
The Serological Cross-Detection of Bat-Borne Hantaviruses: A Valid Strategy or Taking Chances?

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phylogenetic Analyses
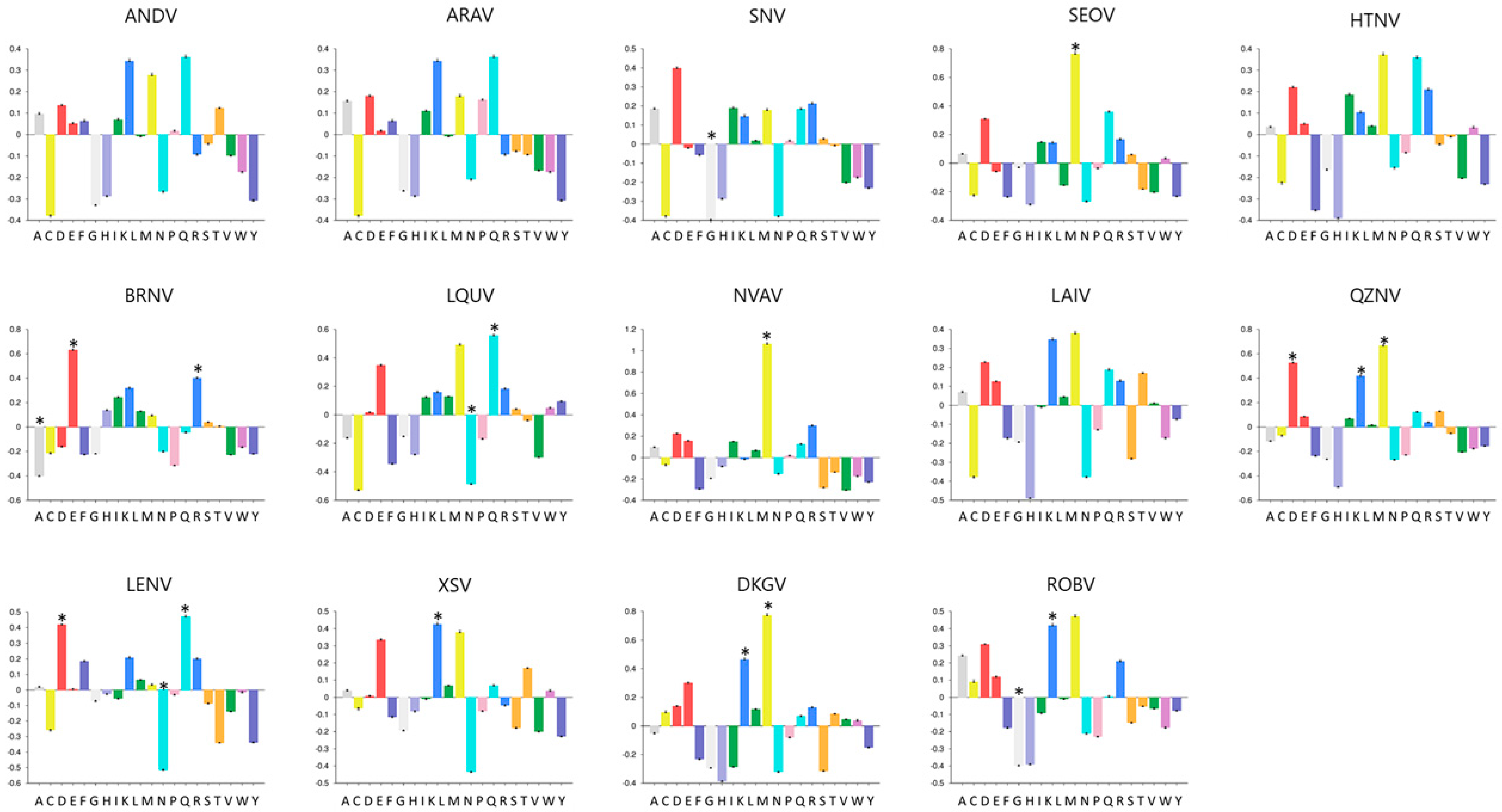
2.2. Amino Acid Composition Analysis
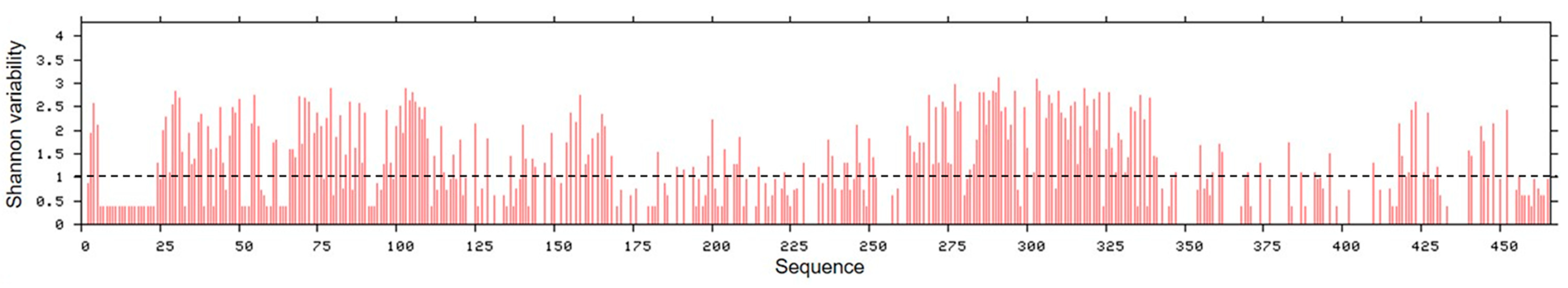
2.3. Hantavirus NP Amino Acid Variability Analysis
2.4. Analysis of Secondary Structure and Solvent Accessibility
2.5. Three-Dimensional (3D) Structure Modeling
2.6. Conformational B-Cell Epitope Prediction
2.7. Conservancy Analysis of the Selected Epitopes
3. Results
3.1. Phylogenetic Analyses
