
Key Factors That Enable the Pandemic Potential of RNA Viruses and Inter-Species Transmission: A Systematic Review


Abstract
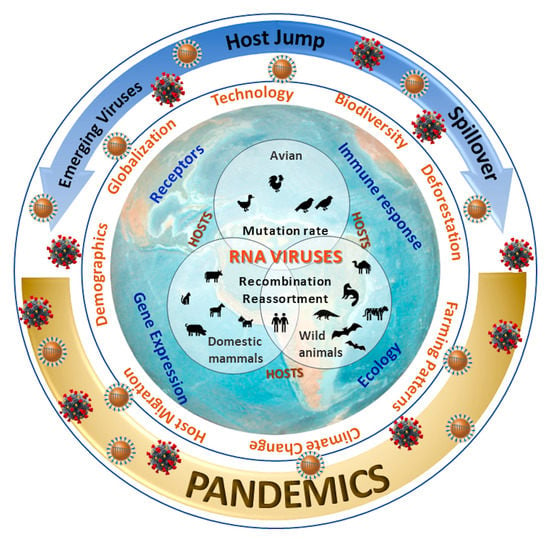
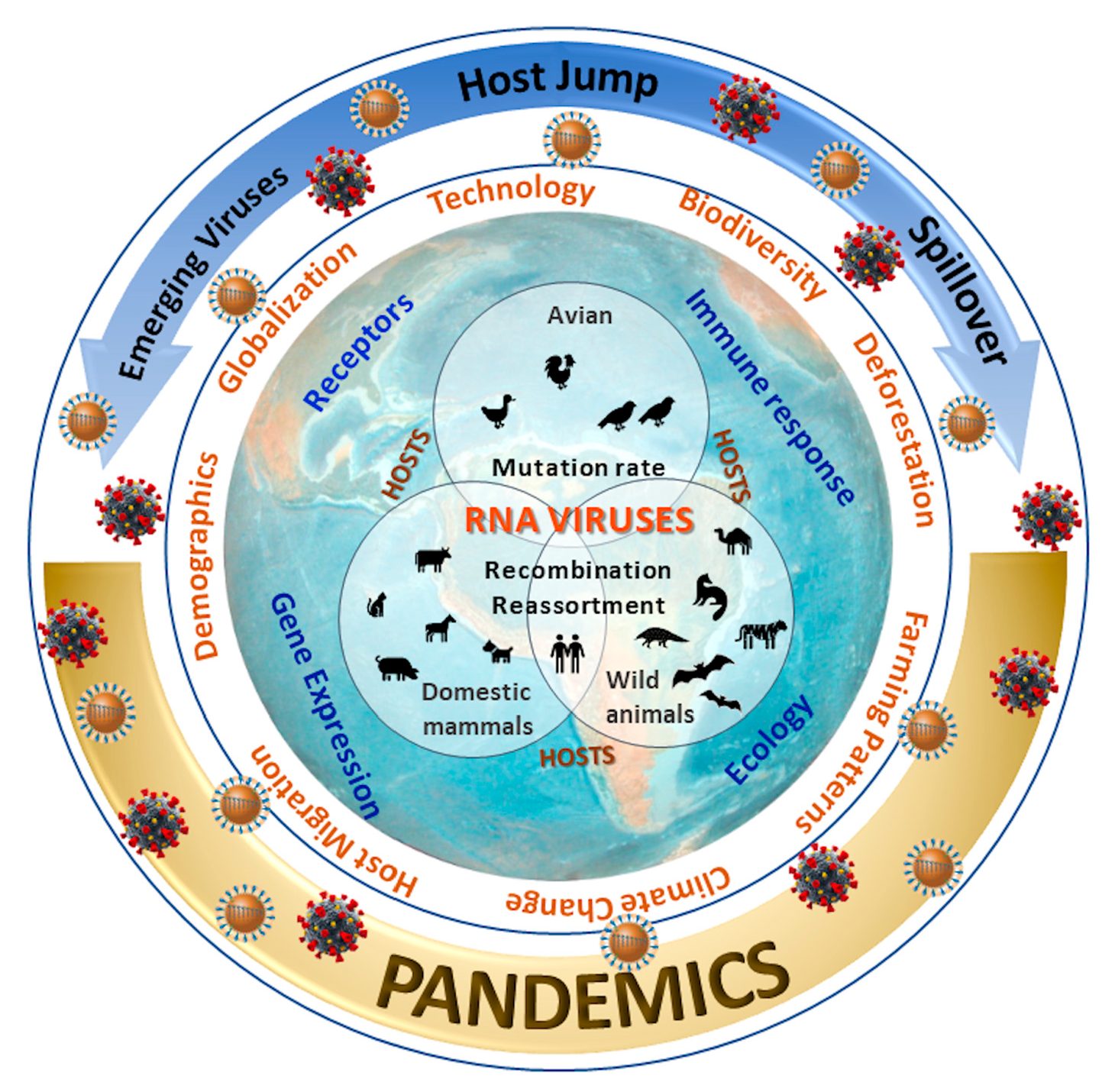
:
1. Introduction
2. Materials and Methods
2.1. Search Strategy and Identification of Relevant Studies
2.2. Charting of Data and Statistical Analysis
3. Results
3.1. Factors Associated with the Virus and its Hosts

{kind=link}
{kind=link}
| Factor | Ref | |
|---|---|---|
| Virus | Changes in cell membrane binding proteins. e.g.,: Glycoprotein S mutations in SARS-CoV-2 (D614G of region S1-S2 near furin cleavage site). | [19] |
| Changes in other internal viral proteins. e.g.,: mutations at central region (intrinsically disordered protein regions—IDRs) of nucleocapsid protein in SARS-CoV-2. | [20] | |
| Modification of cell-entry approach by the virus. e.g.,: HA protein activation in H17 viruses by changing the pH conditions in the cell. | [10] | |
| Host | Receptor recognition and affinity with viral proteins. e.g.,: affinity between ACE-2 receptor and S protein of SARS-CoV-2. e.g.,: Sialic acid receptor affinity dependent on saccharide ending SAα-2,3 and SAα-2,6-Gal for influenza viruses. | [14,21] |
| Intrinsic factors associated with variation in disease presentation and severity. e.g.,: host pro-viral genes involved in chromatin remodeling (complex SWI/SNF), histone modification, cellular signaling, and RNA regulation key for development of coronavirus infection. | [22] | |
| Host immune-response evasion by alteration of gene expression. e.g.,: High and Low pathogenic H5N1 induces alternative splicing in genes such as ADAR, CCL19, RIG-I, DDX60, LGP2, MDA5, IRF7, MX1, NLRC5, STAT1, TRIM25, and VIPERIN in ducks. | [23] | |
| Host ecology and behavior. e.g.,: bats and birds have an extraordinary viral richness due to flying capacity, long migrations, trophic diversity, and social structure | [18] | |
| Other factors | Anthropogenic factors: population density, distribution patterns, traveling and international trading, technological advances in agriculture, changes in farming and food consumption patterns (food source or for medicinal purposes). e.g.,: In Asia, live markets are characterized for the trading of wild animals such as squirrels, rats, porcupines, and wild birds and pigs. | [24] |
