
Genetic Characterization of a New HIV-1 Sub-Subtype A in Cabo Verde, Denominated A8

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Amplification of HIV-1 Full-Length Genomes and Phylogenetic Analyses
2.3. Genetic Distances
2.4. Drug Resistance Analysis (DRM)
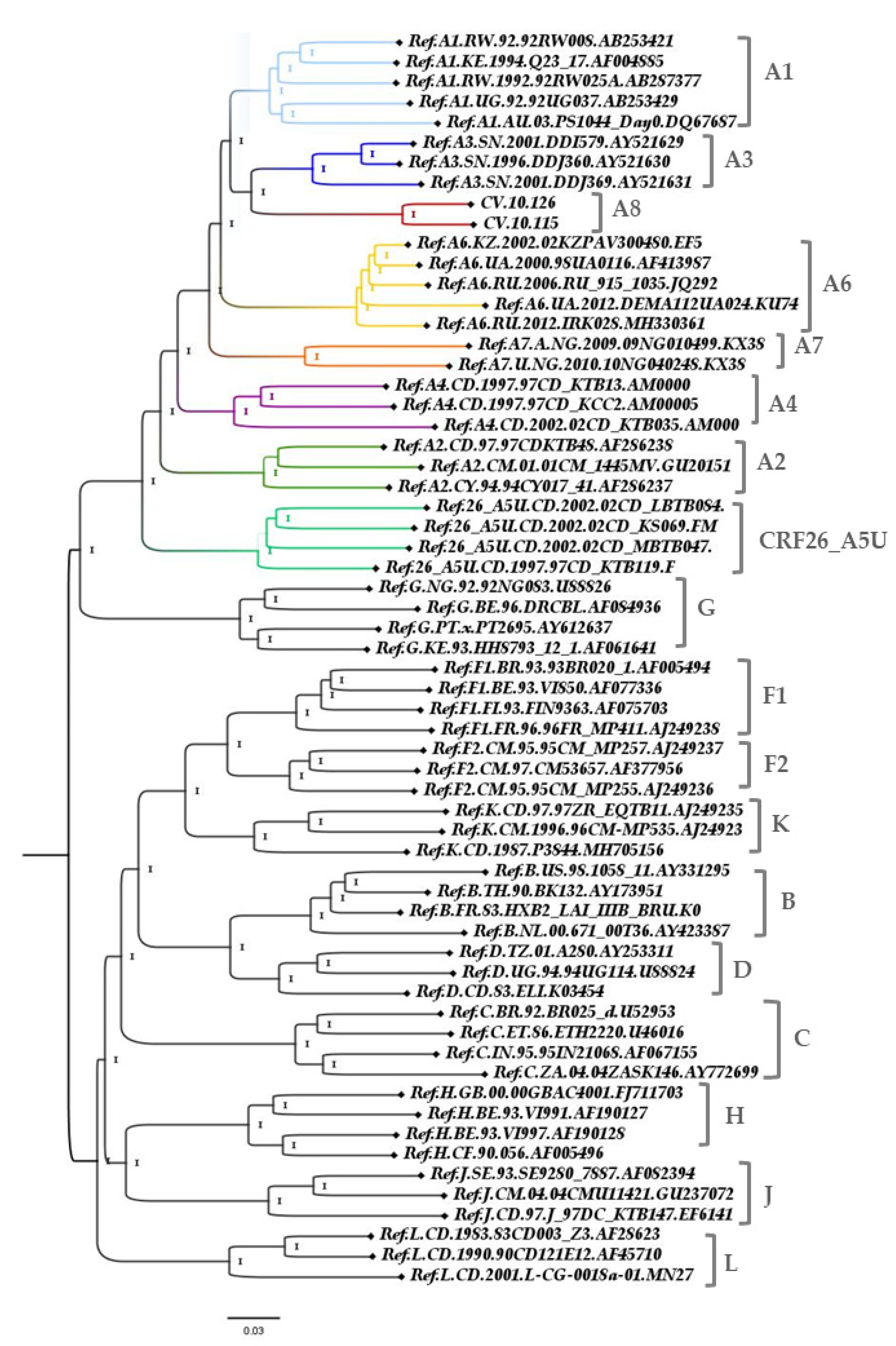
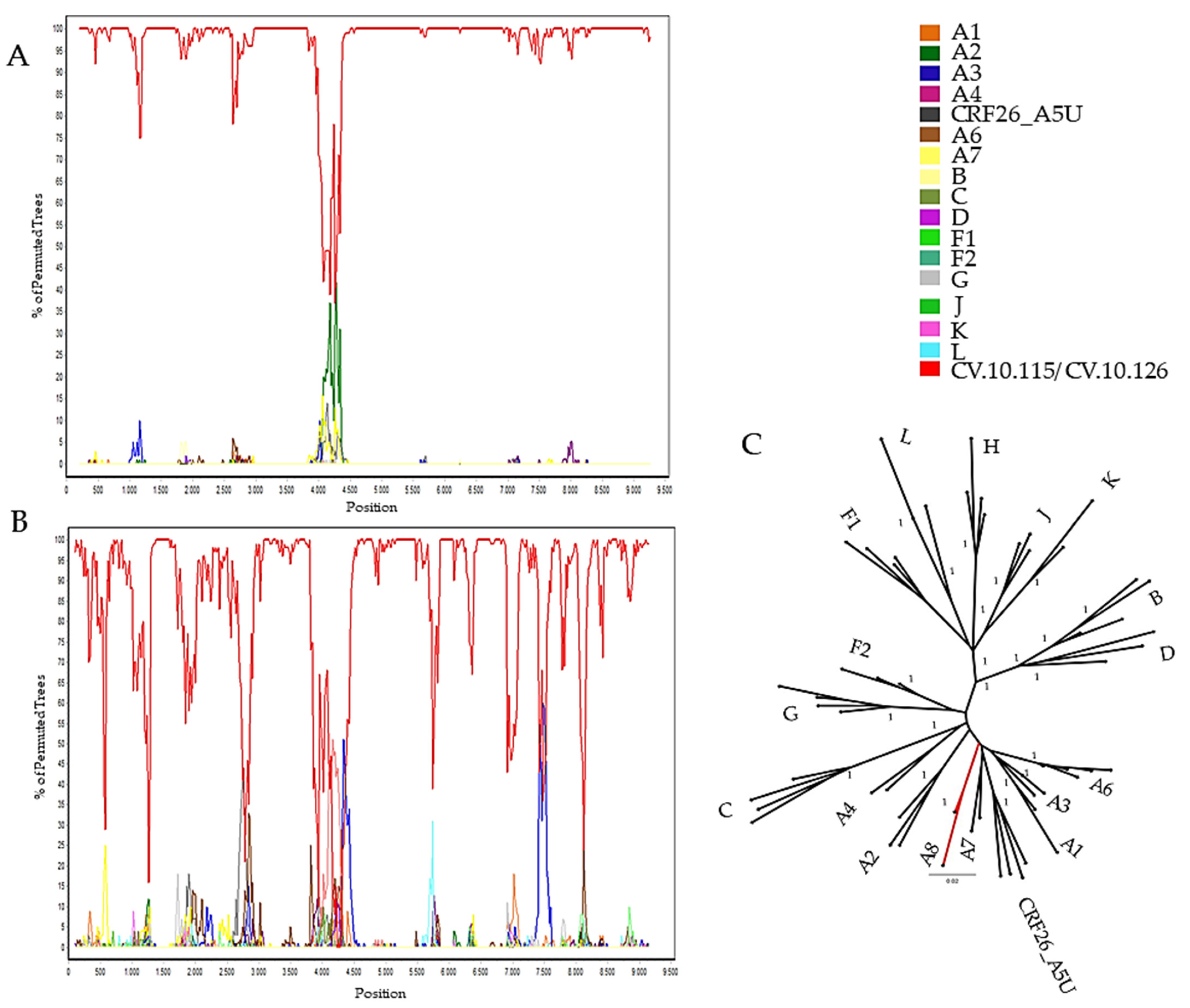
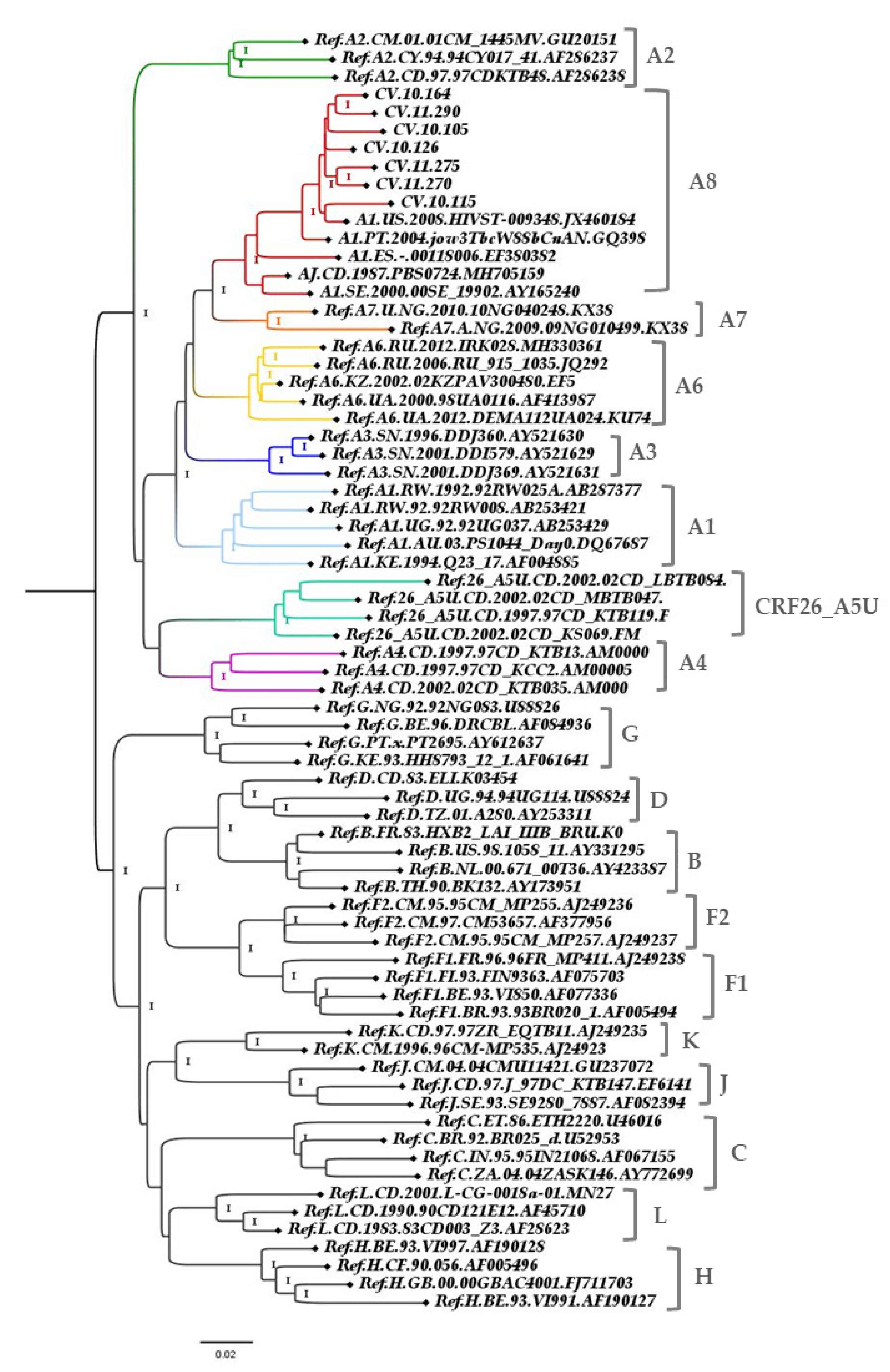
3. Results
3.1. Sociodemographic and Clinical Data
3.2. Genome Amplification and Sequence Analysis
3.3. Genetic Distance
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Plantier, J.C.; Leoz, M.; Dickerson, J.E.; De Oliveira, F.; Cordonnier, F.; Lemée, V.; Damond, F.; Robertson, D.L.; Simon, F. A new human immunodeficiency virus derived from gorillas. Nat. Med. 2009, 15, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Roques, P.; Robertson, D.L.; Souquière, S.; Apetrei, C.; Nerrienet, E.; Barré-Sinoussi, F.; Müller-Trutwin, M.; Simon, F. Phylogenetic characteristics of three new HIV-1 N strains and implications for the origin of group N. Aids 2004, 18, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Simon, F.; Mauclère, P.; Roques, P.; Loussert-Ajaka, I.; Müller-Trutwin, M.C.; Saragosti, S.; Georges-Courbot, M.C.; Barré-Sinoussi, F.; Brun-VÉZINET, F. Identification of a new human immunodeficiency virus type 1 distinct from group M and group O. Nat. Med. 1998, 4, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.L. HIV-1 Nomenclature Proposal. Science 2000, 288, 55–56. [Google Scholar] [CrossRef] [PubMed]
- Los Alamos Database. Available online: http://www.hiv.lanl.gov/ (accessed on 13 November 2020).
