
Exploring Codon Adjustment Strategies towards Escherichia coli-Based Production of Viral Proteins Encoded by HTH1, a Novel Prophage of the Marine Bacterium Hypnocyclicus thermotrophus

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification and Annotation of Prophage Genes
2.2. Taxonomic Analysis of HTH1
2.3. Analysis of Prophage Host Range
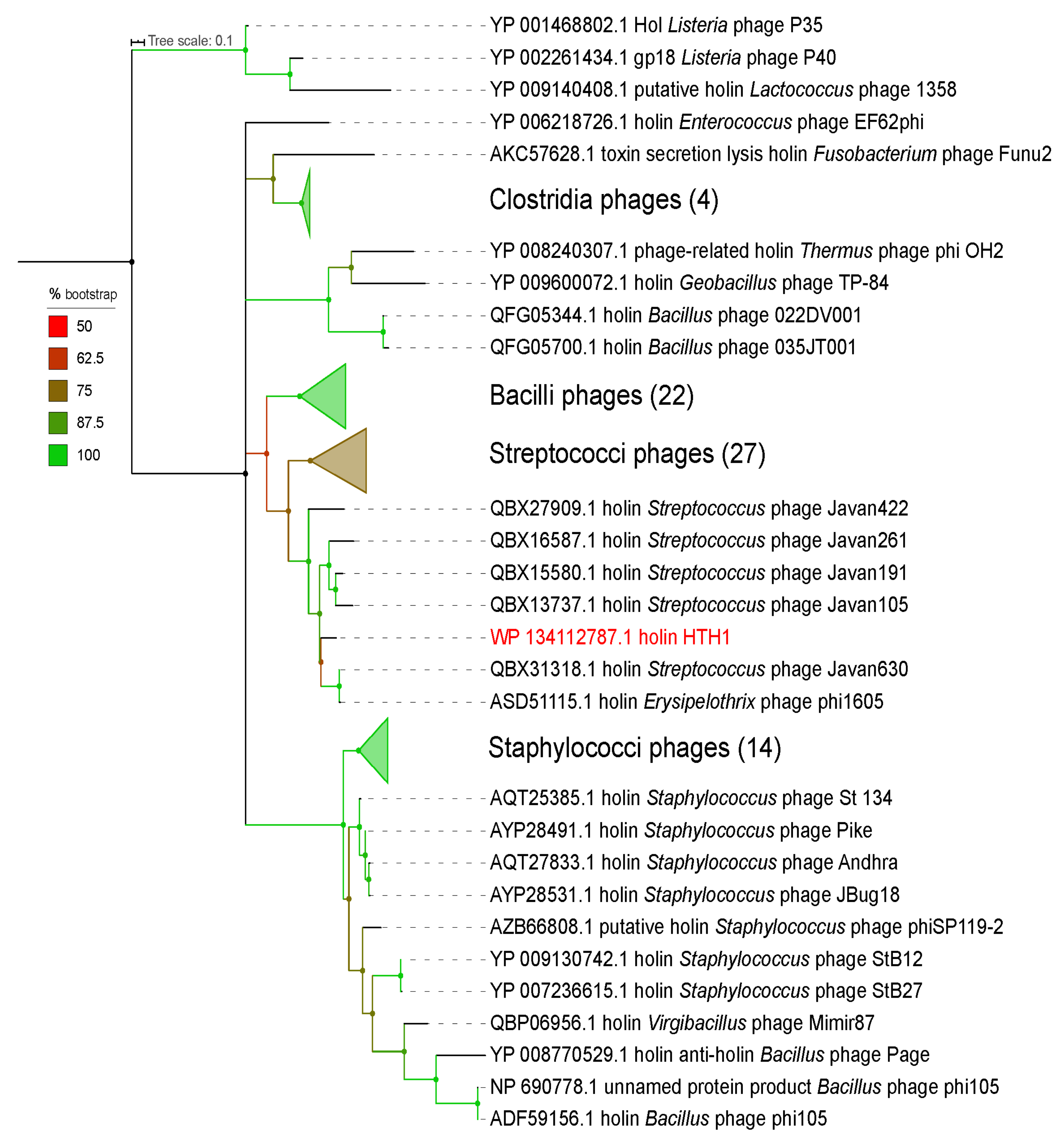
2.4. Phylogeny Analyses
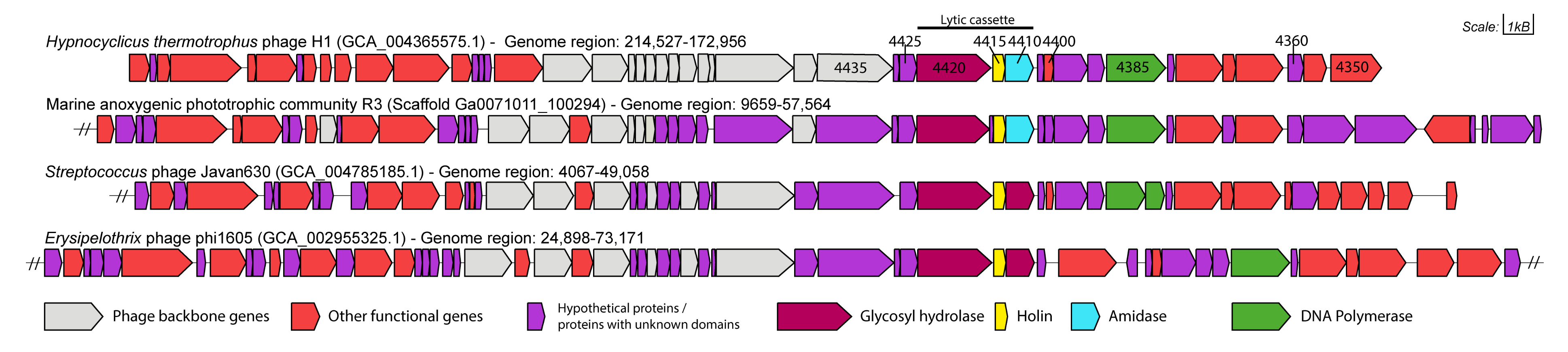
2.5. Gene Neighbourhood Analysis
2.6. Selection of Genes for Expression Trials
2.7. Preparation of Sequences for Protein Expression of Selected Genes
2.8. Protein Production in E. coli
2.9. Protein Solubility Assessment and Yield Estimation
2.10. Protein Purification
2.11. Protein Thermal Unfolding Assay
3. Results
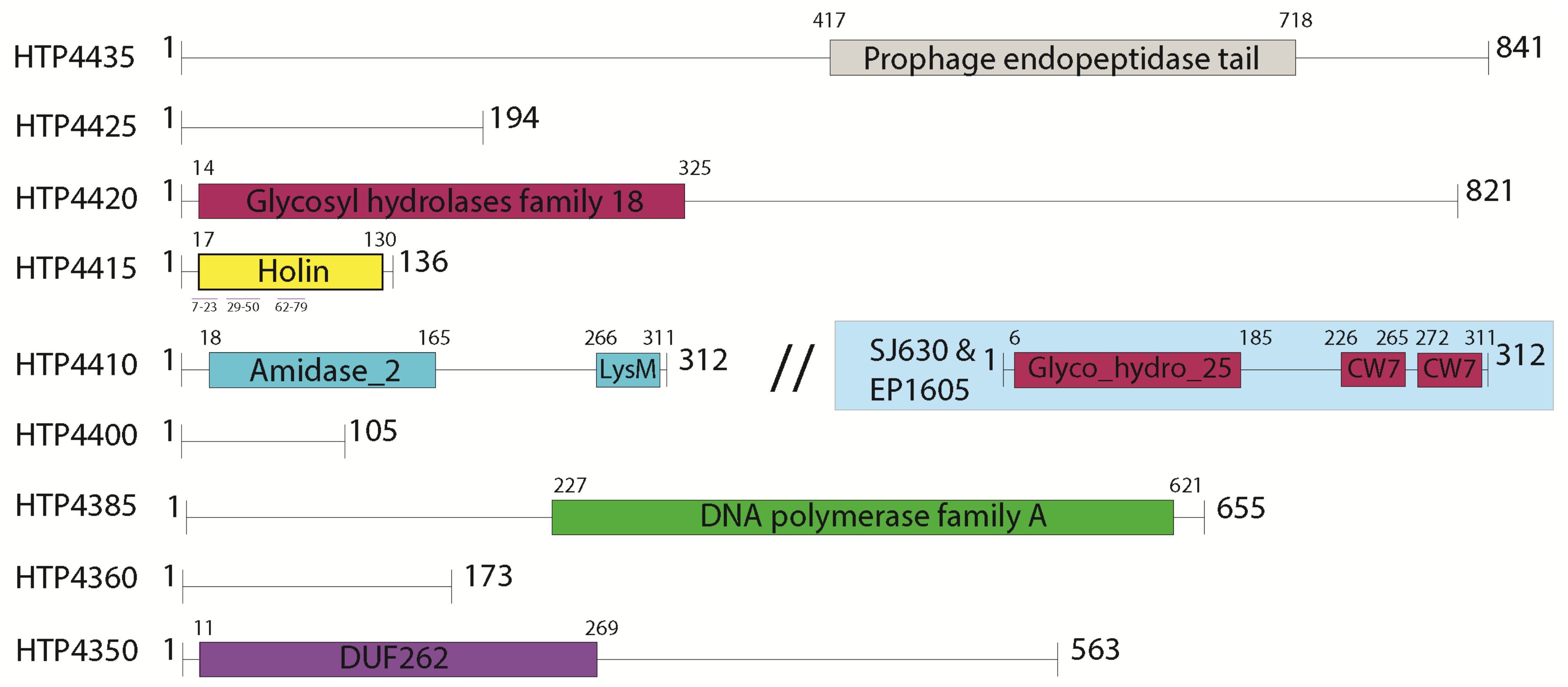
3.1. Functional Annotation and Taxonomy Analysis of HTH1
3.2. Selection of Genes for Expression Trials
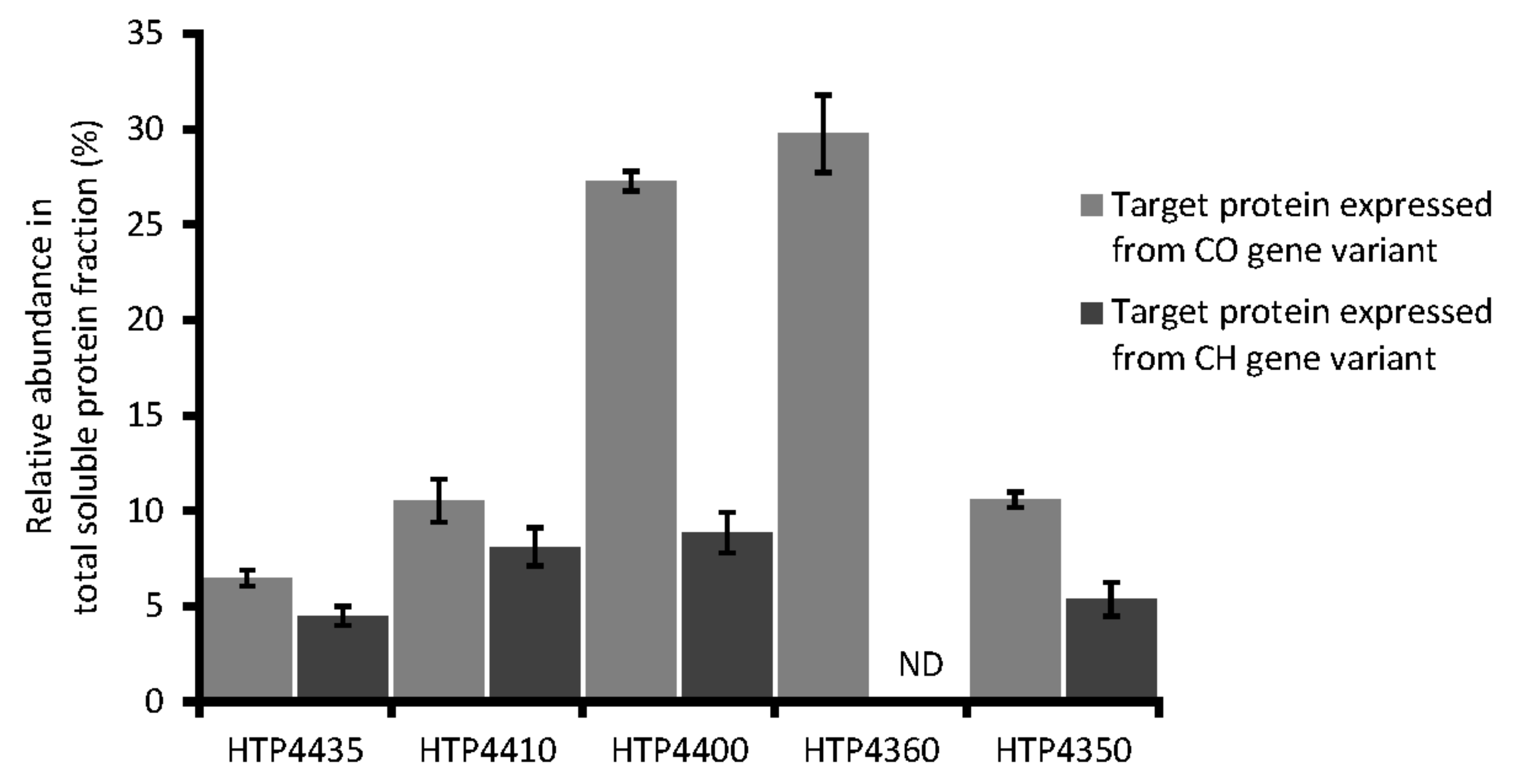
3.3. Expression of Target Codon-Adjusted Gene Variants
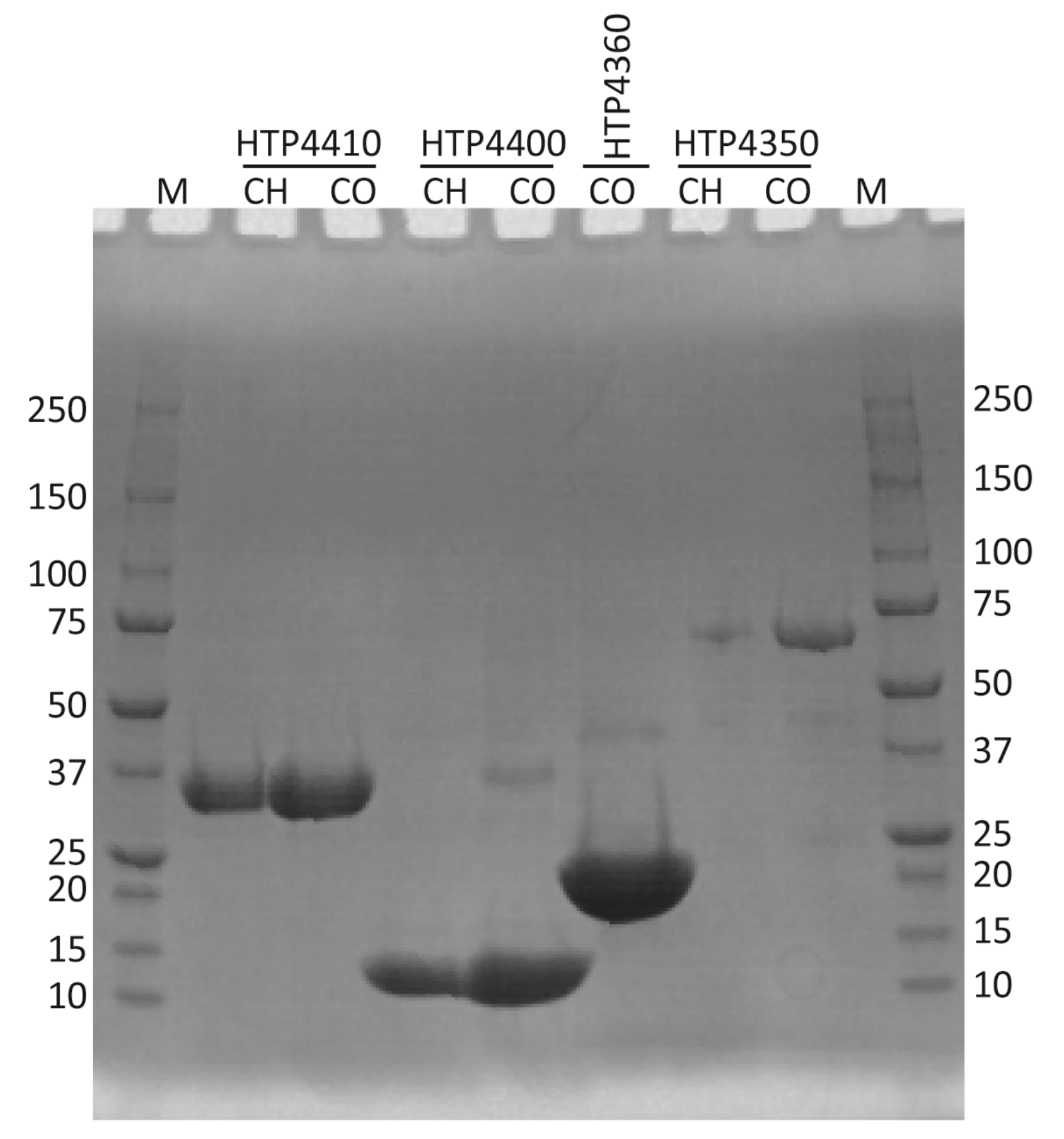
3.4. Protein Purification
3.5. Crystallization and Thermostability of Target Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steen, I.H.; Dahle, H.; Stokke, R.; Roalkvam, I.; Daae, F.-L.; Rapp, H.T.; Pedersen, R.B.; Thorseth, I.H. Novel barite chimneys at the Loki’s Castle vent field shed light on key factors shaping microbial communities and functions in hydrothermal systems. Front. Microbiol. 2016, 6, 1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Takai, K. Theoretical constraints of physical and chemical properties of hydrothermal fluids on variations in chemolithotrophic microbial communities in seafloor hydrothermal systems. Prog. Earth Planet. Sci. 2014, 1, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Corliss, J.B.; Dymond, J.; Gordon, L.I.; Edmond, J.M.; Von Herzen, R.P.; Ballard, R.D.; Green, K.; Williams, D.; Bainbridge, A.; Crane, K.; et al. Submarine thermal springs on the Galápagos Rift. Science 1979, 203, 1073. [Google Scholar] [CrossRef] [PubMed]
- Spiess, F.N.; Macdonald, K.C.; Atwater, T.; Ballard, R.; Carranza, A.; Cordoba, D.; Cox, C.; Diaz Garcia, V.M.; Francheteau, J.; Guerrero, J.; et al. East Pacific Rise: Hot springs and geophysical experiments. Science 1980, 207, 1421–1433. [Google Scholar] [CrossRef]
- Ferrer, M.; Beloqui, A.; Timmis, K.N.; Golyshin, P.N. Metagenomics for mining new genetic resources of microbial communities. J. Mol. Microbiol. Biotechnol. 2009, 16, 109–123. [Google Scholar] [CrossRef]
- Ferrer, M.; Golyshina, O.; Beloqui, A.; Golyshin, P.N. Mining enzymes from extreme environments. Curr. Opin. Microbiol. 2007, 10, 207–214. [Google Scholar] [CrossRef]
- Vester, J.K.; Glaring, M.A.; Stougaard, P. Improved cultivation and metagenomics as new tools for bioprospecting in cold environments. Extremophiles 2015, 19, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Stokke, R.; Reeves, E.P.; Dahle, H.; Fedøy, A.-E.; Viflot, T.; Lie Onstad, S.; Vulcano, F.; Pedersen, R.B.; Eijsink, V.G.H.; Steen, I.H. Tailoring hydrothermal vent biodiversity toward improved biodiscovery using a novel in situ enrichment strategy. Front. Microbiol. 2020, 11, 249. [Google Scholar] [CrossRef]