| Environmental factors: loss of biodiversity and of habitat for wildlife, deforestation, and climate change, associated with an increase in wildlife-human interaction rate. e.g.,: Brazilian regions diminishing the migratory behavior of bats, which impacted the viral dynamics of the Hendra virus. | [6] |
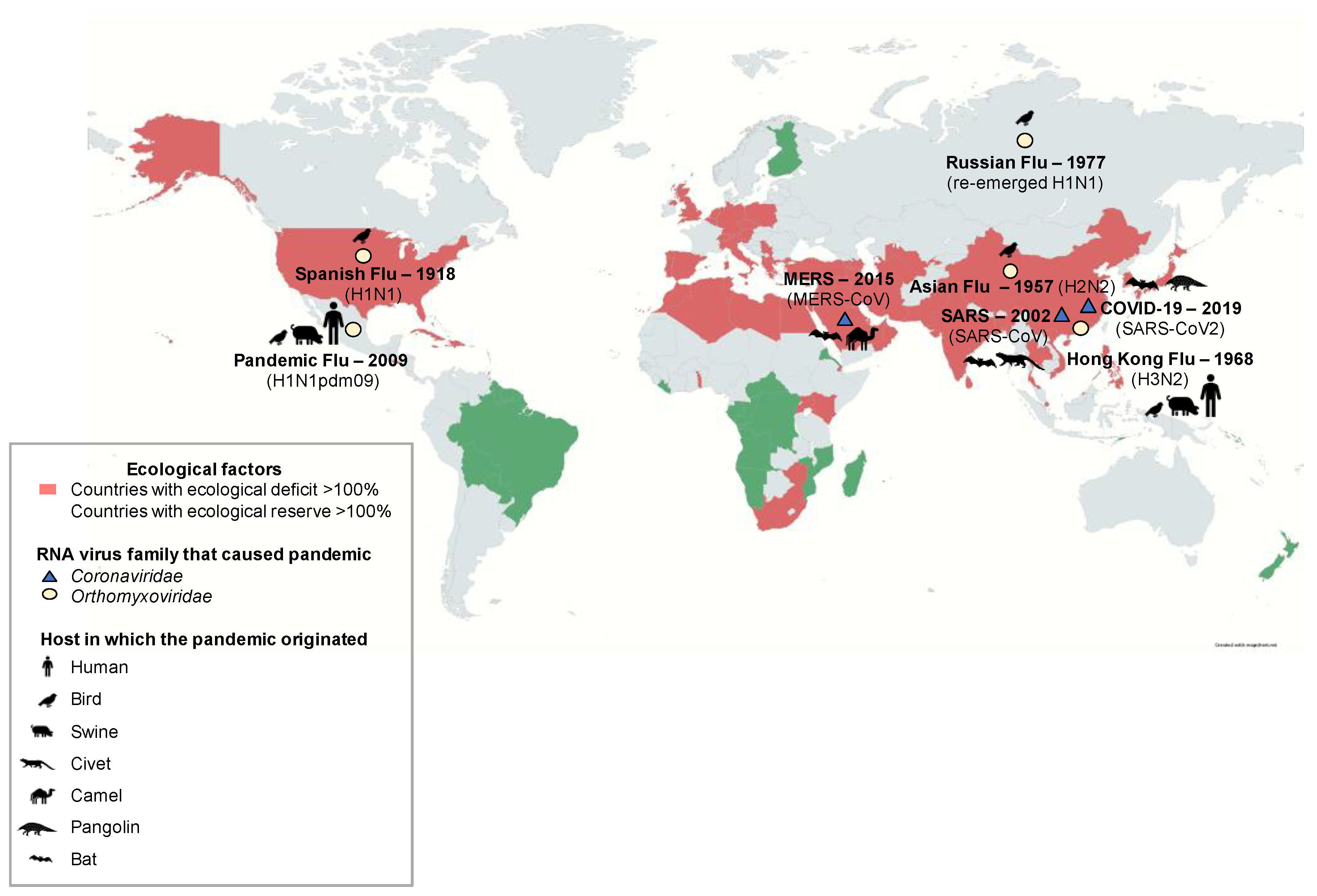
3.2. Other Factors That Affect the Interspecies Transmission
3.3. Case Study: Influenza Virus in Latin America
3.3.1. Factors Associated with the Virus and its Hosts
3.3.2. Other Factors That Affect the Interspecies Transmission
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Domingo, E.; Holland, J.J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.T.; Whitfield, Z.J.; Andino, R. Mechanisms and Concepts in RNA Virus Population Dynamics and Evolution. Annu. Rev. Virol. 2018, 5, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Reid, A.H.; Lourens, R.M.; Wang, R.; Jin, G.; Fanning, T.G. Characterization of the 1918 influenza virus polymerase genes. Nature 2005, 437, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Vijaykrishna, D.; Mukerji, R.; Smith, G.J.D. RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion. PLoS Pathog. 2015, 11, e1004902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilke, C.O.; Lenski, R.E.; Adami, C. Compensatory mutations cause excess of antagonistic epistasis in RNA secondary structure folding. BMC Evol. Biol. 2003, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Priyadarsini, S.L.; Suresh, M.; Huisingh, D. What can we learn from previous pandemics to reduce the frequency of emerging infectious diseases like COVID-19? Glob. Transit. 2020, 2, 202–220. [Google Scholar] [CrossRef] [PubMed]
- van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef]
- Westgeest, K.B.; Russell, C.A.; Lin, X.; Spronken, M.I.J.; Bestebroer, T.M.; Bahl, J.; van Beek, R.; Skepner, E.; Halpin, R.A.; de Jong, J.C.; et al. Genomewide analysis of reassortment and evolution of human influenza A(H3N2) viruses circulating between 1968 and 2011. J. Virol. 2014, 88, 2844–2857. [Google Scholar] [CrossRef] [Green Version]
- Scholtissek, C.; Stech, J.; Krauss, S.; Webster, R.G. Cooperation between the hemagglutinin of avian viruses and the matrix protein of human influenza A viruses. J. Virol. 2002, 76, 1781–1786. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Shi, Y.; Lu, X.; He, J.; Gao, F.; Yan, J.; Qi, J.; Gao, G.F. Bat-Derived Influenza Hemagglutinin H17 Does Not Bind Canonical Avian or Human Receptors and Most Likely Uses a Unique Entry Mechanism. Cell Rep. 2013, 3, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worobey, M.; Han, G.