3.2. NPs Vary in Length and Amino Acid Composition Profiles among Hantaviruses
3.3. C-Terminal Region of NPs Were More Conserved among the Studied Hantavirus
3.4. Hantavirus NPs Share Similar Secondary Structures and Solvent Accessibility
3.5. Validation of Phylogroup II Hantavirus NPs Models
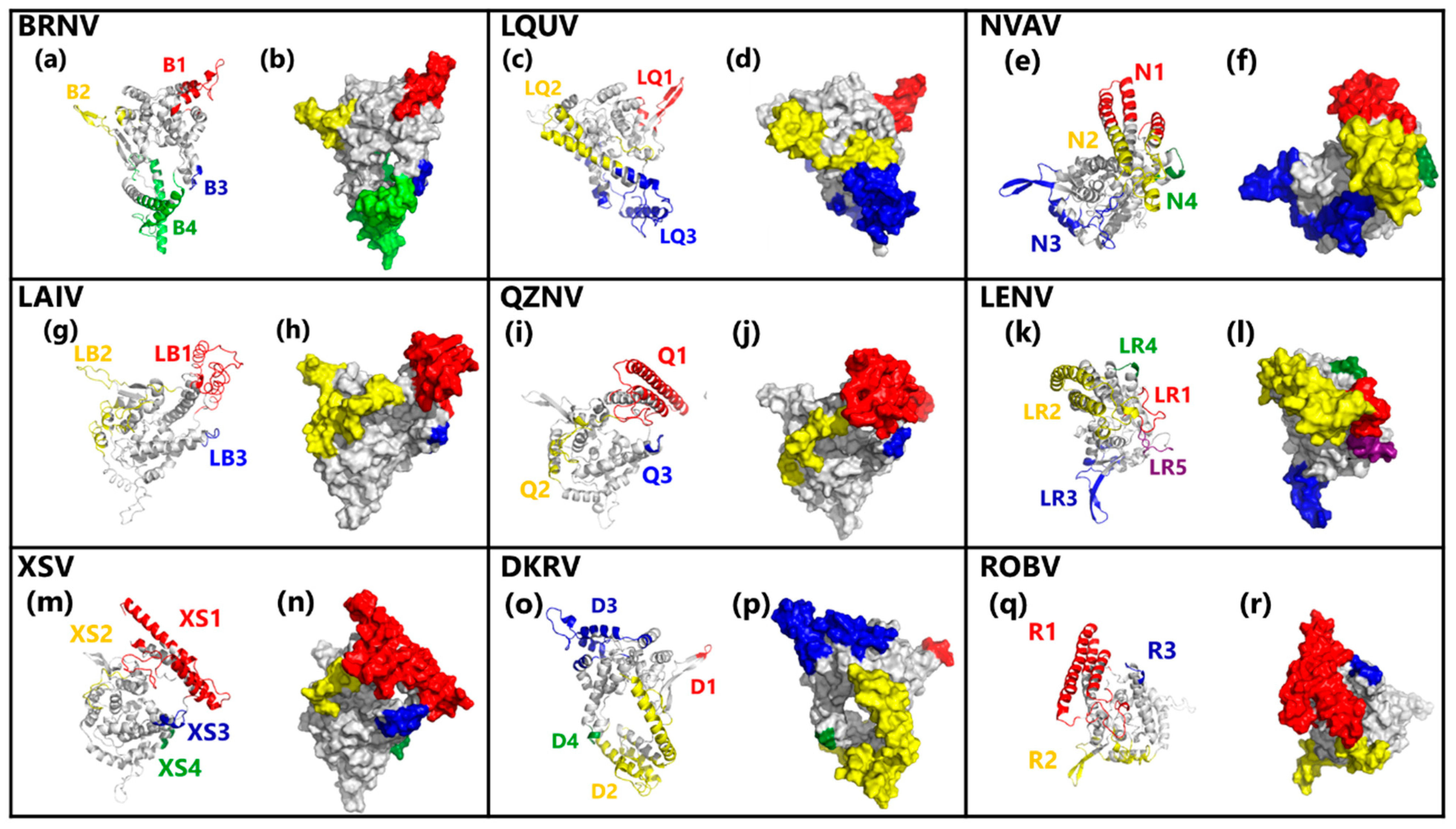
3.6. Predicted Conformational B-Cell Epitopes in Hantavirus NPs Models
3.7. Most of Conformational B-Cell Epitopes Are Not Conserved among Hantavirus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- ASM Mammal Diversity Database. Available online: Mammaldiversity.org (accessed on 27 July 2020).