- Delbarre-Ladrat, C.; Salas, M.L.; Sinquin, C.; Zykwinska, A.; Colliec-Jouault, S. Bioprospecting for exopolysaccharides from deep-sea hydrothermal vent bacteria: Relationship between bacterial diversity and chemical diversity. Microorganisms 2017, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Zykwinska, A.; Marchand, L.; Bonnetot, S.; Sinquin, C.; Colliec-Jouault, S.; Delbarre-Ladrat, C. Deep-sea hydrothermal vent bacteria as a source of glycosaminoglycan-mimetic exopolysaccharides. Molecules 2019, 24, 1703. [Google Scholar] [CrossRef] [Green Version]
- Luo, E.; Aylward, F.O.; Mende, D.R.; Delong, E.F. Bacteriophage distributions and temporal variability in the ocean’s interior. MBio 2017, 8, e01903-17. [Google Scholar] [CrossRef] [Green Version]
- Ray, J.; Dondrup, M.; Modha, S.; Steen, I.H.; Sandaa, R.A.; Clokie, M. Finding a needle in the virus metagenome haystack-micro-metagenome analysis captures a snapshot of the diversity of a bacteriophage armoire. PLoS ONE 2012, 7, e34238. [Google Scholar] [CrossRef] [Green Version]
- Gregory, A.C.; Zayed, A.A.; Conceição-Neto, N.; Temperton, B.; Bolduc, B.; Alberti, A.; Ardyna, M.; Arkhipova, K.; Carmichael, M.; Cruaud, C.; et al. Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell 2019, 177, 1109.e14–1123.e14. [Google Scholar] [CrossRef] [Green Version]
- Castelán-Sánchez, H.G.; Lopéz-Rosas, I.; García-Suastegui, W.A.; Peralta, R.; Dobson, A.D.W.; Batista-García, R.A.; Dávila-Ramos, S. Extremophile deep-sea viral communities from hydrothermal vents: Structural and functional analysis. Mar. Genom. 2019, 46, 16–28. [Google Scholar] [CrossRef]
- Lossouarn, J.; Dupont, S.; Gorlas, A.; Mercier, C.; Bienvenu, N.; Marguet, E.; Forterre, P.; Geslin, C. An abyssal mobilome: Viruses, plasmids and vesicles from deep-sea hydrothermal vents. Res. Microbiol. 2015, 166, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F. Dark matter of the biosphere: The amazing world of bacteriophage diversity. J. Virol. 2015, 89, 8107–8110. [Google Scholar] [CrossRef] [Green Version]
- Youle, M.; Haynes, M.; Rohwer, F. Scratching the surface of biology’s dark matter. In Viruses: Essential Agents of Life; Springer: Dordrecht, The Netherlands, 2012; pp. 61–81. ISBN 9789400748996. [Google Scholar]
- Fernandes, S.; São-José, C. Enzymes and mechanisms employed by tailed bacteriophages to breach the bacterial cell barriers. Viruses 2018, 10, 396. [Google Scholar] [CrossRef] [Green Version]
- Briers, Y.; Walmagh, M.; Van Puyenbreck, V.; Cornelissen, A.; Cenens, W.; Aertsen, A.; Oliveira, H.; Azeredo, J.; Verween, G.; Pirnay, J.P.; et al. Engineered endolysin-based “Artilysins” to combat multidrug-resistant gram-negative pathogens. MBio 2014, 5, 1379–1393. [Google Scholar] [CrossRef] [Green Version]
- Lwoff, A. Lysogeny. Bacteriol. Rev. 1953, 17, 269. [Google Scholar] [CrossRef]