-Z.; Rambaut, A. A synchronized global sweep of the internal genes of modern avian influenza virus. Nature 2014, 508, 254–257. [Google Scholar] [CrossRef]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Ar Gouilh, M.; Puechmaille, S.J.; Diancourt, L.; Vandenbogaert, M.; Serra-Cobo, J.; Lopez Roïg, M.; Brown, P.; Moutou, F.; Caro, V.; Vabret, A.; et al. SARS-CoV related Betacoronavirus and diverse Alphacoronavirus members found in western old-world. Virology 2018, 517, 88–97. [Google Scholar] [CrossRef]
- Barbosa, A.; Varsani, A.; Morandini, V.; Grimaldi, W.; Vanstreels, R.E.T.; Diaz, J.I.; Boulinier, T.; Dewar, M.; González-Acuña, D.; Gray, R.; et al. Risk assessment of SARS-CoV-2 in Antarctic wildlife. Sci. Total Environ. 2020, 143352. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Boon, S.S.; Wang, M.H.; Chan, R.W.Y.; Chan, P.K.S. Genomic and evolutionary comparison between SARS-CoV-2 and other human coronaviruses. J. Virol. Methods 2020, 114032. [Google Scholar] [CrossRef]
- Clayton, B.A.; Middleton, D.; Bergfeld, J.; Haining, J.; Arkinstall, R.; Wang, L.; Marsh, G.A. Transmission routes for Nipah virus from Malaysia and Bangladesh. Emerg. Infect. Dis. 2012, 18, 1983–1993. [Google Scholar] [CrossRef]
- Leopardi, S.; Holmes, E.C.; Gastaldelli, M.; Tassoni, L.; Priori, P.; Scaravelli, D.; Zamperin, G.; De Benedictis, P. Interplay between co-divergence and cross-species transmission in the evolutionary history of bat coronaviruses. Infect. Genet. Evol. 2018, 58, 279–289. [Google Scholar] [CrossRef]
- Nieto-Rabiela, F.; Wiratsudakul, A.; Suzán, G.; Rico-Chávez, O. Viral networks and detection of potential zoonotic viruses in bats and rodents: A worldwide analysis. Zoonoses Public Health 2019, 66, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Laha, S.; Chakraborty, J.; Das, S.; Manna, S.K.; Biswas, S.; Chatterjee, R. Characterizations of SARS-CoV-2 mutational profile, spike protein stability and viral transmission. Infect. Genet. Evol. 2020, 85, 104445. [Google Scholar] [CrossRef] [PubMed]
- Barik, S. Genus-specific pattern of intrinsically disordered central regions in the nucleocapsid protein of coronaviruses. Comput. Struct. Biotechnol. J. 2020, 18, 1884–1890. [Google Scholar] [CrossRef]