- Yamaguchi, J.; Vallari, A.; McArthur, C.; Sthreshley, L.; Cloherty, G.A.; Berg, M.G.; Rodgers, M.A. Brief Report: Complete Genome Sequence of CG-0018a-01 Establishes HIV-1 Subtype L. J. Acquir. Immune Defic. Syndr. 2020, 83, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Désiré, N.; Cerutti, L.; Le Hingrat, Q.; Perrier, M.; Emler, S.; Calvez, V.; Descamps, D.; Marcelin, A.G.; Hué, S.; Visseaux, B. Characterization update of HIV-1 M subtypes diversity and proposal for subtypes A and D sub-subtypes reclassification. Retrovirology 2018, 15, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, N.; Bazepeo, S.E.; Mulanga, C.; Delaporte, E.; Peeters, M. Genetic characterization of eight full-length HIV type 1 genomes from the democratic republic of Congo (DRC) reveal a new subsubtype, A5, in the a radiation that predominates in the recombinant structure of CRF26-A5U. AIDS Res. Hum. Retrovir. 2009, 25, 823–832. [Google Scholar] [CrossRef] [PubMed]
- UNAIDS. Available online: https://unaids.org.br/ (accessed on 8 January 2021).
- De Pina-Araujo, I.I.M.; Guimarães, M.L.; Bello, G.; Vicente, A.C.P.; Morgado, M.G. Profile of the HIV epidemic in Cape Verde: Molecular epidemiology and drug resistance mutations among HIV-1 and HIV-2 infected patients from distinct islands of the archipelago. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Reis, M.N.D.G.; Bello, G.; Guimarães, M.L.; Stefani, M.M.A. Characterization of HIV-1 CRF90_BF1 and putative novel CRFs_BF1 in Central West, North and Northeast Brazilian regions. PLoS ONE 2017, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A. FigTree v. 1.4.4. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 17 March 2021).
- Basic Local Alignment Search Tool (BLAST). Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 30 November 2020).
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gifford, R.J.; Liu, T.F.; Rhee, S.Y.; Kiuchi, M.; Hue, S.; Pillay, D.; Shafer, R.W. The calibrated population resistance tool: Standardized genotypic estimation of transmitted HIV-1 drug resistance. Bioinformatics 2009, 25, 1197–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.F.; Shafer, R.W. Web resources for HIV type 1 genotypic-resistance test interpretation. Clin. Infect. Dis. 2006, 42, 1608–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemelaar, J.; Elangovan, R.; Yun, J.; Dickson-Tetteh, L.; Kirtley, S.; Gouws-Williams, E.; Ghys, P.D.; Abimiku, A.G.; Agwale, S.; Archibald, C.; et al. Global and regional epidemiology of HIV-1 recombinants in 1990–2015: A systematic review and global survey. Lancet HIV 2020, 7, e772–e781. [Google Scholar] [CrossRef]
- Gao, F.; Vidal, N.; Li, Y.; Trask, S.A.; Chen, Y.; Kostrikis, L.G.; Ho, D.D.; Kim, J.; Oh, M.D.; Choe, K.; et al. Evidence of two distinct subsubtypes within the HIV-1 subtype A radiation. AIDS Res. Hum. Retrovir. 2001, 17, 675–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, B.T.; Leitner, T.; Paraskevis, D.; Peeters, M. Primate immunodeficiency virus classification and nomenclature: Review. Infect. Genet. Evol. 2016, 46, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Tongo, M.; Harkins, G.W.; Dorfman, J.R.; Billings, E.; Tovanabutra, S.; de Oliveira, T.; Martin, D.P. Unravelling the complicated evolutionary and dissemination history of HIV-1M subtype A lineages. Virus Evol. 2018, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Schlösser, M.; Kartashev, V.V.; Mikkola, V.H.; Shemshura, A.; Saukhat, S.; Kolpakov, D.; Suladze, A.; Tverdokhlebova, T.; Hutt, K.; Heger, E.; et al. HIV-1 sub-subtype a6: Settings for normalised identification and molecular epidemiology in the Southern Federal District, Russia. Viruses 2020, 12, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pina-Araujo, I.I.M.; Delatorre, E.; Guimarães, M.L.; Morgado, M.G.; Bello, G. Origin and population dynamics of a novel HIV-1 subtype G clade circulating in Cape Verde and Portugal. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Economic Community of West African States ECOWAS. Available online: https://ine.cv/wp-content/uploads/2016/10/IMC-2013-Migracoes.pdf. (accessed on 8 January 2021).