- Hobbs, Z.; Abedon, S.T. Diversity of phage infection types and associated terminology: The problem with “Lytic or lysogenic”. FEMS Microbiol. Lett. 2016, 363, 47. [Google Scholar] [CrossRef] [Green Version]
- Williamson, S.J.; Cary, S.C.; Williamson, K.E.; Helton, R.R.; Bench, S.R.; Winget, D.; Wommack, K.E. Lysogenic virus-host interactions predominate at deep-sea diffuse-flow hydrothermal vents. ISME J. 2008, 2, 1112–1121. [Google Scholar] [CrossRef]
- Levin, B.R.; Lenski, R.E. Coevolution in bacteria and their viruses and plasmids. In Coevolution; Futuyama, D.J., Statkin, M., Eds.; Sinauer Associates: Sunderland, MA, USA, 1983. [Google Scholar]
- Sandaa, R.A. Burden or benefit? Virus-host interactions in the marine environment. Res. Microbiol. 2008, 159, 374–381. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Davidson, A.R. When a virus is not a parasite: The beneficial effects of prophages on bacterial fitness. J. Microbiol. 2014, 52, 235–242. [Google Scholar] [CrossRef]
- Engler, M.J.; Richardson, C.C. DNA Ligases. Enzymes 1982, 15, 3–29. [Google Scholar] [CrossRef]
- Dale, R.M.K.; McClure, B.A.; Houchins, J.P. A rapid single-stranded cloning strategy for producing a sequential series of overlapping clones for use in DNA sequencing: Application to sequencing the corn mitochondrial 18 S rDNA. Plasmid 1985, 13, 31–40. [Google Scholar] [CrossRef]
- Doherty, A.J.; Ashford, S.R.; Subramanya, H.S.; Wigley, D.B. Bacteriophage T7 DNA ligase: Overexpression, purification, crystallization, and characterization. J. Biol. Chem. 1996, 271, 11083–11089. [Google Scholar] [CrossRef] [Green Version]
- Hori, K.; Mark, D.F.; Richardson, C.C. Deoxyribonucleic acid polymerase of bacteriophage T7. Characterization of the exonuclease activities of the gene 5 protein and the reconstituted polymerase. J. Biol. Chem. 1979, 254, 11598–11604. [Google Scholar] [CrossRef]
- Engler, M.J.; Lechner, R.L.; Richardson, C.C. Two forms of the DNA polymerase of bacteriophage T7. J. Biol. Chem. 1983, 258, 11165–11173. [Google Scholar] [CrossRef]
- Sayers, J.R.; Eckstein, F. A single-strand specific endonuclease activity copurifies with overexpressed T5 D15 exonuclease. Nucleic Acids Res. 1991, 19, 4127–4132. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.R.; Son, N.; Lee, J.; Lee, D.W.; Sohn, E.J.; Hwang, I. Production of bacteriophage-encoded endolysin, LysP11, in Nicotiana benthamiana and its activity as a potent antimicrobial agent against Erysipelothrix rhusiopathiae. Plant Cell Rep. 2019, 38, 1485–1499. [Google Scholar] [CrossRef]
- Fischetti, V.A. Bacteriophage endolysins: A novel anti-infective to control Gram-positive pathogens. Int. J. Med. Microbiol. 2010, 300, 357–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witzenrath, M.; Schmeck, B.; Doehn, J.M.; Tschernig, T.; Zahlten, J.; Loeffler, J.M.; Zemlin, M.; Müller, H.; Gutbier, B.; Schütte, H.; et al. Systemic use of the endolysin Cpl-1 rescues mice with fatal pneumococcal pneumonia. Crit. Care Med. 2009, 37, 642–649. [Google Scholar] [CrossRef]
- Gupta, R.; Prasad, Y. P-27/HP Endolysin as antibacterial agent for antibiotic resistant Staphylococcus aureus of human infections. Curr. Microbiol. 2011, 63, 39–45. [Google Scholar] [CrossRef]
