
Geographic Distribution and Phylogeny of Soricine Shrew-Borne Seewis Virus and Altai Virus in Russia

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Trapping and Sample Collection
2.2. RNA Extraction and RT-PCR Analysis
2.3. Genetic and Phylogenetic Analysis
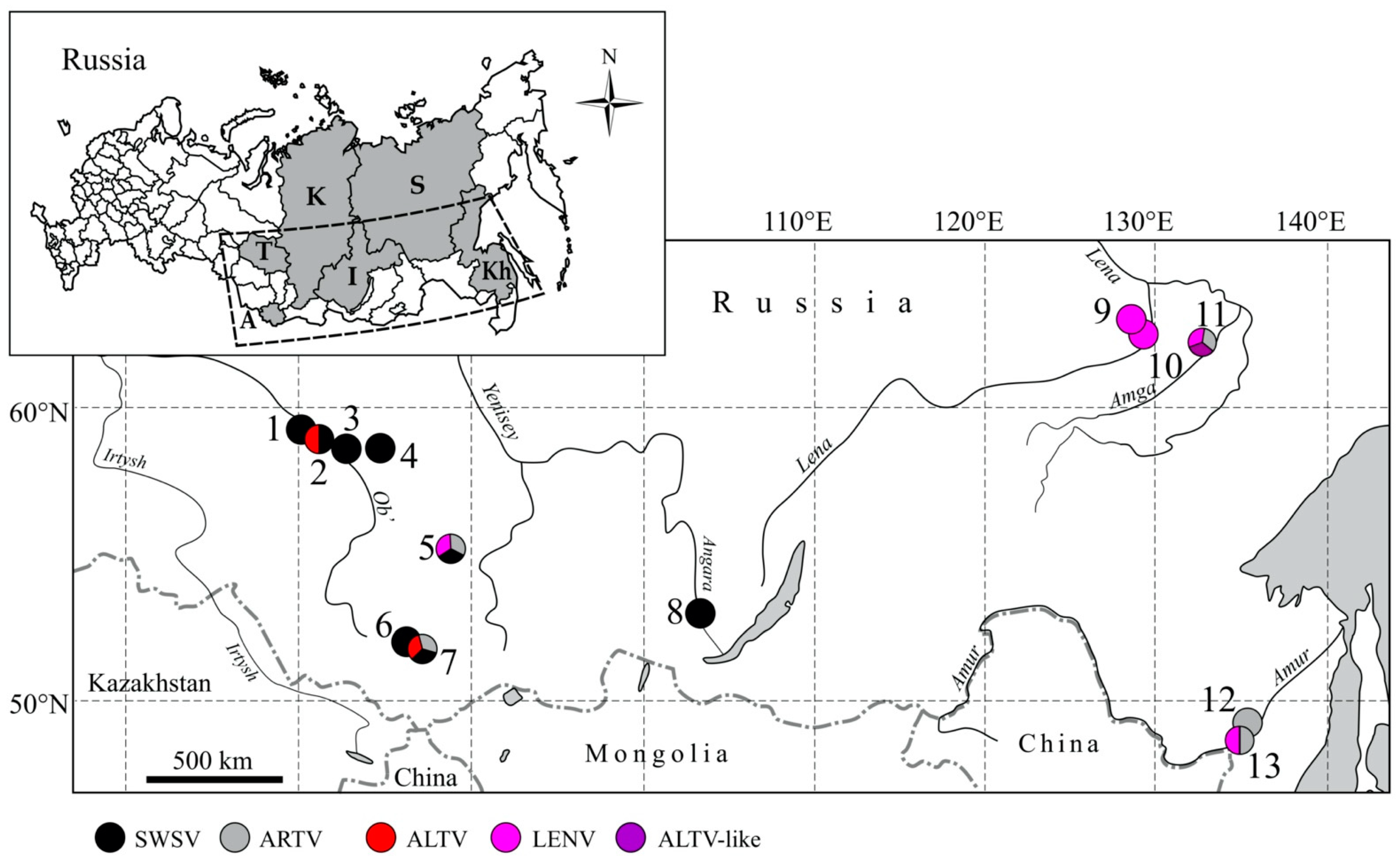
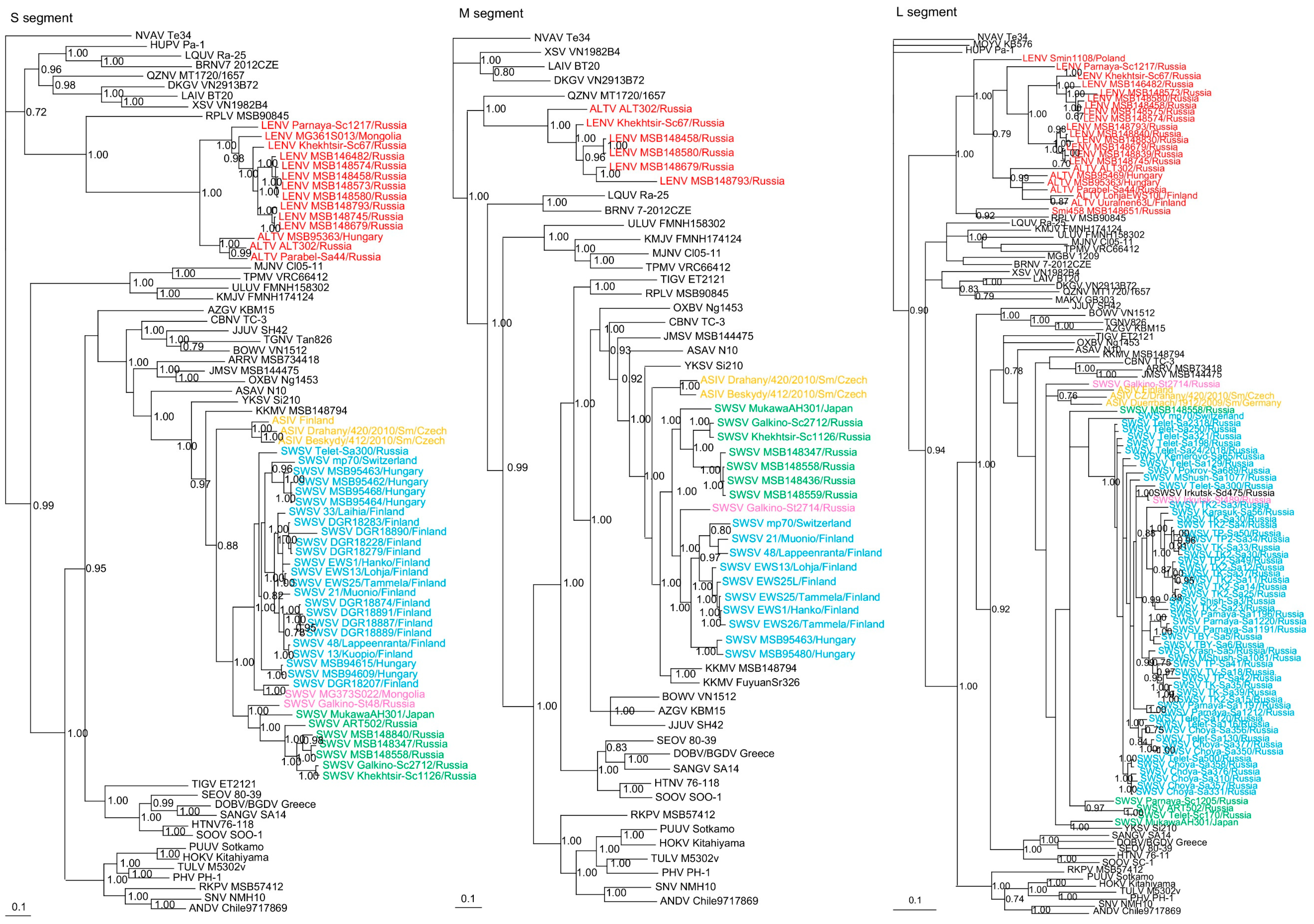
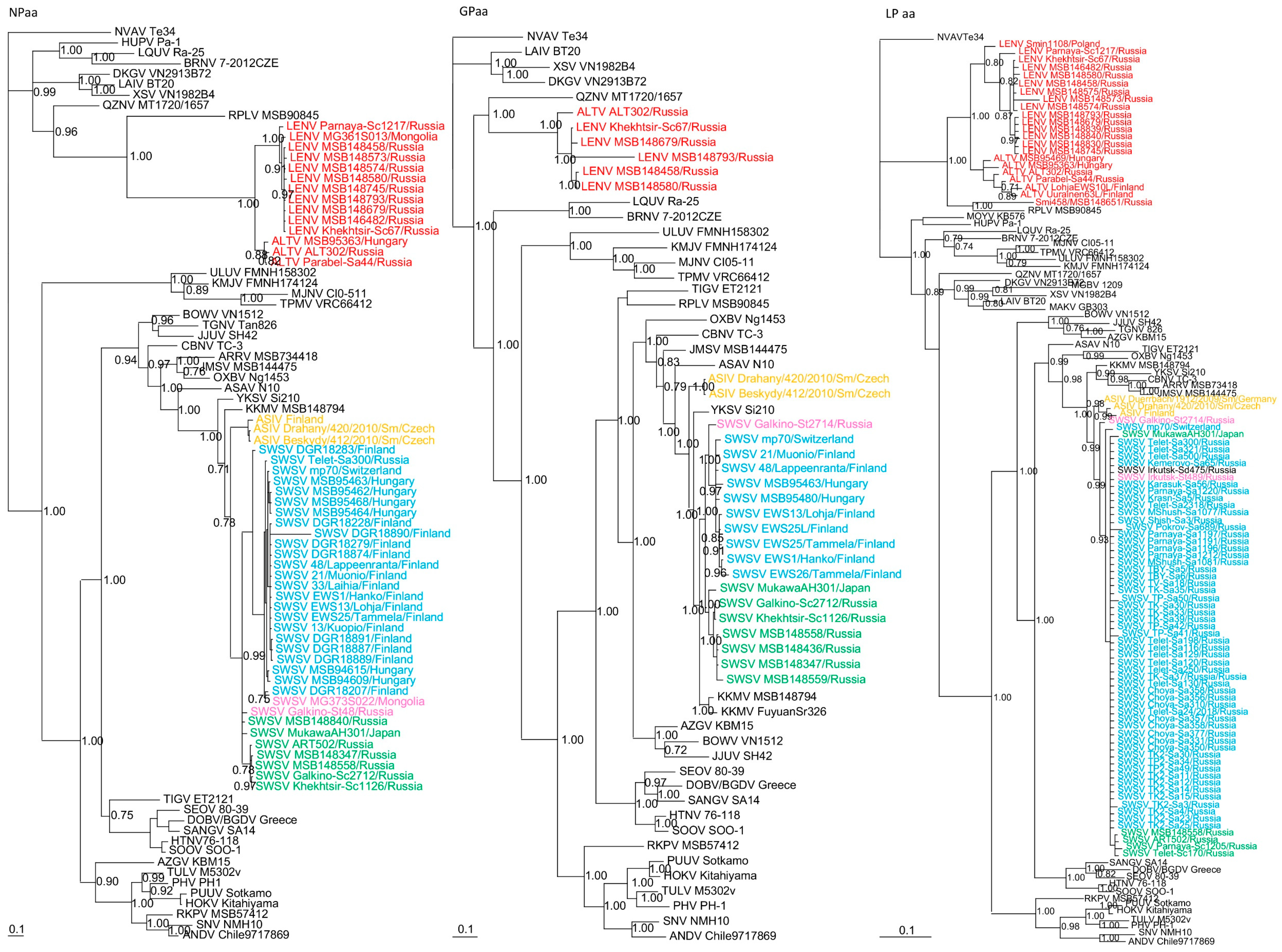
3. Results
3.1. Genetic Analysis
3.2. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Nucleotide Sequences
References
- Jonsson, C.B.; Figueiredo, L.T.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010, 23, 412–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avšič-Županc, T.; Saksida, A.; Korva, M. Hantavirus infections. Clin. Microbiol. Infect. 2019, 21S, e6–e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, P.; Adkins, S.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, E.; Blair, C.D.; Briese, T.; et al. Taxonomy of the order Bunyavirales: Second update 2018. Arch. Virol. 2019, 164, 927–941. [Google Scholar] [CrossRef] [Green Version]
