [21,69] |
| Matrix protein | It is secreted by infected cells in a soluble form and induces the secretion of inflammatory cytokines | [23,24] |
| M2-1 protein | An epitope from this protein stimulates a protective CTL response | [18,73] |
| M2-2 protein | Prevents the homodimerization process of IRF7, resulting in the lack of IFN-α induction from the TLR7 signaling pathway Forms a complex with MyD88 and inhibits TLR-driven signaling | [18,74,75] |
| Small hydrophobic protein | Blocks the phosphorylation process of STAT1, reducing the transcription levels of ISGs Inhibits the TLR7 signaling pathway, decreasing IFN expression It might be involved in decreasing the activation of CD4+ T cells | [67,68,76,77] |
| Glycoprotein | Might participate in reducing the activation of CD4+ T cells Forms a complex with RIG-I to avoid viral sensing Contributes to neutrophil recruitment via enhanced secretion of CXCL2, CCL3, CCL4, IL-17, and TNF | [27,28,29,77] |
2.2. Intrinsic Host Components and Factors Recognizing the Genetic Material of hMPV
2.3. Intrinsic Host Components and Factors Recognizing the Proteins of hMPV
3. Components and Cells of the Adaptive Immune System Responding to hMPV
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Van Den Hoogen, B.G.; De Jong, J.C.; Groen, J.; Kuiken, T.; De Groot, R.; Fouchier, R.A.M.; Osterhaus, A.D.M.E. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 2001, 7, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Van Den Hoogen, B.G.; Osterhaus, D.M.E.; Fouchier, R.A.M. Clinical impact and diagnosis of human metapneumovirus infection. Pediatr. Infect. Dis. J. 2004, 23, S25–S32. [Google Scholar] [CrossRef]
- Jagusic, M.; Slovic, A.; Ivancic-Jelecki, J.; Ljubin-Sternak, S.; Vilibić-Čavlek, T.; Tabain, I.; Forcic, D. Molecular epidemiology of human respiratory syncytial virus and human metapneumovirus in hospitalized children with acute respiratory infections in Croatia, 2014–2017. Infect. Genet. Evol. 2019, 76, 104039. [Google Scholar] [CrossRef]
- Oumei, H.; Xuefeng, W.; Jianping, L.; Kunling, S.; Rong, M.; Zhenze, C.; Li, D.; Huimin, Y.; Lining, W.; Zhaolan, L.; et al. Etiology of community-acquired pneumonia in 1500 hospitalized children. J. Med. Virol. 2018, 90, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Oza, S.; Hogan, D.; Chu, Y.; Perin, J.; Zhu, J.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of under-5 mortality in 2000–2015: An updated systematic analysis with implications for the Sustainable Development Goals. Lancet 2016, 388, 3027–3035. [Google Scholar] [CrossRef] [Green Version]
- Divarathna, M.V.M.; Rafeek, R.A.M.; Noordeen, F. A review on epidemiology and impact of human metapneumovirus infections in children using TIAB search strategy on PubMed and PubMed Central articles. Rev. Med. Virol. 2019, 30, e2090. [Google Scholar] [CrossRef]
- Edwards, K.M.; Zhu, Y.; Griffin, M.R.; Weinberg, G.A.; Hall, C.B.; Szilagyi, P.G.; Staat, M.A.; Iwane, M.; Prill, M.M.; Williams, J. V Burden of human metapneumovirus infection in young children. N. Engl. J. Med. 2013, 368, 633–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, J.A.; Erdman, D.D.; Weinberg, G.A.; Edwards, K.; Hall, C.B.; Walker, F.J.; Iwane, M.; Anderson, L.J. Human Metapneumovirus Infection among Children Hospitalized with Acute Respiratory Illness. Emerg. Infect. Dis. 2004, 10, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.V.; Edwards, K.M.; Weinberg, G.A.; Griffin, M.R.; Hall, C.B.; Zhu, Y.; Szilagyi, P.G.; Wang, C.K.; Yang, C.; Silva, D.; et al. Population-Based Incidence of Human Metapneumovirus Infection among Hospitalized Children. J. Infect. Dis. 2010, 201, 1890–1898. [Google Scholar] [CrossRef] [Green Version]
- Van den Hoogen, B.G.; van Doornum, G.J.J.; Fockens, J.C.; Cornelissen, J.J.; Beyer, W.E.P.; de Groot, R.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Prevalence and Clinical Symptoms of Human Metapneumovirus Infection in Hospitalized Patients. J. Infect. Dis. 2003, 188, 1571–1577. [Google Scholar] [CrossRef] [Green Version]
- Bohmwald, K.; Gálvez, N.M.S.; Ríos, M.; Kalergis, A.M. Neurologic Alterations Due to Respiratory Virus Infections. Front. Cell. Neurosci. 2018, 12, 386. [Google Scholar] [CrossRef]
- Hata, M.; Ito, M.; Kiyosawa, S.; Kimpara, Y.; Tanaka, S.; Yamashita, T.; Hasegawa, A.; Kobayashi, S.; Koyama, N.; Minagawa, H. A fatal case of encephalopathy possibly associated with human metapneumovirus infection. Jpn. J. Infect. Dis. 2007, 60, 328. [Google Scholar]