{kind=link}
{kind=link}
{kind=link}
| Sample | Island | Year of Diagnosis | Year of Collection | Gender | Age | ARV Status | DRM | GenBank Accession |
|---|---|---|---|---|---|---|---|---|
| CV.10.105 | Santiago | - | 2010 | Female | 19 | treated | NNRTI-E138A | KJ395593 (pol)/MW353966 (partial genome) |
| CV.10.115 | Santiago | 2001 | 2010 | Female | 41 | treated | NNRTI-M230I | KJ395594 (pol)/MW353967 (complete genome) |
| CV.10.126 | Santiago | 2007 | 2010 | Female | 44 | treated | None | KJ395596 (pol)/MW353968 (complete genome) |
| CV.10.164 | Santiago | 2009 | 2010 | Male | 40 | naive | None | KJ395613 (pol) |
| CV.11.270 | Santiago | 1995 | 2011 | Female | 52 | treated | None | KJ395697 (pol)/MW353969 (partial genome) |
| CV.11.275 | Santiago | - | 2011 | Female | 45 | naive | None | KJ395701 (pol) |
| CV.11.290 | Sal | 2009 | 2011 | Male | 44 | naive | None | KJ395705 (pol)/MW353970 (partial genome) |
| Genetic Distance Average (%) | |||||||
|---|---|---|---|---|---|---|---|
| Intragroup | Inter-Sub-Subtype A X A8 | ||||||
| A8 (n = 2) | A1 (n = 5) | A2 (n = 5) | A3 (n = 3) | A4 (n = 3) | A6 (n = 5) | A7 (n = 2) | |
| Complete Genome (803 → 9496) | 5.2 | 12.2 (11.9–12.6) | 14.8 (14.1–15.4) | 11.5 (11.2–12.0) | 14.0 (13.3–14.7) | 12.8 (12.2–13.8) | 14.0 (13.6–14.5) |
| GAG (803 → 2280) | 4.2 | 9.9 (8.5–11.9) | 12.1 (11.0–13.8) | 9.6 (8.5–10.3) | 11.9 (11.5–12.8) | 10.2 (9.7–13.0) | 11.3 (10.5–12.3) |
| POL (2065 → 5100) | 4.0 | 8.8 (8.5–9.4) | 10.8 (10.5–11.1) | 8.1 (7.6–8.8) | 9.9 (9.2–10.6) | 8.6 (7.8–9.7) | 8.5 (7.9–9.2) |
| ENV (6223 → 8804) | 7.4 | 16.0 (14.9–16.8) | 18.4 (17.5–19.5) | 14.7 (14.1–15.0) | 19.0 (17.6–20.4) | 17.7 (16.8–17.4) | 18.8 (18.4–19.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendes Da Silva, R.K.; Monteiro de Pina Araujo, I.I.; Venegas Maciera, K.; Gonçalves Morgado, M.; Lindenmeyer Guimarães, M. Genetic Characterization of a New HIV-1 Sub-Subtype A in Cabo Verde, Denominated A8. Viruses 2021, 13, 1093. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061093
Mendes Da Silva RK, Monteiro de Pina Araujo II, Venegas Maciera K, Gonçalves Morgado M, Lindenmeyer Guimarães M. Genetic Characterization of a New HIV-1 Sub-Subtype A in Cabo Verde, Denominated A8. Viruses. 2021; 13(6):1093. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061093
Chicago/Turabian StyleMendes Da Silva, Rayana Katylin, Isabel Inês Monteiro de Pina Araujo, Karine Venegas Maciera, Mariza Gonçalves Morgado, and Monick Lindenmeyer Guimarães. 2021. "Genetic Characterization of a New HIV-1 Sub-Subtype A in Cabo Verde, Denominated A8" Viruses 13, no. 6: 1093. https://0-doi-org.brum.beds.ac.uk/10.3390/v13061093