- Wang, L.F.; Anderson, D.E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 2019, 34, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Yanagihara, R. Genetic Diversity and Geographic Distribution of Bat-borne Hantaviruses. Curr. Issues Mol. Biol. 2020, 39, 1–28. [Google Scholar] [CrossRef]
- Guterres, A.; Oliveira, R.C.; Fernandes, J.; de Lemos, E.R.; Schrago, C.G. New bunya-like viruses: Highlighting their relations. Infect. Genet. Evol. 2017, 49, 164–173. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Amarasinghe, G.K.; Anthony, S.J.; Avšič-Županc, T.; Ayllón, M.A.; Bahl, J.; Balkema-Buschmann, A.; et al. 2020 taxonomic update for phylum Negarnaviricota (Riboviria: Orthornavirae), including the large orders Bunyavirales and Mononegavirales. Arch. Virol. 2020, 165, 3023–3072. [Google Scholar] [CrossRef]
- De Oliveira, R.C.; Guterres, A.; Fernandes, J.; D’Andrea, P.S.; Bonvicino, C.R.; de Lemos, E.R. Hantavirus reservoirs: Current status with an emphasis on data from Brazil. Viruses 2014, 6, 1929–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Gu, S.H.; Yashina, L.N.; Cook, J.A.; Yanagihara, R. Highly Divergent Genetic Variants of Soricid-Borne Altai Virus (Hantaviridae) in Eurasia Suggest Ancient Host-Switching Events. Viruses 2019, 11, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagihara, R.; Gu, S.H.; Arai, S.; Kang, H.J.; Song, J.W. Hantaviruses: Rediscovery and new beginnings. Virus Res. 2014, 187, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Bennett, S.N.; Gu, S.H.; Kang, H.J.; Arai, S.; Yanagihara, R. Reconstructing the evolutionary origins and phylogeography of hantaviruses. Trends Microbiol. 2014, 22, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, P.T.; Drexler, J.F.; Kallies, R.; Ličková, M.; Bokorová, S.; Maganga, G.D.; Szemes, T.; Leroy, E.M.; Krüger, D.H.; Drosten, C.; et al. Phylogenetic analysis of a newfound bat-borne hantavirus supports a laurasiatherian host association for ancestral mammalian hantaviruses. Infect. Genet. Evol. 2016, 41, 113–119. [Google Scholar] [CrossRef]
- Sumibcay, L.; Kadjo, B.; Gu, S.H.; Kang, H.J.; Lim, B.K.; Cook, J.A.; Song, J.W.; Yanagihara, R. Divergent lineage of a novel hantavirus in the banana pipistrelle (Neoromicia nanus) in Cote d’Ivoire. Virol. J. 2012, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.; Witkowski, P.T.; Auste, B.; Nowak, K.; Weber, N.; Fahr, J.; Mombouli, J.V.; Wolfe, N.D.; Drexler, J.F.; Drosten, C.; et al. Hantavirus in Bat, Sierra Leone. Emerg. Infect. Dis. 2012, 18, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.P.; Lin, X.D.; Wang, W.; Tian, J.H.; Cong, M.L.; Zhang, H.L.; Wang, M.R.; Zhou, R.H.; Wang, J.B.; Li, M.H.; et al. Phylogeny and origins of hantaviruses harbored by bats, insectivores, and rodents. PLoS Pathog. 2013, 9, e1003159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Kikuchi, F.; Bawm, S.; Sơn, N.T.; Lin, K.S.; Tú, V.T.; Aoki, K.; Tsuchiya, K.; Tanaka-Taya, K.; Morikawa, S.; et al. Molecular Phylogeny of Mobatviruses (Hantaviridae) in Myanmar and Vietnam. Viruses 2019, 11, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vapalahti, O.; Kallio-Kokko, H.; Narvanen, A.; Julkunen, I.; Lundkvist, A.; Plyusnin, A.; Lehväslaiho, H.; Brummer-Korvenkontio, M.; Vaheri, A.; Lankinen, H. Human B-cell epitopes of Puumala virus nucleocapsid protein, the major antigen in early serological response. J. Med. Virol. 1995, 46, 293–303. [Google Scholar] [CrossRef]