- Wan, H.; Perez, D.R. Quail carry sialic acid receptors compatible with binding of avian and human influenza viruses. Virology 2006, 346, 278–286. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.-M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2020. [Google Scholar] [CrossRef]
- Huang, Y.; Feng, H.; Huang, L.; Yi, K.; Rong, E.; Chen, X.; Li, J.; Wang, Z.; Zhu, P.; Xiao-juan, L.; et al. Transcriptomic analyses reveal new genes and networks response to H5N1 influenza viruses in duck (Anas platyrhynchos). J. Integr. Agric. 2019, 18, 1460–1472. [Google Scholar] [CrossRef]
- Pruvot, M.; Khammavong, K.; Milavong, P.; Philavong, C.; Reinharz, D.; Mayxay, M.; Rattanavong, S.; Horwood, P.; Dussart, P.; Douangngeun, B.; et al. Toward a quantification of risks at the nexus of conservation and health: The case of bushmeat markets in Lao PDR. Sci. Total Environ. 2019, 676, 732–745. [Google Scholar] [CrossRef]
- Ding, N.-Z.; Xu, D.-S.; Sun, Y.-Y.; He, H.-B.; He, C.-Q. A permanent host shift of rabies virus from Chiroptera to Carnivora associated with recombination. Sci. Rep. 2017, 7, 289. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.K.; Hitchens, P.L.; Pandit, P.S.; Rushmore, J.; Evans, T.S.; Young, C.C.W.; Doyle, M.M. Global shifts in mammalian population trends reveal key predictors of virus spillover risk. Proc. R. Soc. B Biol. Sci. 2020, 287. [Google Scholar] [CrossRef] [Green Version]
- Dawood, A.A. Glicosilación, sitios de unión de ligandos y variaciones antigénicas entre la glicoproteína de membrana del COVID-19 y los coronavirus asociados. Vacunas 2020. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Ghorbani, A.; Samarfard, S.; Ramezani, A.; Izadpanah, K.; Afsharifar, A.; Eskandari, M.H.; Karbanowicz, T.P.; Peters, J.R. Quasi-species nature and differential gene expression of severe acute respiratory syndrome coronavirus 2 and phylogenetic analysis of a novel Iranian strain. Infect. Genet. Evol. 2020, 85, 104556. [Google Scholar] [CrossRef] [PubMed]
- Yin, C. Genotyping coronavirus SARS-CoV-2: Methods and implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef] [PubMed]
- Lemmin, T.; Kalbermatter, D.; Harder, D.; Plattet, P.; Fotiadis, D. Structures and dynamics of the novel S1/S2 protease cleavage site loop of the SARS-CoV-2 spike glycoprotein. J. Struct. Biol. X 2020, 4, 100038. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, V.; Aarnio, M.; Alava, M.; Alopaeus, V.; Atanasova, N.; Auvinen, M.; Balasubramanian, N.; Bordbar, H.; Erästö, P.; Grande, R.; et al. Modelling aerosol transport and virus exposure with numerical simulations in relation to SARS-CoV-2 transmission by inhalation indoors. Saf. Sci. 2020, 130, 104866. [Google Scholar] [CrossRef] [PubMed]