- Arnold, J.C.; Singh, K.K.; Milder, E.; Spector, S.A.; Sawyer, M.H.; Gavali, S.; Glaser, C. Human metapneumovirus associated with central nervous system infection in children. Pediatr. Infect. Dis. J. 2009, 28, 1057–1060. [Google Scholar] [CrossRef]
- Anderson, E.J.; Simões, E.A.F.; Buttery, J.P.; Dennehy, P.H.; Domachowske, J.B.; Jensen, K.; Lieberman, J.M.; Losonsky, G.A.; Yogev, R. Prevalence and characteristics of human metapneumovirus infection among hospitalized children at high risk for severe lower respiratory tract infection. J. Pediatr. Infect. Dis. Soc. 2012, 1, 212–222. [Google Scholar] [CrossRef]
- Haynes, A.K.; Fowlkes, A.L.; Schneider, E.; Mutuc, J.D.; Armstrong, G.L.; Gerber, S.I. Human metapneumovirus circulation in the United States, 2008 to 2014. Pediatrics 2016, 137, e20152927. [Google Scholar] [CrossRef] [Green Version]
- Márquez-Escobar, V.A. Current developments and prospects on human metapneumovirus vaccines. Expert Rev. Vaccines 2017, 16, 419–431. [Google Scholar] [CrossRef]
- Palavecino, C.E.; Céspedes, P.F.; Lay, M.K.; Riedel, C.A.; Kalergis, A.M.; Bueno, S.M. Understanding lung immunopathology caused by the human metapneumovirus: Implications for rational vaccine design. Crit. Rev. Immunol. 2015, 35. [Google Scholar] [CrossRef]
- Van den Hoogen, B.G.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Analysis of the genomic sequence of a human metapneumovirus. Virology 2002, 295, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Soto, J.A.; Gálvez, N.M.S.; Benavente, F.M.; Pizarro-Ortega, M.S.; Lay, M.K.; Riedel, C.; Bueno, S.M.; Gonzalez, P.A.; Kalergis, A.M. Human metapneumovirus: Mechanisms and molecular targets used by the virus to avoid the immune system. Front. Immunol. 2018, 9, 2466. [Google Scholar] [CrossRef] [Green Version]
- Renner, M.; Bertinelli, M.; Leyrat, C.; Paesen, G.C.; Saraiva de Oliveira, L.F.; Huiskonen, J.T.; Grimes, J.M. Nucleocapsid assembly in pneumoviruses is regulated by conformational switching of the N protein. Elife 2016, 5, e12627. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Paesen, G.C.; Grison, C.M.; Granier, S.; Grimes, J.M.; Leyrat, C. Structural dissection of human metapneumovirus phosphoprotein using small angle x-ray scattering. Sci. Rep. 2017, 7, 14865. [Google Scholar] [CrossRef] [Green Version]
- Derdowski, A.; Peters, T.R.; Glover, N.; Qian, R.; Utley, T.J.; Burnett, A.; Williams, J.V.; Spearman, P.; Crowe, J.E. Human metapneumovirus nucleoprotein and phosphoprotein interact and provide the minimal requirements for inclusion body formation. J. Gen. Virol. 2008, 89, 2698–2708. [Google Scholar] [CrossRef]
- Leyrat, C.; Renner, M.; Harlos, K.; Huiskonen, J.T.; Grimes, J.M. Structure and self-assembly of the calcium binding matrix protein of human metapneumovirus. Structure 2014, 22, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnaud-Baule, A.; Reynard, O.; Perret, M.; Berland, J.-L.; Maache, M.; Peyrefitte, C.; Vernet, G.; Volchkov, V.; Paranhos-Baccalà, G. The Human Metapneumovirus Matrix Protein Stimulates the Inflammatory Immune Response In Vitro. PLoS ONE 2011, 6, e17818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schowalter, R.M.; Smith, S.E.; Dutch, R.E. Characterization of Human Metapneumovirus F Protein-Promoted Membrane Fusion: Critical Roles for Proteolytic Processing and Low pH. J. Virol. 2006, 80, 10931–10941. [Google Scholar] [CrossRef] [Green Version]
- Masante, C.; El Najjar, F.; Chang, A.; Jones, A.; Moncman, C.L.; Dutch, R.E. The Human Metapneumovirus Small Hydrophobic Protein Has Properties Consistent with Those of a Viroporin and Can Modulate Viral Fusogenic Activity. J. Virol. 2014, 88, 6423–6433. [Google Scholar] [CrossRef] [Green Version]
- Thammawat, S.; Sadlon, T.A.; Hallsworth, P.G.; Gordon, D.L. Role of Cellular Glycosaminoglycans and Charged Regions of Viral G Protein in Human Metapneumovirus Infection. J. Virol. 2008, 82, 11767–11774. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Liu, T.; Shan, Y.; Li, K.; Garofalo, R.P.; Casola, A. Human metapneumovirus glycoprotein G inhibits innate immune responses. PLoS Pathog. 2008, 88, 6423–6433. [Google Scholar] [CrossRef]
- Cheemarla, N.R.; Guerrero-Plata, A. Human metapneumovirus attachment protein contributes to neutrophil recruitment into the airways of infected mice. Viruses 2017, 9, 310. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.; Shafagati, N. Human metapneumovirus—What we know now. F1000Research 2018, 7, 135. [Google Scholar]