- Padula, P.J.; Rossi, C.M.; Valle, M.O.D.; Martínez, P.V.; Colavecchia, S.B.; Edelstein, A.; Miguel, S.D.L.; Rabinovich, R.D.; Segura, E.L. Development and evaluation of a solid-phase enzyme immunoassay based on Andes hantavirus recombinant nucleoprotein. J. Med. Microbiol. 2000, 49, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Olal, D.; Daumke, O. Structure of the Hantavirus Nucleoprotein Provides Insights into the Mechanism of RNA Encapsidation. Cell Rep. 2016, 14, 2092–2099. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wu, J.; Li, Q.; Wei, Y.; Tan, Z.; Cai, J.; Guo, H.; Yang, L.; Huang, X.; Chen, J.; et al. Seroprevalence, cross antigenicity and circulation sphere of bat-borne hantaviruses revealed by serological and antigenic analyses. PLoS Pathog. 2019, 15, e1007545. [Google Scholar] [CrossRef] [PubMed]
- Sabino-Santos, G., Jr.; Ferreira, F.F.; Da Silva, D.J.F.; Machado, D.M.; Da Silva, S.G.; Sao Bernardo, C.S.; Dos Santos Filho, M.; Levi, T.; Figueiredo, L.T.M.; Peres, C.A.; et al. Hantavirus antibodies among phyllostomid bats from the arc of deforestation in Southern Amazonia, Brazil. Transbound. Emerg. Dis. 2020, 67, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Sabino-Santos, G., Jr.; Maia, F.G.; Vieira, T.M.; De Lara Muylaert, R.; Lima, S.M.; Goncalves, C.B.; Barroso, P.D.; Melo, M.N.; Jonsson, C.B.; Goodin, D.; et al. Evidence of Hantavirus Infection Among Bats in Brazil. Am. J. Trop. Med. Hyg. 2015, 93, 404–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Uversky, V.N.; Dunker, A.K.; Lonardi, S. Composition Profiler: A tool for discovery and visualization of amino acid composition differences. BMC Bioinform. 2017, 8, 211. [Google Scholar] [CrossRef] [Green Version]
- Boeckmann, B.; Blatter, M.C.; Famiglietti, L.; Hinz, U.; Lane, L.; Roechert, B.; Bairoch, A. Protein variety and functional diversity: Swiss-Prot annotation in its biological context. C. R. Biol. 2005, 328, 882–899. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. The mathematical theory of communication. 1963. MD Comput. 1997, 14, 306–317. [Google Scholar]
- Rost, B.; Yachdav, G.; Liu, J. The PredictProtein server. Nucleic Acids Res. 2004, 32, W321–W326. [Google Scholar]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar]
- Song, Y.; Dimaio, F.; Wang, R.Y.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-resolution comparative modeling with Rosetta CM. Structure 2013, 21, 1735–1742. [Google Scholar]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Word, J.M.; Lovell, S.C.; Labean, T.H.; Taylor, H.C.; Zalis, M.E.; Presley, B.K.; Richardson, J.S.; Richardson, D.C. Visualizing and quantifying molecular goodness-of-fit: Small-probe contact dots with explicit hydrogen atoms. J. Mol. Biol. 1999, 285, 1711–1733. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Word, J.M.; Richardson, J.S.; Richardson, D.C. The penultimate rotamer library. Proteins 2000, 40, 389–408. [Google Scholar] [CrossRef]
- Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins 2003, 50, 437–450. [Google Scholar]
- Davis, I.W.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MOLPROBITY: Structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004, 32, W615–W619. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.A.; Sarkar, B.; Islam, S.S. Exploiting the reverse vaccinology approach to design novel subunit vaccines against Ebola virus. Immunobiology 2020, 225, 151949. [Google Scholar] [CrossRef] [PubMed]
- Bui, H.H.; Sidney, J.; Li, W.; Fusseder, N.; Sette, A. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinform. 2007, 8, 361. [Google Scholar] [CrossRef] [Green Version]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Moss, D.S.; Thornton, J.M. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 1993, 231, 1049–1067. [Google Scholar] [CrossRef]