- Bedford, T.; Cobey, S.; Beerli, P.; Pascual, M. Global migration dynamics underlie evolution and persistence of human influenza a (H3N2). PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, L.H. Impact of pandemic control over airport economics: Reconciling public health with airport business through a streamlined approach in pandemic control. J. Air Transp. Manag. 2015, 44–45, 42–53. [Google Scholar] [CrossRef]
- Gouilh, M.A.; Puechmaille, S.J.; Gonzalez, J.-P.; Teeling, E.; Kittayapong, P.; Manuguerra, J.-C. SARS-Coronavirus ancestor’s foot-prints in South-East Asian bat colonies and the refuge theory. Infect. Genet. Evol. 2011, 11, 1690–1702. [Google Scholar] [CrossRef]
- The Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services (IPBES). IPBES-7 Plenary. Available online: https://www.ipbes.net/events/ipbes-7-plenary (accessed on 12 February 2021).
- Global Footprint Network. Available online: https://data.footprintnetwork.org/ (accessed on 12 February 2021).
- Collection 2.0 of Annual Maps of Land Cover, Land Use and Land Use Changes between 1985 to 2018 in the Pan-Amazon. Available online: http://amazonia.mapbiomas.org (accessed on 11 March 2021).
- Global Forest Watch. Available online: https://www.globalforestwatch.org/ (accessed on 11 March 2021).
- Lovejoy, T.E.; Nobre, C. Amazon tipping point: Last chance for action. Sci. Adv. 2019, 5, eaba2949. [Google Scholar] [CrossRef] [Green Version]
- Hess, G. Disease in Metapopulation Models: Implications for Conservation. Ecology 1996, 77, 1617–1632. [Google Scholar] [CrossRef]
- Huang, Z.Y.X.; VAN Langevelde, F.; Estrada-Peña, A.; Suzán, G.; DE Boer, W.F. The diversity-disease relationship: Evidence for and criticisms of the dilution effect. Parasitology 2016, 143, 1075–1086. [Google Scholar] [CrossRef]
- Plowright, R.K.; Foley, P.; Field, H.E.; Dobson, A.P.; Foley, J.E.; Eby, P.; Daszak, P. Urban habituation, ecological connectivity and epidemic dampening: The emergence of Hendra virus from flying foxes (Pteropus spp.). Proc. Biol. Sci. 2011, 278, 3703–3712. [Google Scholar] [CrossRef] [Green Version]