- El Najjar, F.; Cifuentes-Muñoz, N.; Chen, J.; Zhu, H.; Buchholz, U.J.; Moncman, C.L.; Dutch, R.E. Human metapneumovirus Induces Reorganization of the Actin Cytoskeleton for Direct Cell-to-Cell Spread. PLoS Pathog. 2016, 12, e1005922. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.G.; Mainou, B.A.; Johnson, M.; Hastings, A.K.; Schuster, J.E.; Dermody, T.S.; Williams, J.V. Human Metapneumovirus Is Capable of Entering Cells by Fusion with Endosomal Membranes. PLoS Pathog. 2015, 11, e1005303. [Google Scholar] [CrossRef] [Green Version]
- Jumat, M.R.; Nguyen Huong, T.; Wong, P.; Loo, L.H.; Tan, B.H.; Fenwick, F.; Toms, G.L.; Sugrue, R.J. Imaging analysis of human metapneumovirus-infected cells provides evidence for the involvement of F-actin and the raft-lipid microdomains in virus morphogenesis. Virol. J. 2014, 11, 198. [Google Scholar] [CrossRef] [Green Version]
- Palavecino, C.E.; Cespedes, P.F.; Gomez, R.S.; Kalergis, A.M.; Bueno, S.M.; Céspedes, P.F.; Gómez, R.S.; Kalergis, A.M.; Bueno, S.M.; Cespedes, P.F.; et al. Immunization with a Recombinant Bacillus Calmette-Guerin Strain Confers Protective Th1 Immunity against the Human Metapneumovirus. J. Immunol. 2014, 192, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Soto, J.A.; Gálvez, N.M.S.; Rivera, C.A.; Palavecino, C.E.; Céspedes, P.F.; Rey-Jurado, E.; Bueno, S.M.; Kalergis, A.M. Recombinant BCG Vaccines Reduce Pneumovirus-Caused Airway Pathology by Inducing Protective Humoral Immunity. Front. Immunol. 2018, 9, 2875. [Google Scholar] [CrossRef] [Green Version]
- Aerts, L.; Rhéaume, C.; Carbonneau, J.; Lavigne, S.; Couture, C.; Hamelin, M.-È.; Boivin, G. Adjuvant effect of the human metapneumovirus (HMPV) matrix protein in HMPV subunit vaccines. J. Gen. Virol. 2015, 96, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.G.; Erickson, J.J.; Hastings, A.K.; Becker, J.C.; Johnson, M.; Craven, R.E.; Tollefson, S.J.; Boyd, K.L.; Williams, J. V Human Metapneumovirus Virus-Like Particles Induce Protective B and T Cell Responses in a Mouse Model. J. Virol. 2014, 88, 6368–6379. [Google Scholar] [CrossRef] [Green Version]
- Dubois, J.; Pizzorno, A.; Cavanagh, M.-H.; Padey, B.; Nicolas de Lamballerie, C.; Uyar, O.; Venable, M.-C.; Carbonneau, J.; Traversier, A.; Julien, T.; et al. Strain-Dependent Impact of G and SH Deletions Provide New Insights for Live-Attenuated HMPV Vaccine Development. Vaccines 2019, 7, 164. [Google Scholar] [CrossRef] [Green Version]
- Kinder, J.T.; Moncman, C.L.; Barrett, C.; Jin, H.; Kallewaard, N.; Dutch, R.E. Respiratory Syncytial Virus and Human Metapneumovirus Infections in Three-Dimensional Human Airway Tissues Expose an Interesting Dichotomy in Viral Replication, Spread, and Inhibition by Neutralizing Antibodies. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.; Harrod, K.S.; Shieh, W.-J.J.; Zaki, S.; Tripp, R.A. Human Metapneumovirus Persists in BALB/c Mice despite the Presence of Neutralizing Antibodies. J. Virol. 2004, 78, 14003–14011. [Google Scholar] [CrossRef] [Green Version]
- Darniot, M.; Petrella, T.; Aho, S.; Pothier, P.; Manoha, C. Immune response and alteration of pulmonary function after primary human metapneumovirus (hMPV) infection of BALB/c mice. Vaccine 2005, 23, 4473–4480. [Google Scholar] [CrossRef]
- Hamelin, M.-E.; Yim, K.; Kuhn, K.H.; Cragin, R.P.; Boukhvalova, M.; Blanco, J.C.G.; Prince, G.A.; Boivin, G. Pathogenesis of Human Metapneumovirus Lung Infection in BALB/c Mice and Cotton Rats. J. Virol. 2005, 79, 8894–8903. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.V.; Tollefson, S.J.; Johnson, J.E.; Crowe, J.E. The Cotton Rat (Sigmodon hispidus) Is a Permissive Small Animal Model of Human Metapneumovirus Infection, Pathogenesis, and Protective Immunity. J. Virol. 2005, 79, 10944–10951. [Google Scholar] [CrossRef] [Green Version]
- Huck, B.; Neumann-Haefelin, D.; Schmitt-Graeff, A.; Weckmann, M.; Mattes, J.; Ehl, S.; Falcone, V. Human metapneumovirus induces more severe disease and stronger innate immune response in BALB/c mice as compared with respiratory syncytial virus. Respir. Res. 2007, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Van Den Hoogen, B.G.; Herfst, S.; Sprong, L.; Cane, P.A.; Forleo-Neto, E.; De Swart, R.L.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Antigenic and Genetic Variability of Human Metapneumoviruses. Emerg. Infect. Dis. 2004, 10, 658–666. [Google Scholar] [CrossRef]
