
Genetic Insights into Feline Parvovirus: Evaluation of Viral Evolutionary Patterns and Association between Phylogeny and Clinical Variables

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phylogenetic Analysis, Viral Evolution and Phylogeography
2.2. Association between Clinicopathological Data and Italian FPV Strain Phylogenesis
3. Results
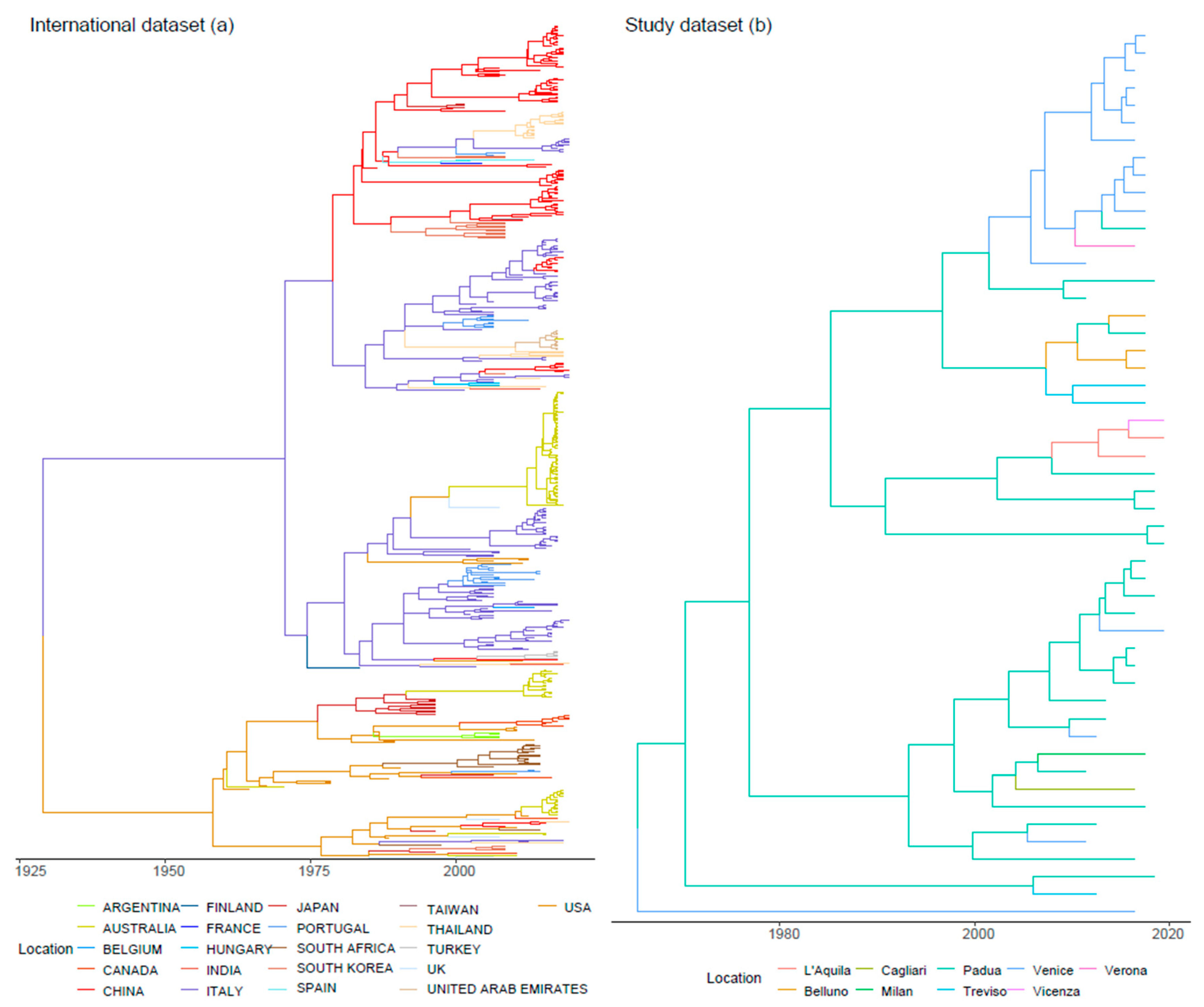
3.1. Datasets
3.2. Phylogenetic Analysis, Viral Evolution, and Phylogeography
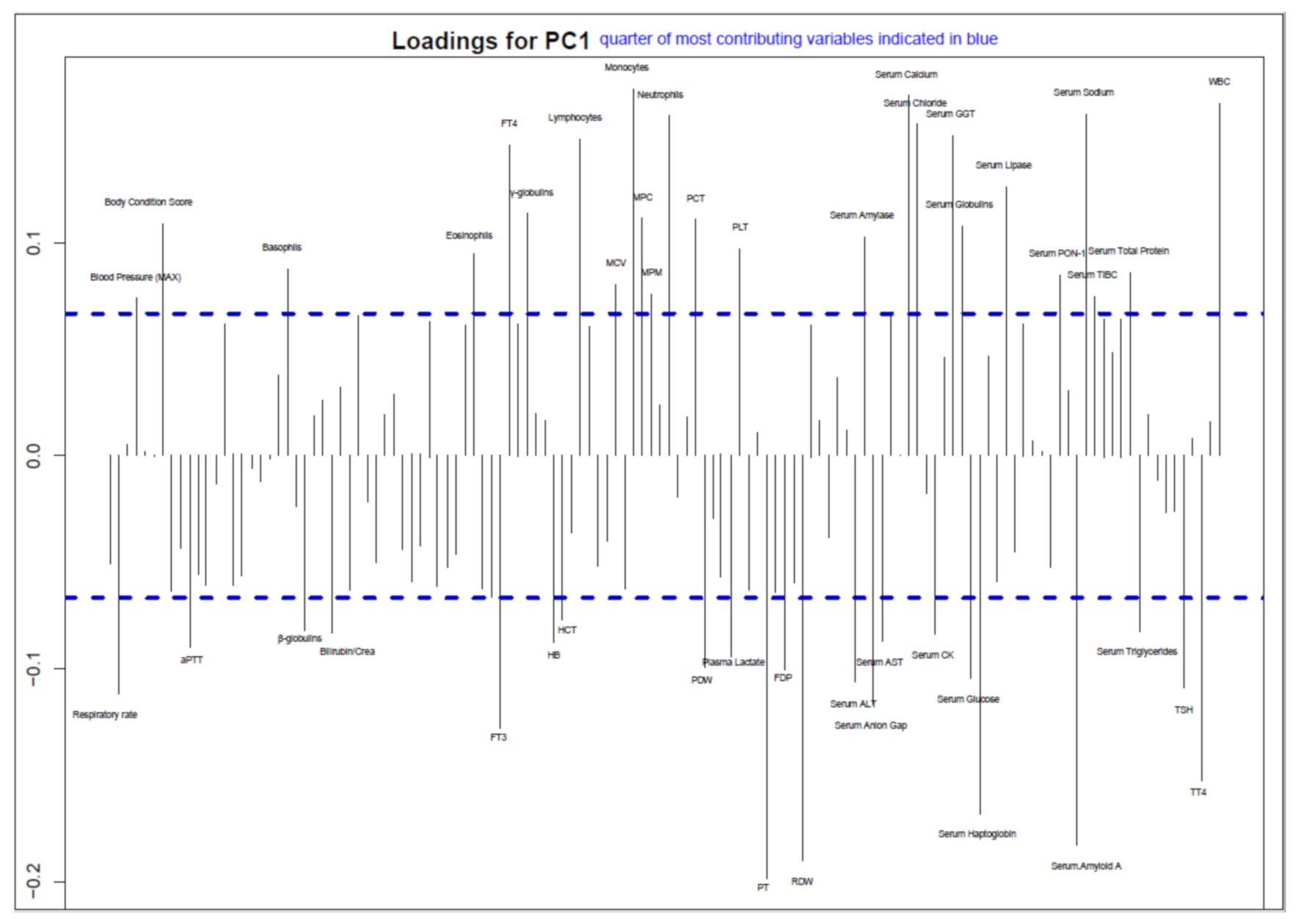
3.3. Association between Clinicopathological Data and Italian FPV Strain Phylogenesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef]
- Squires, R. An update on aspects of viral gastrointestinal diseases of dogs and cats. N. Z. Vet. J. 2003, 51, 252–261. [Google Scholar] [CrossRef]
- Hoelzer, K.; Parrish, C.R. The emergence of parvoviruses of carnivores. Vet. Res. 2010, 41, 39. [Google Scholar] [CrossRef] [Green Version]
- Hoelzer, K.; Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Phylogenetic analysis reveals the emergence, evolution and dispersal of carnivore parvoviruses. J. Gen. Virol. 2008, 89, 2280–2289. [Google Scholar] [CrossRef]
- Miranda, C.; Thompson, G. Canine parvovirus: The worldwide occurrence of antigenic variants. J. Gen. Virol. 2016, 97, 2043–2057. [Google Scholar] [CrossRef] [PubMed]
- DeCaro, N.; Desario, C.; Miccolupo, A.; Campolo, M.; Parisi, A.; Martella, V.; Amorisco, F.; Lucente, M.S.; Lavazza, A.; Buonavoglia, C. Genetic analysis of feline panleukopenia viruses from cats with gastroenteritis. J. Gen. Virol. 2008, 89, 2290–2298. [Google Scholar] [CrossRef] [PubMed]
- Verge, J.; Christoforoni, N. La gastroenterite infectieuse des chats; est-elle due à un virus filtrable. CR Seances Soc. Biol. Fil. 1928, 99, 312. [Google Scholar]