- Lowenstein, C.; Waters, W.F.; Roess, A.; Leibler, J.H.; Graham, J.P. Animal husbandry practices and perceptions of zoonotic infectious disease risks among livestock keepers in a rural parish of quito, Ecuador. Am. J. Trop. Med. Hyg. 2016, 95, 1450–1458. [Google Scholar] [CrossRef] [Green Version]
- Paige, S.B.; Malavé, C.; Mbabazi, E.; Mayer, J.; Goldberg, T.L. Uncovering zoonoses awareness in an emerging disease ‘hotspot’. Soc. Sci. Med. 2015, 129, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, M.J.; De Carvalho, M.H.F.; Hoet, A.E.; Vigilato, M.A.N.; Pompei, J.C.; Cosivi, O.; Del Rio Vilas, V.J. Building the road to a regional zoonoses strategy: A survey of zoonoses programmes in the Americas. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.E. The traveller and emerging infections: Sentinel, courier, transmitter. J. Appl. Microbiol. 2003, 94, 1S–11S. [Google Scholar] [CrossRef] [PubMed]
- Ruan, S.; Wang, W.; Levin, S.A. The effect of global travel on the spread of sars. Math. Biosci. Eng. 2006, 3, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Lin, Y.; Xie, D. Evolutionary and Ecological Dynamics of Transboundary Disease Caused by H5N1 Virus in Southeast Asia. Transbound. Emerg. Dis. 2015, 62, 315–327. [Google Scholar] [CrossRef]
- Caini, S.; Alonso, W.J.; Balmaseda, A.; Bruno, A.; Bustos, P.; Castillo, L.; De Lozano, C.; De Mora, D.; Fasce, R.A.; De Almeida, W.A.F.; et al. Characteristics of seasonal influenza A and B in Latin America: Influenza surveillance data from ten countries. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Gorini da Veiga, A.B.; Kretzmann, N.A.; Corrêa, L.T.; Goshiyama, A.M.; Baccin, T.; Ardenghi, P.; Matias, F.; Gregianini, T.S.; Alves d’Azevedo, P. Viral load and epidemiological profile of patients infected by pandemic influenza A (H1N1) 2009 and seasonal influenza A virus in Southern Brazil. J. Med. Virol. 2012, 84, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Pollett, S.; Ghersi, B.; Silva, M.; Simons, M.P.; Icochea, E.; Gonzalez, A.E.; Segovia, K.; Kasper, M.R.; Montgomery, J.M.; et al. The genetic diversity of influenza A viruses in wild birds in Peru. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.; Cavia, R.; Carbajo, A.E.; Bellomo, C.; Gonzalez Capria, S.; Padula, P. Spatial and temporal analysis of the distribution of hantavirus pulmonary syndrome in Buenos Aires Province, and its relation to rodent distribution, agricultural and demographic variables. Trop. Med. Int. Heal. 2004, 9, 508–519. [Google Scholar] [CrossRef]