- Hermoso, J.A.; García, J.L.; García, P. Taking aim on bacterial pathogens: From phage therapy to enzybiotics. Curr. Opin. Microbiol. 2007, 10, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Zelcbuch, L.; Yahav, S.; Buchshtab, N.; Kahan-Hanum, M.; Vainberg-Slutskin, I.; Weiner, I.; Golembo, M.; Kredo-Russo, S.; Zak, N.; Gahali-Sass, I.; et al. Abstract PR07: Novel phages targeting the intratumor-associated bacteria Fusobacterium nucleatum. Cancer Res. 2020, 80, PR07. [Google Scholar] [CrossRef]
- Liekniņa, I.; Kalniņš, G.; Akopjana, I.; Bogans, J.; Šišovs, M.; Jansons, J.; Rūmnieks, J.; Tārs, K. Production and characterization of novel ssRNA bacteriophage virus-like particles from metagenomic sequencing data. J. Nanobiotechnology 2019, 17, 61. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Wang, L.; Mitsunobu, H.; Lu, X.; Hernandez, A.J.; Yoshida-Takashima, Y.; Nunoura, T.; Tabor, S.; Richardson, C.C. Deep-sea vent phage DNA polymerase specifically initiates DNA synthesis in the absence of primers. Proc. Natl. Acad. Sci. USA 2017, 114, E2310–E2318. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Garciá, J.L.; De Ory, A.; Brussaard, C.P.D.; De Vega, M. Phaeocystis globosa Virus DNA Polymerase X: A “swiss Army knife”, Multifunctional DNA polymerase-lyase-ligase for base excision repair. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [Green Version]
- Kurland, C.G. Codon bias and gene expression. FEBS Lett. 1991, 285, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Makrides, S.C. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol. Rev. 1996, 60, 512–538. [Google Scholar] [CrossRef]
- Rosano, G.L.; Morales, E.S.; Ceccarelli, E.A. New tools for recombinant protein production in Escherichia coli: A 5-year update. Protein Sci. 2019, 28, 1412–1422. [Google Scholar] [CrossRef]
- Costa, S.; Almeida, A.; Castro, A.; Domingues, L. Fusion tags for protein solubility, purification and immunogenicity in Escherichia coli: The novel Fh8 system. Front. Microbiol. 2014, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Kapust, R.B.; Waugh, D.S. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999, 8, 1668–1674. [Google Scholar] [CrossRef] [Green Version]
- Esposito, D.; Chatterjee, D.K. Enhancement of soluble protein expression through the use of fusion tags. Curr. Opin. Biotechnol. 2006, 17, 353–358. [Google Scholar] [CrossRef]
- Bjerga, G.E.K.; Arsın, H.; Larsen, Ø.; Puntervoll, P.; Kleivdal, H.T. A rapid solubility-optimized screening procedure for recombinant subtilisins in E. coli. J. Biotechnol. 2016, 222, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Elena, C.; Ravasi, P.; Castelli, M.E.; Peirú, S.; Menzella, H.G. Expression of codon optimized genes in microbial systems: Current industrial applications and perspectives. Front. Microbiol. 2014, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Mignon, C.; Mariano, N.; Stadthagen, G.; Lugari, A.; Lagoutte, P.; Donnat, S.; Chenavas, S.; Perot, C.; Sodoyer, R.; Werle, B. Codon harmonization–going beyond the speed limit for protein expression. FEBS Lett. 2018, 592, 1554–1564. [Google Scholar] [CrossRef]