- Shackelton, L.A.; Parrish, C.R.; Truyen, U.; Holmes, E.C. High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc. Natl. Acad. Sci. USA 2005, 102, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrs, V.R. Feline Panleukopenia. Vet. Clin. N. Am. Small Anim. Pract. 2019, 49, 651–670. [Google Scholar] [CrossRef]
- Greene, C.E. Feline Enteric Viral Infections. In Infectious Diseases of the Dog and Cat, 4th ed.; Sykes, J., Greene, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 80–91. [Google Scholar]
- Franzo, G.; Tucciarone, C.M.; Casagrande, S.; Caldin, M.; Cortey, M.; Furlanello, T.; Legnardi, M.; Cecchinato, M.; Drigo, M. Canine parvovirus (CPV) phylogeny is associated with disease severity. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Porporato, F.; Horzinek, M.C.; Hofmann-Lehmann, R.; Ferri, F.; Gerardi, G.; Contiero, B.; Vezzosi, T.; Rocchi, P.; Auriemma, E.; Lutz, H.; et al. Survival estimates and outcome predictors for shelter cats with feline panleukopenia virus infection. J. Am. Vet. Med. Assoc. 2018, 253, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Kruse, B.; Unterer, S.; Horlacher, K.; Sauter-Louis, C.; Hartmann, K. Prognostic Factors in Cats with Feline Panleukopenia. J. Vet. Intern. Med. 2010, 24, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Petini, M.; Drigo, M.; Zoia, A. Prognostic value of systemic inflammatory response syndrome and serum concentrations of acute phase proteins, cholesterol, and total thyroxine in cats with panleukopenia. J. Vet. Intern. Med. 2020, 34, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Truyen, U.; Parrish, C.R. Feline panleukopenia virus: Its interesting evolution and current problems in immunoprophylaxis against a serious pathogen. Vet. Microbiol. 2013, 165, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Stuetzer, B.; Hartmann, K. Feline parvovirus infection and associated diseases. Vet. J. 2014, 201, 150–155. [Google Scholar] [CrossRef]