- Pitoiset, C.; Darniot, M.; Huet, F.; Aho, S.L.; Pothier, P.; Manoha, C. Human metapneumovirus genotypes and severity of disease in young children (n = 100) during a 7-year study in Dijon Hospital, France. J. Med. Virol. 2010, 82, 1782–1789. [Google Scholar] [CrossRef]
- Uche, I.K.; Guerrero-Plata, A. Interferon-Mediated Response to Human Metapneumovirus Infection. Viruses 2018, 10, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, A.K.; Erickson, J.J.; Schuster, J.E.; Boyd, K.L.; Tollefson, S.J.; Johnson, M.; Gilchuk, P.; Joyce, S.; Williams, J. V Role of Type I Interferon Signaling in Human Metapneumovirus Pathogenesis and Control of Viral Replication. J. Virol. 2015, 89, 4405–4420. [Google Scholar] [CrossRef] [Green Version]
- Andrade, C.A.; Pacheco, G.A.; Gálvez, N.M.S.; Soto, J.A.; Bueno, S.M.; Kalergis, A.M. Innate Immune Components That Regulate the Pathogenesis and Resolution of hRSV and hMPV Infections. Viruses 2020, 12, 637. [Google Scholar] [CrossRef]
- Loevenich, S.; Malmo, J.; Liberg, A.M.; Sherstova, T.; Li, Y.; Rian, K.; Johnsen, I.B.; Anthonsen, M.W. Cell-type-specific transcription of innate immune regulators in response to hmpv infection. Mediators Inflamm. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Céspedes, P.F.; Gonzalez, P.A.; Kalergis, A.M. Human metapneumovirus keeps dendritic cells from priming antigen-specific naive T cells. Immunology 2013, 139, 366–376. [Google Scholar] [CrossRef]
- Kolli, D.; Bataki, E.L.; Spetch, L.; Guerrero-Plata, A.; Jewell, A.M.; Piedra, P.A.; Milligan, G.N.; Garofalo, R.P.; Casola, A. T Lymphocytes Contribute to Antiviral Immunity and Pathogenesis in Experimental Human Metapneumovirus Infection. J. Virol. 2008, 82, 8560–8569. [Google Scholar] [CrossRef] [Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Soto, J.A.; Gálvez, N.M.S.; Andrade, C.A.; Pacheco, G.A.; Bohmwald, K.; Berrios, R.V.; Bueno, S.M.; Kalergis, A.M. The Role of Dendritic Cells During Infections Caused by Highly Prevalent Viruses. Front. Immunol. 2020, 11, 1513. [Google Scholar] [CrossRef]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef]
- Vassileva, G.; Chen, S.-C.; Zeng, M.; Abbondanzo, S.; Jensen, K.; Gorman, D.; Baroudy, B.M.; Jiang, Y.; Murgolo, N.; Lira, S.A. Expression of a Novel Murine Type I IFN in the Pancreatic Islets Induces Diabetes in Mice. J. Immunol. 2003, 170, 5748–5755. [Google Scholar] [CrossRef] [Green Version]
- Van Pesch, V.; Lanaya, H.; Renauld, J.-C.; Michiels, T. Characterization of the Murine Alpha Interferon Gene Family. J. Virol. 2004, 78, 8219–8228. [Google Scholar] [CrossRef] [Green Version]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Valente, G.; Ozmen, L.; Novelli, F.; Geuna, M.; Palestro, G.; Forni, G.; Garotta, G. Distribution of interferon-γ receptor in human tissues. Eur. J. Immunol. 1992, 22, 2403–2412. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type i interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Arimoto, K.-I.; Miyauchi, S.; Stoner, S.A.; Fan, J.-B.; Zhang, D.-E. Negative regulation of type I IFN signaling. J. Leukoc. Biol. 2018, 103, 1099–1116. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.-Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Dinwiddie, D.L.; Harrod, K.S. Human Metapneumovirus Inhibits IFN-α Signaling through Inhibition of STAT1 Phosphorylation. Am. J. Respir. Cell Mol. Biol. 2008, 38, 661–670. [Google Scholar] [CrossRef]
- Hastings, A.K.; Amato, K.R.; Wen, S.C.; Peterson, L.S.; Williams, J. V Human metapneumovirus small hydrophobic (SH) protein downregulates type I IFN pathway signaling by affecting STAT1 expression and phosphorylation. Virology 2016, 494, 248–256. [Google Scholar] [CrossRef]
- Goutagny, N.; Jiang, Z.; Tian, J.; Parroche, P.; Schickli, J.; Monks, B.G.; Ulbrandt, N.; Ji, H.; Kiener, P.A.; Coyle, A.J.; et al. Cell Type-Specific Recognition of Human Metapneumoviruses (HMPVs) by Retinoic Acid-Inducible Gene I (RIG-I) and TLR7 and Viral Interference of RIG-I Ligand Recognition by HMPV-B1 Phosphoprotein. J. Immunol. 2010, 184, 1168–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmo, J.; Moe, N.; Krokstad, S.; Ryan, L.; Loevenich, S.; Johnsen, I.B.; Espevik, T.; Nordbø, S.A.; Døllner, H.; Anthonsen, M.W. Cytokine profiles in human metapneumovirus infected children: Identification of genes involved in the antiviral response and pathogenesis. PLoS ONE 2016, 11, e0155484. [Google Scholar] [CrossRef] [PubMed]