- Zana, B.; Kemenesi, G.; Buzas, D.; Csorba, G.; Gorfol, T.; Khan, F.A.; Tahir, N.; Zeghbib, S.; Madai, M.; Papp, H.; et al. Molecular Identification of a Novel Hantavirus in Malaysian Bronze Tube-Nosed Bats (Murina aenea). Viruses 2019, 11, 887. [Google Scholar] [CrossRef] [Green Version]
- Raboni, S.M.; Levis, S.; Rosa, E.S.; Bisordi, I.; Delfraro, A.; Lemos, E.; Correia, D.C.; Duarte Dos Santos, C.N. Hantavirus infection in Brazil: Development and evaluation of an enzyme immunoassay and immunoblotting based on N recombinant protein. Diagn. Microbiol. Infect. Dis. 2007, 58, 89–97. [Google Scholar] [CrossRef]
- Schlegel, M.; Tegshduuren, E.; Yoshimatsu, K.; Petraityte, R.; Sasnauskas, K.; Hammerschmidt, B.; Friedrich, R.; Mertens, M.; Groschup, M.H.; Arai, S.; et al. Novel serological tools for detection of Thottapalayam virus, a Soricomorpha-borne hantavirus. Arch. Virol. 2012, 157, 2179–2187. [Google Scholar] [CrossRef]
- Amada, T.; Yoshimatsu, K.; Yasuda, S.P.; Shimizu, K.; Koma, T.; Hayashimoto, N.; Gamage, C.D.; Nishio, S.; Takakura, A.; Arikawa, J. Rapid, whole blood diagnostic test for detecting anti-hantavirus antibody in rats. J. Virol. Methods 2013, 193, 42–49. [Google Scholar] [CrossRef]
- Kalaiselvan, S.; Sankar, S.; Ramamurthy, M.; Ghosh, A.R.; Nandagopal, B.; Sridharan, G. Prediction of B Cell Epitopes Among Hantavirus Strains Causing Hemorragic Fever with Renal Syndrome. J. Cell Biochem. 2017, 118, 1182–1188. [Google Scholar] [CrossRef]
- Kalaiselvan, S.; Sankar, S.; Ramamurthy, M.; Ghosh, A.R.; Nandagopal, B.; Sridharan, G. Prediction of Pan-Specific B-Cell Epitopes from Nucleocapsid Protein of Hantaviruses Causing Hantavirus Cardiopulmonary Syndrome. J. Cell Biochem. 2017, 118, 2320–2324. [Google Scholar] [CrossRef]
- Conte, F.P.; Tinoco, B.C.; Santos Chaves, T.; Oliveira, R.C.; Figueira Mansur, J.; Mohana-Borges, R.; Lemos, E.R.S.; Neves, P.; Rodrigues-Da-Silva, R.N. Identification and validation of specific B-cell epitopes of hantaviruses associated to hemorrhagic fever and renal syndrome. PLoS Negl. Trop. Dis. 2019, 13, e0007915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, M.; Yoshimatsu, K.; Kumperasart, S.; Nakamura, I.; Ogino, M.; Taruishi, M.; Sungdee, A.; Pattamadilok, S.; Ibrahim, I.N.; Erlina, S.; et al. Development of serological assays for Thottapalayam virus, an insectivore-borne Hantavirus. Clin. Vaccine Immunol. 2007, 14, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Yoshimatsu, K.; Arikawa, J. Serological diagnosis with recombinant N antigen for hantavirus infection. Virus Res. 2014, 187, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, M.; Krüger, D.H. The nucleocapsid protein of hantaviruses: Much more than a genome-wrapping protein. Virus Genes 2018, 54, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and Methods for T- and B-Cell Epitope Prediction. J. Immunol. Res. 2017, 2680160. [Google Scholar] [CrossRef] [Green Version]
- Van Regenmortel, M.H. What is a B-cell epitope? Methods Mol. Biol. 2009, 524, 3–20. [Google Scholar]
- Levitt, M. Nature of the protein universe. Proc. Natl. Acad. Sci. USA 2009, 106, 11079–11084. [Google Scholar] [CrossRef] [Green Version]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, D.; Luthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [PubMed]
- Von Grotthuss, M.; Pas, J.; Wyrwicz, L.; Ginalski, K.; Rychlewski, L. Application of 3D-Jury, GRDB, and Verify3D in fold recognition. Proteins 2003, 53 (Suppl. S6), 418–423. [Google Scholar] [CrossRef] [PubMed]