- Palekar, R.; Rodriguez, A.; Avila, C.; Barrera, G.; Barrera, M.; Brenes, H.; Bruno, A.; El Omeiri, N.; Fasce, R.; Ferreira de Almeida, W.; et al. Patterns of influenza B circulation in Latin America and the Caribbean, 2010–2017. PLoS ONE 2019, 14, e0219595. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Codeço, C.; Luz, P. Seasonal dynamics of influenza in Brazil: The latitude effect. BMC Infect. Dis. 2018, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, J.A.; Resende, P.; Araya, J.L.; Barrera, G.B.; Baumeister, E.; Caicedo, A.B.; Coppola, L.; de Mello, W.A.; de Mora, D.; dos Santos, M.C.; et al. Genetic evolution of influenza viruses among selected countries in Latin America, 2017–2018. PLoS ONE 2020, 15. [Google Scholar] [CrossRef]
- Pereda, A.; Rimondi, A.; Cappuccio, J.; Sanguinetti, R.; Angel, M.; Ye, J.; Sutton, T.; Dibárbora, M.; Olivera, V.; Craig, M.I.; et al. Evidence of reassortment of pandemic H1N1 influenza virus in swine in Argentina: Are we facing the expansion of potential epicenters of influenza emergence? Influenza Other Respi. Viruses 2011, 5, 409–412. [Google Scholar] [CrossRef]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New World Bats Harbor Diverse Influenza A Viruses. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [Green Version]
- De Araujo, J.; Thomazelli, L.M.; Henriques, D.A.; Lautenschalager, D.; Ometto, T.; Dutra, L.M.; Aires, C.C.; Favorito, S.; Durigon, E.L. Detection of hantavirus in bats from remaining rain forest in São Paulo, Brazil. BMC Res. Notes 2012, 5. [Google Scholar] [CrossRef] [Green Version]
- Milazzo, M.L.; Duno, G.; Utrera, A.; Richter, M.H.; Duno, F.; de Manzione, N.; Fulhorst, C.F. Natural host relationships of hantaviruses native to western Venezuela. Vector Borne Zoonotic Dis. 2010, 10, 605–611. [Google Scholar] [CrossRef]
- Prist, P.R.; Uriarte, M.; Tambosi, L.R.; Prado, A.; Pardini, R.; D’Andrea, P.S.; Metzger, J.P. Landscape, environmental and social predictors of Hantavirus risk in São Paulo, Brazil. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, B.R.; Loureiro, N.; Strecht, L.; Gentile, R.; Oliveira, R.C.; Guterres, A.; Fernandes, J.; Mattos, L.H.B.V.; Raboni, S.M.; Rubio, G.; et al. Population ecology of hantavirus rodent hosts in Southern Brazil. Am. J. Trop. Med. Hyg. 2014, 91, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saasa, N.; Sánchez-Hernández, C.; de Lourdes Romero-Almaraz, M.; Guerrero-Ibarra, E.; Almazán-Catalán, A.; Yoshida, H.; Miyashita, D.; Ishizuka, M.; Sanada, T.; Seto, T.; et al. Ecology of hantaviruses in Mexico: Genetic identification of rodent host species and spillover infection. Virus Res. 2012, 168, 88–96. [Google Scholar] [CrossRef]