- Yanagihara, R.; Gu, S.H.; Arai, S.; Kang, H.J.; Song, J.-W. Hantaviruses: Rediscovery and new beginnings. Virus Res. 2014, 187, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Yanagihara, R. Genetic diversity and geographic distribution of bat-borne hantaviruses. Curr. Issues Mol. Biol. 2020, 39, 1–28. [Google Scholar] [CrossRef]
- Vapalahti, O.; Lundkvist, A.; Fedorov, V.; Conroy, C.J.; Hirvonen, S.; Plyusnina, A.; Nemirov, K.; Fredga, K.; Cook, J.A.; Niemimaa, J.; et al. Isolation and characterization of a hantavirus from Lemmus sibiricus: Evidence for host switch during hantavirus evolution. J. Virol. 1999, 73, 5586–5592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Bennett, S.N.; Dizney, L.; Sumibcay, L.; Arai, S.; Ruedas, L.A.; Song, J.W.; Yanagihara, R. Host switch during evolution of a genetically distinct hantavirus in the American shrew mole (Neurotrichus gibbsii). Virology 2009, 388, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Ling, J.; Sironen, T.; Voutilainen, L.; Hepojoki, S.; Niemimaa, J.; Isoviita, V.M.; Vaheri, A.; Henttonen, H.; Vapalahti, O. Hantaviruses in Finnish soricomorphs: Evidence for two distinct hantaviruses carried by Sorex araneus suggesting ancient host-switch. Infect. Genet. Evol. 2014, 27, 51–61. [Google Scholar] [CrossRef]