- Prisant, M.G.; Williams, C.J.; Chen, V.B.; Richardson, J.S.; Richardson, D.C. New tools in MolProbity validation: CaBLAM for CryoEM backbone, UnDowser to rethink "waters," and NGL Viewer to recapture online 3D graphics. Protein Sci. 2020, 29, 315–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Shimizu, K.; Nishigami, K.; Tsuda, Y.; Sarathukumara, Y.; Muthusinghe, D.S.; Gamage, C.D.; Granathne, L.; Lokupathirage, S.M.W.; Nanayakkara, N.; et al. Serological methods for detection of infection with shrew-borne hantaviruses: Thottapalayam, Seewis, Altai, and Asama viruses. Arch. Virol. 2021, 166, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Yon, L.; Duff, J.P.; Agren, E.O.; Erdelyi, K.; Ferroglio, E.; Godfroid, J.; Hars, J.; Hestvik, G.; Horton, D.; Kuiken, T.; et al. Recent Changes in Infectious Diseases in European Wildlife. J. Wildl. Dis. 2019, 55, 3–43. [Google Scholar] [CrossRef] [PubMed]
- Tischler, N.D.; Galeno, H.; Rosemblatt, M.; Valenzuela, P.D. Human and rodent humoral immune responses to Andes virus structural proteins. Virology 2005, 334, 319–326. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Taxonomic Data | Ecological Data | Nucleocapsid Protein Data | ||||||
|---|---|---|---|---|---|---|---|---|
| Hantavirus | Phylogroup | Genus | Reservoir Order | Reservoir Families | Reservoir Species | Virus Distribution | Length | Molecular Weight (Da) |
| Andes virus (ANDV) | V | Orthohantavirus | Rodentia | Cricetidae | Oligoryzomys longicaudatus | Argentina, Chile | 428 | 48,039.69 |
| Araraquara virus (ARAV) | V | Orthohantavirus | Rodentia | Cricetidae | Necromys lasiurus | Brazil | 428 | 47,878.46 |
| Sin Nombre virus (SNV) | V | Orthohantavirus | Rodentia | Cricetidae | Peromyscus maniculatus | USA, Canada, and Mexico | 428 | 48,174.57 |
| Seoul virus (SEOV) | IV | Orthohantavirus | Rodentia | Muridae | Rattus rattus and R. norvegicus | Worldwide | 429 | 47,930.31 |
| Hantaan virus (HTNV) | IV | Orthohantavirus | Rodentia | Muridae | Apodemus agrarius | China, Korea, Japan, and Russia | 429 | 48,152.61 |
| Brno virus (BRNV) | II | Loanvirus | Chiroptera | Vespertilionidae | Nyctalus noctula | Czech Republic | 423 | 48,699.39 |
| Longquan virus (LQUV) | II | Loanvirus | Chiroptera | Rhinolophidae | Rhinolophus sinicus; R. affinis and R. monoceros | China | 423 | 48,193.69 |
| Nova virus (NVAV) | II | Mobatvirus | Soricomorpha | Talpidae | Talpa europaea | Belgium, France, Hungary, and Poland | 428 | 48,418.26 |
| Laibin virus (LAIV) | II | Mobatvirus | Chiroptera | Emballonuridae | Taphozous melanopogon | China, Myanmar | 427 | 48,108.92 |
| Quezon virus (QZNV) | II | Mobatvirus | Chiroptera | Pteropodidae | Rousettus amplexicaudatus | Philippines | 429 | 48,441.9 |
| Lena River virus (LENV) | II | Mobatvirus | Soricomorpha | Soricidae | Sorex caecutiens | Russia | 448 | 50,532.14 |
| Xuan Son virus (XSV) | II | Mobatvirus | Chiroptera | Hipposideridae | Hipposideros pomon; H. cineraceus | Vietnam | 427 | 48,136.93 |
| Dakrong virus (DKGV) | II | Unclassified | Chiroptera | Hipposideridae | Aselliscus stoliczkanus | Vietnam | 427 | 48,612.78 |
| Robina virus (ROBV) | II | Orthohantavirus | Chiroptera | Pteropodidae | Pteropus alecto | Australia | 429 | 48,525.26 |
| Hantavirus | Poor Rotamers | Favored Rotamers | Ramachandran Outliers | Ramachandran Favored | Z-Score |
|---|---|---|---|---|---|
| Brno | 0 | 377 | 1 | 412 | 1.15 ± 0.39 |
| 0.00% | 99.21% | 0.24% | 97.86% | ||
| Longquan | 0 | 369 | 1 | 414 | 0.66 ± 0.40 |
| 0.00% | 99.73% | 0.24% | 98.34% | ||
| Nova | 0 | 365 | 0 | 418 | 0.88 ± 0.38 |
| 0.00% | 99.46% | 0.00% | 98.12% | ||
| Laibin | 1 | 364 | 0 | 413 | 0.59 ± 0.38 |
| 0.27% | 99.18% | 0.00% | 97.18% | ||
| Quezon | 0 | 375 | 0 | 420 | 1.04 ± 0.38 |
| 0.00% | 99.47% | 0.00% | 98.36% | ||
| Lena River | 0 | 378 | 2 | 438 | −0.08 ± 0.37 |