- Decaro, N.; Buonavoglia, C.; Barrs, V. Canine parvovirus vaccination and immunisation failures: Are we far from disease eradication? Vet. Microbiol. 2020, 247, 108760. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Elia, G.; Martella, V.; Desario, C.; Campolo, M.; Di Trani, L.; Tarsitano, E.; Tempesta, M.; Buonavoglia, C. A real-time PCR assay for rapid detection and quantitation of canine parvovirus type 2 in the feces of dogs. Vet. Microbiol. 2005, 105, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Pérez, R.; Calleros, L.; Marandino, A.; Sarute, N.; Iraola, G.; Grecco, S.; Blanc, H.; Vignuzzi, M.; Isakov, O.; Shomron, N.; et al. Phylogenetic and Genome-Wide Deep-Sequencing Analyses of Canine Parvovirus Reveal Co-Infection with Field Variants and Emergence of a Recent Recombinant Strain. PLoS ONE 2014, 9, e111779. [Google Scholar] [CrossRef] [Green Version]
- Tucciarone, C.M.; Franzo, G.; Mazzetto, E.; Legnardi, M.; Caldin, M.; Furlanello, T.; Cecchinato, M.; Drigo, M. Molecular insight into Italian canine parvovirus heterogeneity and comparison with the worldwide scenario. Infect. Genet. Evol. 2018, 66, 171–179. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian Population Dynamics Inference: A Coalescent-Based Model for Multiple Loci. Mol. Biol. Evol. 2012, 30, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.Y.W.; Shapiro, B. Skyline-plot methods for estimating demographic history from nucleotide sequences. Mol. Ecol. Resour. 2011, 11, 423–434. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J. TreeAnnotator, Version 1.7. 5. 2012. Available online: Beast.bio.ed.ac.uk/TreeAnnotator (accessed on 15 April 2013).