- Claassens, N.J.; Siliakus, M.F.; Spaans, S.K.; Creutzburg, S.C.A.A.; Nijsse, B.; Schaap, P.J.; Quax, T.E.F.F.; Van Der Oost, J. Improving heterologous membrane protein production in Escherichia coli by combining transcriptional tuning and codon usage algorithms. PLoS ONE 2017, 12, e0184355. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.Y.; Xu, T.T.; Zhao, Q.J.; Li, C.L. Codon optimization enhances the expression of porcine β-defensin-2 in Escherichia coli. Genet. Mol. Res. 2015, 14, 4978–4988. [Google Scholar] [CrossRef]
- Burgess-Brown, N.A.; Sharma, S.; Sobott, F.; Loenarz, C.; Oppermann, U.; Gileadi, O. Codon optimization can improve expression of human genes in Escherichia coli: A multi-gene study. Protein Expr. Purif. 2008, 59, 94–102. [Google Scholar] [CrossRef]
- Fei, D.; Zhang, H.; Diao, Q.; Jiang, L.; Wang, Q.; Zhong, Y.; Fan, Z.; Ma, M. Codon optimization, expression in Escherichia coli, and immunogenicity of recombinant Chinese Sacbrood Virus (CSBV) structural proteins VP1, VP2, and VP3. PLoS ONE 2015, 10, e0128486. [Google Scholar] [CrossRef]
- Roalkvam, I.; Bredy, F.; Baumberger, T.; Pedersen, R.B.; Steen, I.H. Hypnocyclicus thermotrophus gen. Nov., sp. nov. isolated from a microbial mat in a hydrothermal vent field. Int. J. Syst. Evol. Microbiol. 2015, 65, 4521–4525. [Google Scholar] [CrossRef]
- Marques, A.F.A.; Roerdink, D.L.; Baumberger, T.; de Ronde, C.E.J.; Ditchburn, R.G.; Denny, A.; Thorseth, I.H.; Okland, I.; Lilley, M.D.; Whitehouse, M.J.; et al. The seven sisters hydrothermal system: First record of shallow hybrid mineralization hosted in mafic volcaniclasts on the arctic mid-ocean ridge. Minerals 2020, 10, 439. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition-Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLOS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.; Tavares, P.; Petit, M.A.; Guérois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 2014, 15, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I. phyloT: A Phylogenetic Tree Generator. Available online: https://phylot.biobyte.de/index.cgi (accessed on 5 June 2020).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Fu, Y.X.; Li, W.H. Maximum likelihood estimation of the heterogeneity of substitution rate among nucleotide sites. Mol. Biol. Evol. 1995, 12, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paez-Espino, D.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Szeto, E.; Pillay, M.; Huang, J.; Markowitz, V.M.; Nielsen, T.; et al. IMG/VR: A database of cultured and uncultured DNA Viruses and retroviruses. Nucleic Acids Res. 2016, 45. [Google Scholar] [CrossRef] [PubMed]