- Kang, H.J.; Gu, S.H.; Yashina, L.N.; Cook, J.A.; Yanagihara, R. Highly divergent genetic variants of soricid-borne Altai virus (Hantaviridae) in Eurasia suggest ancient host-switching events. Viruses 2019, 11, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liphardt, S.W.; Kang, H.J.; Dizney, L.J.; Ruedas, L.A.; Cook, J.A.; Yanagihara, R. Complex history of codiversification and host switching of a newfound soricid-borne orthohantavirus in North America. Viruses 2019, 11, 637. [Google Scholar] [CrossRef] [Green Version]
- Guterres, A.; de Oliveira, R.C.; Fernandes, J.; de Lemos, E.R.S. The mystery of the phylogeographic structural pattern in rodent-borne hantaviruses. Mol. Phylogenet. Evol. 2019, 136, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Schmaljohn, A.L.; Anderson, K.; Schmaljohn, C.S. Complete nucleotide sequences of the M and S segments of two hantavirus isolates from California: Evidence for reassortment in nature among viruses related to hantavirus pulmonary syndrome. Virology 1995, 206, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Sironen, T.; Henttonen, H.; Plyusnin, A. Analysis of Puumala hantavirus in a bank vole population in northern Finland: Evidence for co-circulation of two genetic lineages and frequent reassortment between strains. J. Gen. Virol. 