- Pancham, K.; Perez, G.F.; Huseni, S.; Jain, A.; Kurdi, B.; Rodriguez-Martinez, C.E.; Preciado, D.; Rose, M.C.; Nino, G. Premature infants have impaired airway antiviral IFNγ responses to human metapneumovirus compared to respiratory syncytial virus. Pediatr. Res. 2015, 78, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Melendi, G.A.; Laham, F.R.; Monsalvo, A.C.; Casellas, J.M.; Israele, V.; Polack, N.R.; Kleeberger, S.R.; Polack, F.P. Cytokine Profiles in the Respiratory Tract During Primary Infection With Human Metapneumovirus, Respiratory Syncytial Virus, or Influenza Virus in Infants. Pediatrics 2007, 120, e410–e415. [Google Scholar] [CrossRef]
- Melendi, G.A.; Zavala, F.; Buchholz, U.J.; Boivin, G.; Collins, P.L.; Kleeberger, S.R.; Polack, F.P. Mapping and Characterization of the Primary and Anamnestic H-2d-Restricted Cytotoxic T-Lymphocyte Response in Mice against Human Metapneumovirus. J. Virol. 2007, 81, 11461–11467. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, Y.; Sakai, M.; Funayama, M.; Itoh, M.; Gotoh, B. Human Metapneumovirus M2-2 Protein Acts as a Negative Regulator of Alpha Interferon Production by Plasmacytoid Dendritic Cells. J. Virol. 2017, 91, e00579-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Liu, G.; Go, J.; Kolli, D.; Zhang, G.; Bao, X. Human metapneumovirus M2-2 protein inhibits innate immune response in monocyte-derived dendritic cells. PLoS ONE 2014, 9, e91865. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Kolli, D.; Esham, D.; Velayutham, T.S.; Casola, A. Human metapneumovirus small hydrophobic protein inhibits interferon induction in plasmacytoid dendritic cells. Viruses 2018, 10, 278. [Google Scholar] [CrossRef] [Green Version]
- Le Nouen, C.; Hillyer, P.; Brock, L.G.; Winter, C.C.; Rabin, R.L.; Collins, P.L.; Buchholz, U.J. Human Metapneumovirus SH and G Glycoproteins Inhibit Macropinocytosis-Mediated Entry into Human Dendritic Cells and Reduce CD4+ T Cell Activation. J. Virol. 2014, 88, 6453–6469. [Google Scholar] [CrossRef] [Green Version]
- McMichael, T.M.; Zhang, Y.; Kenney, A.D.; Zhang, L.; Zani, A.; Lu, M.; Chemudupati, M.; Li, J.; Yount, J.S. IFITM3 Restricts Human Metapneumovirus Infection. J. Infect. Dis. 2018, 218, 1582–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.E.; Busse, D.C.; Binter, S.; Weston, S.; Diaz Soria, C.; Laksono, B.M.; Clare, S.; Van Nieuwkoop, S.; Van den Hoogen, B.G.; Clement, M.; et al. Interferon-Induced Transmembrane Protein 1 Restricts Replication of Viruses That Enter Cells via the Plasma Membrane. J. Virol. 2018, 93, e02003–e02018. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef]
- Koganti, R.; Suryawanshi, R.; Shukla, D. Heparanase, cell signaling, and viral infections. Cell. Mol. Life Sci. 2020, 77, 5059–5077. [Google Scholar] [CrossRef]
- Brosseau, C.; Colas, L.; Magnan, A.; Brouard, S. CD9 tetraspanin: A new pathway for the regulation of inflammation? Front. Immunol. 2018, 9, 2316. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Schlender, J.; Guenthner-Biller, M.; Rothenfusser, S.; Endres, S.; Conzelmann, K.-K.; Hartmann, G. Replication-Dependent Potent IFN-α Induction in Human Plasmacytoid Dendritic Cells by a Single-Stranded RNA Virus. J. Immunol. 2004, 173, 5935–5943. [Google Scholar] [CrossRef] [Green Version]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern recognition receptors and the host cell death molecular machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.; McCarthy, J.; O’Driscoll, C.; Melgar, S. Pattern recognition receptors-Molecular orchestrators of inflammation in inflammatory bowel disease. Cytokine Growth Factor Rev. 2013, 24, 91–104. [Google Scholar] [CrossRef]
- Patra, M.C.; Choi, S. Recent progress in the development of Toll-like receptor (TLR) antagonists. Expert Opin. Ther. Pat. 2016, 26, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Liu, K.; Cheng, A.; Wang, M.; Cui, M.; Huang, J.; Zhu, D.; Chen, S.; Liu, M.; Zhao, X.; et al. SOCS Proteins Participate in the Regulation of Innate Immune Response Caused by Viruses. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Zhao, Y.; Zhang, Z.Y.; Mao, H.W.; Tu, W.W.; Zhao, X.D. Respiratory Syncytial Virus Infection Induces Higher Toll-Like Receptor-3 Expression and TNF-α Production Than Human Metapneumovirus Infection. PLoS ONE 2013, 8, e73488. [Google Scholar] [CrossRef] [Green Version]