- Villar, E.; Vannier, T.; Vernette, C.; Lescot, M.; Cuenca, M.; Alexandre, A.; Bachelerie, P.; Rosnet, T.; Pelletier, E.; Sunagawa, S.; et al. The Ocean Gene Atlas: Exploring the biogeography of plankton genes online. Nucleic Acids Res. 2018, 46, W289–W295. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.J.; de Crécy-Lagard, V.; Zallot, R. Gene Graphics: A genomic neighborhood data visualization web application. Bioinformatics 2017, 34, 1406–1408. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Clements, J.; Arndt, W.; Miller, B.L.; Wheeler, T.J.; Schreiber, F.; Bateman, A.; Eddy, S.R. HMMER web server: 2015 update. Nucleic Acids Res. 2015, 43, W30–W38. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [Green Version]
- Angov, E.; Legler, P.; Mease, R. Adjustment of codon usage frequencies by codon harmonization improves protein expression and folding. Methods Mol. Biol. 2011, 705, 1–13. [Google Scholar] [CrossRef]
- Angov, E. Codon usage: Nature’s roadmap to expression and folding of proteins. Biotechnol. J. 2011, 6, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Novagen Inc. pET System Manual. In TB055; Merck: Darmstadt, Germany, 2006; pp. 19–24. [Google Scholar]
- Hochuli, E.; Bannwarth, W.; Döbeli, H.; Gentz, R.; Stüber, D. Genetic approach to facilitate purification of recombinant proteins with a novel metal chelate adsorbent. Nat. Biotechnol. 1988, 6, 1321–1325. [Google Scholar] [CrossRef]
- Kristensen, D.M.; Waller, A.S.; Yamada, T.; Bork, P.; Mushegian, A.R.; Koonin, E.V. Orthologous gene clusters and taxon signature genes for viruses of prokaryotes. J. Bacteriol. 2013, 195, 941–950. [Google Scholar] [CrossRef] [Green Version]
- Rezaei Javan, R.; Ramos-Sevillano, E.; Akter, A.; Brown, J.; Brueggemann, A.B. Prophages and satellite prophages are widespread in Streptococcus and may play a role in pneumococcal pathogenesis. Nat. Commun. 2019, 10, 4852. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, K.; Manson McGuire, A.; Priest, M.E.; Abouelleil, A.; Cerqueira, G.C.; Lo, R.; Earl, A.M.; Allen-Vercoe, E. Complete genome sequences and analysis of the Fusobacterium nucleatum subspecies animalis 7-1 bacteriophage ɸFunu1 and ɸFunu2. Anaerobe 2016, 38, 125–129. [Google Scholar] [CrossRef] [Green Version]
- Machuca, P.; Daille, L.; Vinés, E.; Berrocal, L.; Bittner, M. Isolation of a novel bacteriophage specific for the periodontal pathogen Fusobacterium nucleatum. Appl. Environ. Microbiol. 2010, 76, 7243–7250. [Google Scholar] [CrossRef] [Green Version]
- Kabwe, M.; Brown, T.L.; Dashper, S.; Speirs, L.; Ku, H.; Petrovski, S.; Chan, H.T.; Lock, P.; Tucci, J. Genomic, morphological and functional characterisation of novel bacteriophage FNU1 capable of disrupting Fusobacterium nucleatum biofilms. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum—Symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef]
- Quax, T.E.F.; Claassens, N.J.; Söll, D.; van der Oost, J. Codon bias as a means to fine-tune gene expression. Mol. Cell 2015, 59, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Porath, J. Immobilized metal ion affinity chromatography. Protein Expr. Purif. 1992, 3, 263–281. [Google Scholar] [CrossRef]
- Razdan, A. Affinity Chromatography Handbook, Vol. 2: Tagged Proteins; GE Healthcare Bio-Sciences AB: Uppsala, Sweden, 2000; Available online: https://www.cytivalifesciences.com/en/us/support/handbooks (accessed on 9 January 2021).