| 0.00% | 98.69% | 0.45% | 98.21% | ||
| Xuan Son | 0 | 367 | 0 | 418 | 0.20 ± 0.37 |
| 0.00% | 99.73% | 0.00% | 98.35% | ||
| Dakrong | 0 | 370 | 0 | 418 | 0.43 ± 0.38 |
| 0.00% | 98.93% | 0.00% | 98.35% | ||
| Robina | 0 | 369 | 2 | 420 | 0.79 ± 0.39 |
| 0.00% | 100% | 0.47% | 98.36% | ||
| Goals | <0.3% | >98% | <0.05% | >98% | <2 |
| Virus | Epitope | Length | Conservation among Orthohantavirus | Conservation among Phylogroup II | ||
|---|---|---|---|---|---|---|
| High Similar NPs (≥70%) (%—N/5) | Mean Conservation % (Min–Max) | High Similar NPs (≥70%) (%—N/9) | Mean Conservation % (Min–Max) | |||
| BRNV | B1 | 35 | 0.00 (0/5) | 21.1 (20–22.9) | 11.11 (1/9) | 35.9 (20–100) |
| B2 | 28 | 0.00 (0/5) | 40 (35.7–42.9) | 11.11 (1/9) | 50.4 (21.4–100) | |
| B3 | 103 | 0.00 (0/5) | 22.1 (20.4–24.3) | 11.11 (1/9) | 35.5 (21.4–100) | |
| B4 | 7 | 100.00 (5/5) | 80 (71.4–85.7) | 88.89 (8/9) | 92.1 (57.1–100) | |
| LQUV | LQ1 | 25 | 0.00 (0/5) | 54.4 (48–60) | 11.11 (1/9) | 52.4 (44–100) |
| LQ2 | 37 | 0.00 (0/5) | 22.7 (18.9−29.7) | 11.11 (1/9) | 36.6 (18.9–100) | |
| LQ3 | 88 | 0.00 (0/5) | 44.5 (42–46.6) | 11.11 (1/9) | 55.6 (40.9–100) | |
| NVAV | N1 | 44 | 0.00 (0/5) | 35.5 (22.7–43.2) | 11.11 (1/9) | 31.8 (18.2–100) |
| N2 | 53 | 0.00 (0/5) | 38.9 (32.1–45.3) | 11.11 (1/9) | 42.6 (7.6–100) | |
| N3 | 57 | 0.00 (0/5) | 47 (43.9–49.1) | 11.11 (1/9) | 57.3 (31.6–100) | |
| N4 | 9 | 0.00 (0/5) | 53.3 (44.4–55.6) | 11.11 (1/9) | 66.7 (55.6–100) | |
| LAIV | LB1 | 72 | 0.00 (0/5) | 27.5 (26.4–29.2) | 22.22 (2/9) | 53.1 (33.3–100) |
| LB2 | 65 | 0.00 (0/5) | 39.7 (35.4–44.6) | 33.33 (3/9) | 60.9 (40–100) | |
| LB3 | 8 | 100.00 (5/5) | 80 (75–87.5) | 88.89 (8/9) | 93.1 (62.5–100) | |
| QZNV | Q1 | 95 | 0.00 (0/5) | 14.1 (10.5–18.9) | 22.22 (2/9) | 29.5 (8.4–100) |
| Q2 | 32 | 0.00 (0/5) | 55 (50–56.3) | 22.22 (2/9) | 62.5 (37.5–100) | |
| Q3 | 6 | 40.00 (2/5) | 73.3 (66.7–83.3) | 88.89 (8/9) | 90.7 (50–100) | |
| LENV | LR1 | 10 | 0.00 (0/5) | 50 (50–50) | 11.11 (1/9) | 61.9 (57.1–100) |
| LR2 | 85 | 0.00 (0/5) | 10.4 (9.4–11.8) | 11.11 (1/9) | 20.7 (7.1–100) | |
| LR3 | 36 | 0.00 (0/5) | 25.6 (22.2–30.6) | 11.11 (1/9) | 30.6 (19.4–100) | |
| LR4 | 4 | 80.00 (4/5) | 80 (50–100) | 100.00 (9/9) | 77.8 (75–100) | |
| LR5 | 12 | 0.00 (0/5) | 41.7 (41.7–41.7) | 11.11 (1/9) | 42.6 (33.3–100) | |
| XSV | XS1 | 99 | 0.00 (0/5) | 20.8 (18.9–24.2) | 22.22 (2/9) | 48.4 (19.2–100) |
| XS2 | 15 | 40.00 (2/5) | 65.3 (60–73.3) | 77.78 (7/9) | 75.6 (46.7–100) | |
| XS3 | 11 | 100.00 (5/5) | 80 (72.7–81.82) | 88.89 (8/9) | 79.8 (54.6–100) | |
| XS4 | 4 | 0.00 (0/5) | 50 (50–50) | 22.22 (2/9) | 58.3 (50–100) | |
| DKGV | D1 | 5 | 0.00 (0/5) | 52 (40–60) | 55.56 (5/9) | 77.8 (60–100) |
| D2 | 118 | 0.00 (0/5) | 28.6 (26.3–30.5) | 33.33 (3/9) | 49.4 (17.8–100) | |
| D3 | 59 | 0.00 (0/5) | 16.3 (15.3–18.6) | 11.11 (1/9) | 40.1 (15.3–100) | |
| D4 | 6 | 40.00 (2/5) | 73.3 (66.7–83.3) | 88.89 (8/9) | 90.7 (50–100) | |
| ROBV | R1 | 102 | 0.00 (0/5) | 15.4 (11.1–20.2) | 22.22 (2/9) | 31.6 (13.1–100) |
| R2 | 63 | 0.00 (0/5) | 45.1 (42.9–47.6) | 22.22 (2/9) | 59.4 (31.8–100) | |
| R3 | 6 | 40.00 (2/5) | 73.4 (66.7–83.3) | 88.89 (8/9) | 90.7 (50–100) | |
| Virus | Epitope | Length | Conservation Degree (%) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rodent-Borne | Bat-Borne | Shrew-Borne | ||||||||||||||
| ANDV | ARAV | SNV | SEOV | HTNV | BRNV | LQUV | LAIV | QZNV | XSV | DKGV | ROBV | LENV | NVAV | |||
| Brno | B1 | 35 | 20 | 20 | 23 | 20 | 23 | 100 | 54 | 20 | 26 | 23 | 29 | 26 | 26 | 20 |
| B2 | 28 | 43 | 43 | 43 | 36 | 36 | 100 | 57 | 43 | 43 | 39 | 54 | 57 | 21 | 39 | |