- Kolli, D.; Bao, X.; Liu, T.; Hong, C.; Wang, T.; Garofalo, R.P.; Casola, A. Human Metapneumovirus Glycoprotein G Inhibits TLR4-Dependent Signaling in Monocyte-Derived Dendritic Cells. J. Immunol. 2011, 187, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Zhao, Y.; Zhang, Z.Y.; Zhao, X.D. Toll-like receptors expression in the lungs of human metapneumovirus infected mice and the effects of polyI:C on viral infection. Bing Du Xue Bao 2010, 26, 1–7. [Google Scholar]
- Eng, H.L.; Hsu, Y.Y.; Lin, T.M. Differences in TLR7/8 activation between monocytes and macrophages. Biochem. Biophys. Res. Commun. 2018, 497, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.F.; Chen, C.Y.; Shih, Y.C.; Liu, H.Y.; Huang, C.M.; Hsueh, Y.P. Endosomal TLR3, TLR7, and TLR8 control neuronal morphology through different transcriptional programs. J. Cell Biol. 2018, 217, 2727–2742. [Google Scholar] [CrossRef]
- Bender, A.T.; Tzvetkov, E.; Pereira, A.; Wu, Y.; Kasar, S.; Przetak, M.M.; Vlach, J.; Niewold, T.B.; Jensen, M.A.; Okitsu, S.L. TLR7 and TLR8 Differentially Activate the IRF and NF-κB Pathways in Specific Cell Types to Promote Inflammation. ImmunoHorizons 2020, 4, 93–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Funami, K.; Oshiumi, H.; Seya, T. Toll-IL-1-receptor-containing adaptor molecule-1: A signaling adaptor linking innate immunity to adaptive immunity. Prog. Mol. Biol. Transl. Sci. 2013, 117, 487–510. [Google Scholar]
- Zainol, M.I.B.; Kawasaki, T.; Monwan, W.; Murase, M.; Sueyoshi, T.; Kawai, T. Innate immune responses through Toll-like receptor 3 require human-antigen-R-mediated Atp6v0d2 mRNA stabilization. Sci. Rep. 2019, 9, 20406–20411. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Barton, G.M.; Medzhitov, R. Toll-like receptor signaling pathways. Science 2003, 300, 1524–1525. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.-M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; García-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 Signaling by RNA Viruses in Innate Immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Bao, X.; Liu, T.; Lai, S.; Li, K.; Garofalo, R.P.; Casola, A. Role of retinoic acid inducible gene-I in human metapneumovirus-induced cellular signalling. J. Gen. Virol. 2008, 89, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Diab, M.; Vitenshtein, A.; Drori, Y.; Yamin, R.; Danziger, O.; Zamostiano, R.; Mandelboim, M.; Bacharach, E.; Mandelboim, O. Suppression of human metapneumovirus (HMPV) infection by the innate sensing gene CEACAM1. Oncotarget 2016, 7, 66468–66479. [Google Scholar] [CrossRef] [Green Version]
- Baños-Lara, M.D.R.; Ghosh, A.; Guerrero-Plata, A. Critical role of MDA5 in the interferon response induced by human metapneumovirus infection in dendritic cells and in vivo. J. Virol. 2013, 87, 1242–1251. [Google Scholar] [CrossRef] [Green Version]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.Q.; Xia, T.; Hu, Y.H.; Sun, M.S.; Yan, S.; Lei, C.Q.; Shu, H.B.; Guo, J.H.; Liu, Y. IFITM3 inhibits virus-triggered induction of type I interferon by mediating autophagosome-dependent degradation of IRF3. Cell. Mol. Immunol. 2018, 15, 858–867. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.Y.J.; Fu, R.M.; Liang, C.; Sloan, R.D. IFITM proteins inhibit HIV-1 protein synthesis. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, G.; Li, S. Tripartite motif proteins: An emerging antiviral protein family. Future Virol. 2019, 14, 107–122. [Google Scholar] [CrossRef]
- Liu, B.; Li, N.L.; Shen, Y.; Bao, X.; Fabrizio, T.; Elbahesh, H.; Webby, R.J.; Li, K. The C-Terminal Tail of TRIM56 Dictates Antiviral Restriction of Influenza A and B Viruses by Impeding Viral RNA Synthesis. J. Virol. 2016, 90, 4369–4382. [Google Scholar] [CrossRef] [Green Version]
- Hage, A.; Rajsbaum, R. To TRIM or not to TRIM: The balance of host-virus interactions mediated by the ubiquitin system. J. Gen. Virol. 2019, 100, 1641–1662. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.K.H.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Vergnolle, N. Protease-activated receptors as drug targets in inflammation and pain. Pharmacol. Ther. 2009, 123, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Gieseler, F.; Ungefroren, H.; Settmacher, U.; Hollenberg, M.D.; Kaufmann, R. Proteinase-activated receptors (PARs)—Focus on receptor-receptor- interactions and their physiological and pathophysiological impact. Cell Commun. Signal. 2013, 11, 86. [Google Scholar] [CrossRef] [Green Version]