- Freitag-Pohl, S.; Jasilionis, A.; Håkansson, M.; Svensson, L.A.; Kovačič, R.; Welin, M.; Watzlawick, H.; Wang, L.; Altenbuchner, J.; Płotka, M.; et al. Crystal structures of the Bacillus subtilis prophage lytic cassette proteins XepA and YomS. Acta Crystallogr. Sect. D Struct. Biol. 2019, 75, 1028–1039. [Google Scholar] [CrossRef] [Green Version]
- Weinbauer, M.G.; Brettar, I.; Höfle, M.G. Lysogeny and virus-induced mortality of bacterioplankton in surface, deep, and anoxic marine waters. Limnol. Oceanogr. 2003, 48, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, E.; Brockhurst, M.A. Ecological and evolutionary benefits of temperate phage: What does or doesn’t kill you makes you stronger. BioEssays 2017, 39, 1700112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, J.H. Prophages in marine bacteria: Dangerous molecular time bombs or the key to survival in the seas? ISME J. 2008, 2, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Grose, J.H.; Casjens, S.R. Bacteriophage Diversity. In Encyclopedia of Virology; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Amarillas, L.; Rubí-Rangel, L.; Chaidez, C.; González-Robles, A.; Lightbourn-Rojas, L.; León-Félix, J. Isolation and characterization of phiLLS, a novel phage with potential biocontrol agent against multidrug-resistant Escherichia coli. Front. Microbiol. 2017, 8, 1355. [Google Scholar] [CrossRef]
- Bokma, E.; Van Koningsveld, G.A.; Jeronimus-Stratingh, M.; Beintema, J.J. Hevamine, a chitinase from the rubber tree Hevea brasiliensis, cleaves peptidoglycan between the C-1 of N-acetylglucosamine and C-4 of N-acetylmuramic acid and therefore is not a lysozyme. FEBS Lett. 1997, 411, 161–163. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.J.; Sørbotten, A.; Synstad, B.; Sikorski, P.; Sørlie, M.; Vårum, K.M.; Eijsink, V.G.H. Endo/exo mechanism and processivity of family 18 chitinases produced by Serratia marcescens. FEBS J. 2006, 273, 491–503. [Google Scholar] [CrossRef]
- Saier, M.H.; Reddy, B.L. Holins in bacteria, eukaryotes, and archaea: Multifunctional xenologues with potential biotechnological and biomedical applications. J. Bacteriol. 2015, 197, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Cahill, J.; Young, R. Phage Lysis: Multiple genes for multiple barriers. In Advances in Virus Research; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 103, pp. 33–70. ISBN 9780128177228. [Google Scholar]
- Kongari, R.; Rajaure, M.; Cahill, J.; Rasche, E.; Mijalis, E.; Berry, J.; Young, R. Phage spanins: Diversity, topological dynamics and gene convergence. BMC Bioinform. 2018, 19, 326. [Google Scholar] [CrossRef] [Green Version]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yi, L.; Marek, P.; Iverson, B.L. Commercial proteases: Present and future. FEBS Lett. 2013, 587, 1155–1163. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.; Ismail, A.; Dinu, C.; Chapman, J.; Ismail, A.E.; Dinu, C.Z. Industrial applications of enzymes: Recent Advances, techniques, and outlooks. Catalysts 2018, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Gomes, J.; Steiner, W. The biocatalytic potential of extremophiles and extremozymes. Food Technol. Biotechnol. 2004, 42, 223–225. [Google Scholar]
- Aschenbrenner, J.; Marx, A. DNA polymerases and biotechnological applications. Curr. Opin. Biotechnol. 2017, 48, 187–195. [Google Scholar] [CrossRef]
- Vieille, C.; Burdette, D.S.; Zeikus, J.G. Thermozymes. Biotechnol. Annu. Rev. 1996, 2, 1–83. [Google Scholar]
- Ishino, S.; Ishino, Y. DNA polymerases as useful reagents for biotechnology - The history of developmental research in the field. Front. Microbiol. 2014, 5, 465. [Google Scholar] [CrossRef] [Green Version]
- Aevarsson, A.; Kaczorowska, A.-K.; Adalsteinsson, B.T.; Ahlqvist, J.; Al-Karadaghi, S.; Altenbuchner, J.; Arsın, H.; Átlasson, Ú.Á.; Brandt, D.; Cichowicz-Cieślak, M.; et al. Going to extremes—A metagenomic journey into the dark matter of life. FEMS Microbiol. Lett. 2021. [Google Scholar] [CrossRef]
- GenScript GenSmartTM Codon Optimization Tool-GenScript. Available online: https://www.genscript.com/gensmart-free-gene-codon-optimization.html (accessed on 15 September 2020).