| B3 | 103 | 20 | 21 | 20 | 24 | 24 | 100 | 43 | 22 | 25 | 21 | 27 | 24 | 30 | 26 | |
| B4 | 7 | 86 | 86 | 71 | 86 | 71 | 100 | 100 | 100 | 100 | 86 | 100 | 100 | 86 | 57 | |
| Longquan | LQ1 | 25 | 48 | 48 | 56 | 60 | 60 | 56 | 100 | 44 | 52 | 44 | 48 | 60 | 24 | 44 |
| LQ2 | 37 | 19 | 19 | 19 | 27 | 30 | 22 | 100 | 41 | 24 | 43 | 35 | 24 | 19 | 22 | |
| LQ3 | 88 | 47 | 47 | 42 | 43 | 44 | 69 | 100 | 47 | 50 | 41 | 48 | 51 | 52 | 42 | |
| Nova | N1 | 44 | 43 | 43 | 43 | 25 | 23 | 32 | 27 | 18 | 23 | 18 | 20 | 27 | 20 | 100 |
| N2 | 53 | 45 | 43 | 42 | 32 | 32 | 8 | 13 | 49 | 51 | 47 | 42 | 45 | 28 | 100 | |
| N3 | 57 | 49 | 49 | 44 | 47 | 46 | 44 | 47 | 56 | 60 | 60 | 61 | 56 | 32 | 100 | |
| N4 | 9 | 56 | 56 | 56 | 44 | 56 | 56 | 67 | 67 | 67 | 56 | 67 | 67 | 56 | 100 | |
| Laibin | LB1 | 72 | 28 | 26 | 26 | 28 | 29 | 21 | 28 | 100 | 57 | 78 | 69 | 60 | 32 | 33 |
| LB2 | 65 | 37 | 38 | 35 | 43 | 45 | 40 | 45 | 100 | 57 | 82 | 82 | 62 | 29 | 52 | |
| LB3 | 8 | 88 | 88 | 75 | 75 | 75 | 100 | 100 | 100 | 100 | 88 | 100 | 100 | 88 | 63 | |
| Quezon | Q1 | 95 | 11 | 11 | 12 | 19 | 19 | 9 | 8 | 15 | 100 | 14 | 14 | 71 | 22 | 13 |
| Q2 | 32 | 50 | 50 | 56 | 56 | 63 | 53 | 56 | 53 | 100 | 66 | 59 | 81 | 38 | 56 | |
| Q3 | 6 | 83 | 83 | 67 | 67 | 67 | 100 | 100 | 100 | 100 | 83 | 100 | 100 | 83 | 50 | |
| Lena River | LR1 | 10 | 50 | 50 | 50 | 50 | 50 | 57 | 57 | 57 | 57 | 57 | 57 | 57 | 100 | 57 |
| LR2 | 85 | 9 | 9 | 11 | 12 | 11 | 7 | 11 | 11 | 13 | 9 | 12 | 13 | 100 | 11 | |
| LR3 | 36 | 22 | 22 | 25 | 28 | 31 | 25 | 22 | 19 | 22 | 19 | 22 | 22 | 100 | 22 | |
| LR4 | 4 | 75 | 75 | 100 | 50 | 100 | 75 | 75 | 75 | 75 | 75 | 75 | 75 | 100 | 75 | |
| LR5 | 12 | 42 | 42 | 42 | 42 | 42 | 33 | 42 | 33 | 33 | 33 | 33 | 42 | 100 | 33 | |
| Xuan Son | XS1 | 99 | 20 | 19 | 18 | 22 | 24 | 19 | 29 | 78 | 40 | 100 | 70 | 43 | 22 | 33 |
| XS2 | 15 | 60 | 60 | 60 | 73 | 73 | 67 | 73 | 80 | 80 | 100 | 87 | 73 | 47 | 73 | |
| XS | 11 | 82 | 82 | 73 | 82 | 82 | 73 | 82 | 91 | 82 | 100 | 82 | 82 | 73 | 55 | |
| XS4 | 4 | 50 | 50 | 50 | 50 | 50 | 50 | 75 | 50 | 50 | 100 | 50 | 50 | 50 | 50 | |
| Dakrong | D1 | 5 | 60 | 60 | 60 | 40 | 40 | 60 | 60 | 100 | 80 | 100 | 100 | 60 | 60 | 80 |
| D2 | 118 | 29 | 28 | 26 | 30 | 31 | 18 | 20 | 75 | 51 | 70 | 100 | 48 | 25 | 37 | |
| D3 | 59 | 19 | 17 | 15 | 15 | 15 | 24 | 25 | 46 | 34 | 47 | 100 | 47 | 15 | 22 | |
| D4 | 6 | 83 | 83 | 67 | 67 | 67 | 100 | 100 | 100 | 100 | 83 | 100 | 100 | 83 | 50 | |
| Robina | R1 | 102 | 11 | 12 | 13 | 20 | 20 | 13 | 13 | 17 | 73 | 17 | 16 | 100 | 22 | 13 |
| R2 | 63 | 43 | 44 | 48 | 44 | 46 | 51 | 48 | 60 | 73 | 59 | 63 | 100 | 32 | 49 | |
| R3 | 6 | 83 | 83 | 67 | 67 | 67 | 100 | 100 | 100 | 100 | 83 | 100 | 100 | 83 | 50 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira, R.C.; Fernandes, J.; de Sampaio Lemos, E.R.; de Paiva Conte, F.; Rodrigues-da-Silva, R.N. The Serological Cross-Detection of Bat-Borne Hantaviruses: A Valid Strategy or Taking Chances? Viruses 2021, 13, 1188. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071188
de Oliveira RC, Fernandes J, de Sampaio Lemos ER, de Paiva Conte F, Rodrigues-da-Silva RN. The Serological Cross-Detection of Bat-Borne Hantaviruses: A Valid Strategy or Taking Chances? Viruses. 2021; 13(7):1188. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071188
Chicago/Turabian Stylede Oliveira, Renata Carvalho, Jorlan Fernandes, Elba Regina de Sampaio Lemos, Fernando de Paiva Conte, and Rodrigo Nunes Rodrigues-da-Silva. 2021. "The Serological Cross-Detection of Bat-Borne Hantaviruses: A Valid Strategy or Taking Chances?" Viruses 13, no. 7: 1188. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071188