- Khoufache, K.; Berri, F.; Nacken, W.; Vogel, A.B.; Delenne, M.; Camerer, E.; Coughlin, S.R.; Carmeliet, P.; Lina, B.; Rimmelzwaan, G.F.; et al. PAR1 contributes to influenza A virus pathogenicity in mice. J. Clin. Invest. 2013, 123, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Aerts, L.; Hamelin, M.È.; Rhéaume, C.; Lavigne, S.; Couture, C.; Kim, W.J.; Susan-Resiga, D.; Prat, A.; Seidah, N.G.; Vergnolle, N.; et al. Modulation of Protease Activated Receptor 1 Influences Human Metapneumovirus Disease Severity in a Mouse Model. PLoS ONE 2013, 8, e72529. [Google Scholar]
- Sutherland, M.R.; Friedman, H.M.; Pryzdial, E.L.G. Thrombin enhances herpes simplex virus infection of cells involving protease-activated receptor 1. J. Thromb. Haemost. 2007, 5, 1055–1061. [Google Scholar] [CrossRef]
- Cabello-Gutiérrez, C.; Manjarrez-Zavala, M.E.; Huerta-Zepeda, A.; Cime-Castillo, J.; Monroy-Martínez, V.; Biruete-Correa, B.; Ruiz-Ordaz, B.H. Modification of the cytoprotective protein C pathway during Dengue virus infection of human endothelial vascular cells. Thromb. Haemost. 2009, 101, 916–928. [Google Scholar] [CrossRef]
- Antoniak, S.; Owens, A.P.; Baunacke, M.; Williams, J.C.; Lee, R.D.; Weithäuser, A.; Sheridan, P.A.; Malz, R.; Luyendyk, J.P.; Esserman, D.A.; et al. PAR-1 contributes to the innate immune response during viral infection. J. Clin. Invest. 2013, 123, 1310–1322. [Google Scholar] [CrossRef] [Green Version]
- Lê, V.B.; Riteau, B.; Alessi, M.C.; Couture, C.; Jandrot-Perrus, M.; Rhéaume, C.; Hamelin, M.È.; Boivin, G. Protease-activated receptor 1 inhibition protects mice against thrombin-dependent respiratory syncytial virus and human metapneumovirus infections. Br. J. Pharmacol. 2018, 175, 388–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barral, P.M.; Sarkar, D.; Su, Z.Z.; Barber, G.N.; DeSalle, R.; Racaniello, V.R.; Fisher, P.B. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: Key regulators of innate immunity. Pharmacol. Ther. 2009, 124, 219–234. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Miller, D.J.; Bowman, E.R.; Nagarkar, D.R.; Schneider, D.; Zhao, Y.; Linn, M.J.; Goldsmith, A.M.; Bentley, J.K.; Sajjan, U.S.; et al. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog. 2011, 7, e1002070. [Google Scholar] [CrossRef]
- McCartney, S.A.; Thackray, L.B.; Gitlin, L.; Gilfillan, S.; Virgin IV, H.W.; Colonna, M. MDA-5 recognition of a murine norovirus. PLoS Pathog. 2008, 4, e1000108. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spann, K.M.; Loh, Z.; Lynch, J.P.; Ullah, A.; Zhang, V.; Baturcam, E.; Werder, R.B.; Khajornjiraphan, N.; Rudd, P.; Loo, Y.M.; et al. IRF-3, IRF-7, and IPS-1 promote host defense against acute human metapneumovirus infection in neonatal mice. Am. J. Pathol. 2014, 184, 1795–1806. [Google Scholar] [CrossRef]
- Alcover, A.; Alarcón, B.; Di Bartolo, V. Cell Biology of T Cell Receptor Expression and Regulation. Annu. Rev. Immunol. 2018, 36, 103–125. [Google Scholar] [CrossRef]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 112, 1557–1569. [Google Scholar] [CrossRef] [Green Version]
- Mak, T.W.; Saunders, M.E.; Jett, B.D. Primer to The Immune Response, 2nd ed.; Elsevier: Amsterdam, The Netherland, 2014; ISBN 9780123854612. [Google Scholar]
- Laydon, D.J.; Bangham, C.R.M.; Asquith, B. Estimating T-cell repertoire diversity: Limitations of classical estimators and a new approach. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerome, K.R. Viral Modulation of T-Cell Receptor Signaling. J. Virol. 2008, 82, 4194–4204. [Google Scholar] [CrossRef] [Green Version]
- Céspedes, P.F.; Palavecino, C.E.; Kalergis, A.M.; Bueno, S.M. Modulation of host immunity by the human metapneumovirus. Clin. Microbiol. Rev. 2016, 29, 795–818. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Qin, T.; Zhao, X.; Dong, S.; Zhu, J.; Peng, D.; Zhong, J.; Li, T.; Chen, X. Skewed balance of regulatory T cell and inflammatory T cell in IL-17 defect with human metapneumovirus infection. Cell. Immunol. 2018, 331, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Hastings, A.K.; Gilchuk, P.; Joyce, S.; Williams, J.V. Novel HLA-A2-restricted human metapneumovirus epitopes reduce viral titers in mice and are recognized by human T cells. Vaccine 2016, 34, 2663–2670. [Google Scholar] [CrossRef] [Green Version]