- Alonso, W.J.; Viboud, C.; Simonsen, L.; Hirano, E.W.; Daufenbach, L.Z.; Miller, M.A. Seasonality of influenza in Brazil: A traveling wave from the amazon to the subtropics. Am. J. Epidemiol. 2007, 165, 1434–1442. [Google Scholar] [CrossRef]
- Hamrick, P.N.; Aldighieri, S.; Machado, G.; Leonel, D.G.; Vilca, L.M.; Uriona, S.; Schneider, M.C. Geographic patterns and environmental factors associated with human yellow fever presence in the Americas. PLoS Negl. Trop. Dis. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Morales, A.J.; Gallego, V.; Escalera-Antezana, J.P.; Méndez, C.A.; Zambrano, L.I.; Franco-Paredes, C.; Suárez, J.A.; Rodriguez-Enciso, H.D.; Balbin-Ramon, G.J.; Savio-Larriera, E.; et al. COVID-19 in Latin America: The implications of the first confirmed case in Brazil. Travel Med. Infect. Dis. 2020, 35, 101613. [Google Scholar] [CrossRef] [PubMed]
- Ministerio de Ambiente y Desarrollo Sostenible Pacto de Leticia por la Amazonía. Available online: https://www.minambiente.gov.co/images/2019/PLAN_DE_ACCION_PACTO_DE_LETICIA_POR_LA_AMAZONIA.pdf (accessed on 25 February 2021).
- Allen, T.; Murray, K.A.; Zambrana-Torrelio, C.; Morse, S.S.; Rondinini, C.; Di Marco, M.; Breit, N.; Olival, K.J.; Daszak, P. Global hotspots and correlates of emerging zoonotic diseases. Nat. Commun. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez-Munoz, S.; Upegui-Porras, N.; Gomez, A.P.; Ramirez-Nieto, G. Key Factors That Enable the Pandemic Potential of RNA Viruses and Inter-Species Transmission: A Systematic Review. Viruses 2021, 13, 537. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040537
Alvarez-Munoz S, Upegui-Porras N, Gomez AP, Ramirez-Nieto G. Key Factors That Enable the Pandemic Potential of RNA Viruses and Inter-Species Transmission: A Systematic Review. Viruses. 2021; 13(4):537. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040537
Chicago/Turabian StyleAlvarez-Munoz, Santiago, Nicolas Upegui-Porras, Arlen P. Gomez, and Gloria Ramirez-Nieto. 2021. "Key Factors That Enable the Pandemic Potential of RNA Viruses and Inter-Species Transmission: A Systematic Review" Viruses 13, no. 4: 537. https://0-doi-org.brum.beds.ac.uk/10.3390/v13040537