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [Green Version]
- Hackathon, R.; Bolker, B.; Butler, M.; Cowan, P.; de Vienne, D.; Eddelbuettel, D.; Holder, M.; Jombart, T.; Kembel, S.; Michonneau, F. Phylobase: Base Package for Phylogenetic Structures and Comparative Data. R Package Version 0.6, 5. 2013. Available online: https://cran.r-project.org/web/packages/phylobase/index.html (accessed on 8 February 2021).
- Jombart, T.; Balloux, F.; Dray, S. Adephylo: New tools for investigating the phylogenetic signal in biological traits. Bioinformatics 2010, 26, 1907–1909. [Google Scholar] [CrossRef] [PubMed]
- Keck, F.; Rimet, F.; Bouchez, A.; François, K. Phylosignal: An R package to measure, test, and explore the phylogenetic signal. Ecol. Evol. 2016, 6, 2774–2780. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Bell, J.; Cavanagh, K.; Tilley, L.; Smith, F.W.K. Veterinary Medical Guide to Dog and Cat Breeds; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- FEDIAF. Number of Pet Cats in Europe from 2010 to 2019. Statista. 2020. Available online: https://0-www-statista-com.brum.beds.ac.uk/statistics/516041/cat-population-europe-europe/ (accessed on 8 February 2021).
- Lauzi, S.; Stranieri, A.; Giordano, A.; Luzzago, C.; Zehender, G.; Paltrinieri, S.; Ebranati, E. Origin and transmission of Feline coronavirus type I in domestic cats from Northern Italy: A phylogeographic approach. Vet. Microbiol. 2020, 244, 108667. [Google Scholar] [CrossRef]
- Myrrha, L.W.; Silva, F.M.F.; Vidigal, P.M.P.; Resende, M.; Bressan, G.C.; Fietto, J.L.R.; Santos, M.R.; Silva, L.M.N.; Assao, V.S.; Nior, A.S.-J.; et al. Feline coronavirus isolates from a part of Brazil: Insights into molecular epidemiology and phylogeny inferred from the 7b gene. J. Vet. Med. Sci. 2019, 81, 1455–1460. [Google Scholar] [CrossRef]
- Cano-Ortiz, L.; Junqueira, D.M.; Comerlato, J.; Costa, C.S.; Zani, A.; Duda, N.B.; Tochetto, C.; Dos Santos, R.N.; Da Costa, F.V.A.; Roehe, P.M.; et al. Phylodynamics of the Brazilian feline immunodeficiency virus. Infect. Genet. Evol. 2017, 55, 166–171. [Google Scholar] [CrossRef]
- Pedersen, N.C. A review of feline infectious peritonitis virus infection: 1963–2008. J. Feline Med. Surg. 2009, 11, 225–258. [Google Scholar] [CrossRef] [PubMed]
- Coyne, K.P.; Christley, R.; Pybus, O.G.; Dawson, S.; Gaskell, R.M.; Radford, A.D. Large-Scale Spatial and Temporal Genetic Diversity of Feline Calicivirus. J. Virol. 2012, 86, 11356–11367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clegg, S.; Coyne, K.; Dawson, S.; Spibey, N.; Gaskell, R.; Radford, A. Canine parvovirus in asymptomatic feline carriers. Vet. Microbiol. 2012, 157, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.E.; Melian, C.; Nichols, R. Measurement of serum concentrations of free thyroxine, total thyroxine, and total triiodothyronine in cats with hyperthyroidism and cats with nonthyroidal disease. J. Am. Vet. Med. Assoc. 2001, 218, 529–536. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tucciarone, C.M.; Franzo, G.; Legnardi, M.; Lazzaro, E.; Zoia, A.; Petini, M.; Furlanello, T.; Caldin, M.; Cecchinato, M.; Drigo, M. Genetic Insights into Feline Parvovirus: Evaluation of Viral Evolutionary Patterns and Association between Phylogeny and Clinical Variables. Viruses 2021, 13, 1033. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061033
Tucciarone CM, Franzo G, Legnardi M, Lazzaro E, Zoia A, Petini M, Furlanello T, Caldin M, Cecchinato M, Drigo M. Genetic Insights into Feline Parvovirus: Evaluation of Viral Evolutionary Patterns and Association between Phylogeny and Clinical Variables. Viruses. 2021; 13(6):1033. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061033
Chicago/Turabian StyleTucciarone, Claudia Maria, Giovanni Franzo, Matteo Legnardi, Elena Lazzaro, Andrea Zoia, Matteo Petini, Tommaso Furlanello, Marco Caldin, Mattia Cecchinato, and Michele Drigo. 2021. "Genetic Insights into Feline Parvovirus: Evaluation of Viral Evolutionary Patterns and Association between Phylogeny and Clinical Variables" Viruses 13, no. 6: 1033. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061033