- Chen, F.; Wu, P.; Deng, S.; Zhang, H.; Hou, Y.; Hu, Z.; Zhang, J.; Chen, X.; Yang, J.-R. Dissimilation of synonymous codon usage bias in virus–host coevolution due to translational selection. Nat. Ecol. Evol. 2020, 4, 589–600. [Google Scholar] [CrossRef]
- Angov, E.; Hillier, C.J.; Kincaid, R.L.; Lyon, J.A. Heterologous protein expression is enhanced by harmonizing the codon usage frequencies of the target gene with those of the expression host. PLoS ONE 2008, 3, e2189. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Lord, H.; Knutson, N.; Wikström, M. Nano differential scanning fluorimetry for comparability studies of therapeutic proteins. Anal. Biochem. 2020, 593, 113581. [Google Scholar] [CrossRef] [PubMed]
- Pellizza, L.; Smal, C.; Rodrigo, G.; Arán, M. Codon usage clusters correlation: Towards protein solubility prediction in heterologous expression systems in E. coli. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Gould, N.; Hendy, O.; Papamichail, D. Computational tools and algorithms for designing customized synthetic genes. Front. Bioeng. Biotechnol. 2014, 2, 41. [Google Scholar] [CrossRef] [Green Version]
- Sen, A.; Kargar, K.; Akgün, E.; Pınar, M.C. Codon optimization: A mathematical programing approach. Bioinformatics 2020, 36, 4012–4020. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identifier | Proposed Protein Function | CAI for Expression Host | CHI for Expression Host | Codon Native Gene Sequence CAI for Native Host | Soluble Produced Protein Yield * (mg/L) | |||
|---|---|---|---|---|---|---|---|---|
| CO Gene Variant | CH Gene Variant | CO Gene Variant | CH Gene Variant | Expressed from CO Gene Variant | Expressed from CH Gene Variant | |||
| HTP4435 | Endopeptidase tail | 0.89 | 0.64 | 0.61 | 0.48 | 0.50 | 18.7 ± 1.3 | 13.8 ± 1.5 |
| HTP4425 | Hypothetical protein | 0.87 | 0.67 | 0.59 | 0.48 | 0.51 | ND | ND |
| HTP4420 | Glycosyl hydrolase 18 | 0.89 | 0.66 | 0.60 | 0.45 | 0.51 | ND | ND |
| HTP4415 | Holin, toxin secretion/phage lysis | 0.87 | 0.58 | 0.60 | 0.47 | 0.40 | ND | ND |
| HTP4410 | N-acetylmuramoyl-L-alanine amidase | 0.88 | 0.64 | 0.61 | 0.47 | 0.46 | 40.7 ± 5 | 30.8 ± 3.4 |
| HTP4400 | rRNA biogenesis protein rrp5, putative | 0.84 | 0.64 | 0.58 | 0.48 | 0.48 | 135.80 ± 2.49 | 27.9 ± 3.2 |
| HTP4385 | DNA Polymerase | 0.86 | 0.62 | 0.60 | 0.46 | 0.47 | ND | ND |
| HTP4360 | hypothetical protein | 0.84 | 0.74 | 0.55 | 0.43 | 0.56 | 151.7 ± 10.2 | ND |
| HTP4350 | DUF262 / DNase | 0.86 | 0.62 | 0.59 | 0.45 | 0.44 | 32.3 ± 1.2 | 15.6 ± 2.7 |
| Identifier | Proposed Protein Function | Protein Purification Yield * (%) | |
|---|---|---|---|
| Target Protein Expressed from CO Gene Variant | Target Protein Expressed from CH Gene Variant | ||
| HTP4410 | N-acetylmuramoyl-L-alanine amidase | 85.6 ± 1.4 | 85.9 ± 1.9 |
| HTP4400 | rRNA biogenesis protein rrp5, putative | 38.6 ± 7.2 | 58 ± 3.7 |
| HTP4360 | hypothetical protein | 75 ± 3.8 | ND |
| HTP4350 | DUF262/DNase | 83.5 ± 5.2 | 92.1 ± 2.1 |
| Target Protein | Proposed Protein Function | Melting Temperature (Tm, °C) | |
|---|---|---|---|
| Target Protein Expressed from CO Gene Variant | Target Protein Expressed from CH Gene Variant | ||
| HTP4410 | N-acetylmuramoyl-L-alanine amidase | 66.23 ± 0.07 * | 73.03 ± 0.10 |
| HTP4400 | rRNA biogenesis protein rrp5, putative | 51.57 ± 0.34 | 55.70 ± 0.22 |
| HTP4350 | DUF262 / DNase | 61.57 ± 1.47 | 65.24 ± 0.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arsın, H.; Jasilionis, A.; Dahle, H.; Sandaa, R.-A.; Stokke, R.; Nordberg Karlsson, E.; Steen, I.H. Exploring Codon Adjustment Strategies towards Escherichia coli-Based Production of Viral Proteins Encoded by HTH1, a Novel Prophage of the Marine Bacterium Hypnocyclicus thermotrophus. Viruses 2021, 13, 1215. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071215
Arsın H, Jasilionis A, Dahle H, Sandaa R-A, Stokke R, Nordberg Karlsson E, Steen IH. Exploring Codon Adjustment Strategies towards Escherichia coli-Based Production of Viral Proteins Encoded by HTH1, a Novel Prophage of the Marine Bacterium Hypnocyclicus thermotrophus. Viruses. 2021; 13(7):1215. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071215
Chicago/Turabian StyleArsın, Hasan, Andrius Jasilionis, Håkon Dahle, Ruth-Anne Sandaa, Runar Stokke, Eva Nordberg Karlsson, and Ida Helene Steen. 2021. "Exploring Codon Adjustment Strategies towards Escherichia coli-Based Production of Viral Proteins Encoded by HTH1, a Novel Prophage of the Marine Bacterium Hypnocyclicus thermotrophus" Viruses 13, no. 7: 1215. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071215