- Herd, K.A.; Nelson, M.; Mahalingam, S.; Tindle, R.W. Pulmonary infection of mice with human metapneumovirus induces local cytotoxic T-cell and immunoregulatory cytokine responses similar to those seen with human respiratory syncytial virus. J. Gen. Virol. 2010, 91, 1302–1310. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.J.; Gilchuk, P.; Hastings, A.K.; Tollefson, S.J.; Johnson, M.; Downing, M.B.; Boyd, K.L.; Johnson, J.E.; Kim, A.S.; Joyce, S.; et al. Viral acute lower respiratory infections impair CD8+ T cells through PD-1. J. Clin. Invest. 2012, 122, 2967–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Rey-Jurado, E.; Bohmwald, K.; Gálvez, N.M.S.; Becerra, D.; Porcelli, S.A.; Carreño, L.J.; Kalergis, A.M. Contribution of NKT cells to the immune response and pathogenesis triggered by respiratory viruses. Virulence 2020, 11, 580–593. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.J.; Rogers, M.C.; Hastings, A.K.; Tollefson, S.J.; Williams, J.V. Programmed Death-1 Impairs Secondary Effector Lung CD8 + T Cells during Respiratory Virus Reinfection. J. Immunol. 2014, 193, 5108–5117. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef] [Green Version]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Fujita, A.; Kan-o, K.; Tonai, K.; Yamamoto, N.; Ogawa, T.; Fukuyama, S.; Nakanishi, Y.; Matsumoto, K. Inhibition of PI3Kδ Enhances Poly I:C-Induced Antiviral Responses and Inhibits Replication of Human Metapneumovirus in Murine Lungs and Human Bronchial Epithelial Cells. Front. Immunol. 2020, 11, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agresta, L.; Hoebe, K.H.N.; Janssen, E.M. The Emerging Role of CD244 Signaling in Immune Cells of the Tumor Microenvironment. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Kolli, D.; Bao, X.; Casola, A. Human metapneumovirus antagonism of innate immune responses. Viruses 2012, 4, 3551–3571. [Google Scholar] [CrossRef] [Green Version]
- Yuseff, M.I.; Pierobon, P.; Reversat, A.; Lennon-Duménil, A.M. How B cells capture, process and present antigens: A crucial role for cell polarity. Nat. Rev. Immunol 2013, 13, 475–486. [Google Scholar] [CrossRef]
- Bar-Peled, Y.; Diaz, D.; Pena-Briseno, A.; Murray, J.; Huang, J.; Tripp, R.A.; Mousa, J.J. A Potent Neutralizing Site III-Specific Human Antibody Neutralizes Human Metapneumovirus In Vivo. J. Virol. 2019, 93, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulbrandt, N.D.; Ji, H.; Patel, N.K.; Barnes, A.S.; Wilson, S.; Kiener, P.A.; Suzich, J.A.; McCarthy, M.P. Identification of antibody neutralization epitopes on the fusion protein of human metapneumovirus. J. Gen. Virol. 2008, 89, 3113–3118. [Google Scholar] [CrossRef]
- Chu, C.; Wang, Y.; Zhang, X.; Ni, X.; Cao, J.; Xu, W.; Dong, Z.; Yuan, P.; Wei, W.; Ma, Y.; et al. SAP-regulated T Cell-APC adhesion and ligation-dependent and -independent Ly108-CD3ζ interactions. J. Immunol. 2014, 193, 3860–3871. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Shih, C.; Qi, H. Germinal centers: Ephrin B1-mediated repulsion and signaling control germinal center T cell territoriality and function. Science (80-) 2017, 356, eaai9264. [Google Scholar] [CrossRef] [PubMed]
- Meli, A.P.; Fontés, G.; Avery, D.T.; Leddon, S.A.; Tam, M.; Elliot, M.; Ballesteros-Tato, A.; Miller, J.; Stevenson, M.M.; Fowell, D.J.; et al. The Integrin LFA-1 Controls T Follicular Helper Cell Generation and Maintenance. Immunity 2016, 45, 831–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolduc, A.; Long, E.; Stapler, D.; Cascalho, M.; Tsubata, T.; Koni, P.A.; Shimoda, M. Constitutive CD40L expression on B cells prematurely terminates germinal center response and leads to augmented plasma cell production in T cell areas. J. Immunol. 2010, 185, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gálvez, N.M.S.; Andrade, C.A.; Pacheco, G.A.; Soto, J.A.; Stranger, V.; Rivera, T.; Vásquez, A.E.; Kalergis, A.M. Host Components That Modulate the Disease Caused by hMPV. Viruses 2021, 13, 519. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030519
Gálvez NMS, Andrade CA, Pacheco GA, Soto JA, Stranger V, Rivera T, Vásquez AE, Kalergis AM. Host Components That Modulate the Disease Caused by hMPV. Viruses. 2021; 13(3):519. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030519
Chicago/Turabian StyleGálvez, Nicolás M. S., Catalina A. Andrade, Gaspar A. Pacheco, Jorge A. Soto, Vicente Stranger, Thomas Rivera, Abel E. Vásquez, and Alexis M. Kalergis. 2021. "Host Components That Modulate the Disease Caused by hMPV" Viruses 13, no. 3: 519. https://0-doi-org.brum.beds.ac.uk/10.3390/v13030519