2009, 90, 1923–1931. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.A.; Kim, W.K.; No, J.S.; Lee, S.H.; Lee, S.Y.; Kim, J.H.; Kho, J.H.; Lee, D.; Song, D.H.; Gu, S.H.; et al. Genetic diversity and reassortment of Hantaan virus tripartite RNA genomes in nature, the Republic of Korea. PLoS Negl. Trop. Dis. 2016, 10, e0004650. [Google Scholar] [CrossRef]
- Klempa, B. Reassortment events in the evolution of hantaviruses. Virus Genes 2018, 54, 638–646. [Google Scholar] [CrossRef] [Green Version]
- Laenen, L.; Vergote, V.; Kafetzopoulou, L.E.; Wawina, T.B.; Vassou, D.; Cook, J.A.; Hugot, J.P.; Deboutte, W.; Kang, H.J.; Witkowski, P.T.; et al. A novel hantavirus of the European mole, Bruges virus, is involved in frequent Nova virus coinfections. Genome Biol. Evol. 2018, 10, 45–55. [Google Scholar] [CrossRef]
- Liphardt, S.W.; Kang, H.J.; Arai, S.; Gu, S.H.; Cook, J.A.; Yanagihara, R. Reassortment between divergent strains of Camp Ripley virus (Hantaviridae) in the northern short-tailed shrew (Blarina brevicauda). Front. Cell Infect. Microbiol. 2020, 10, 460. [Google Scholar] [CrossRef]
- Klempa, B.; Avšič-Županc, T.; Clement, J.; Dzagurova, T.K.; Henttonen, H.; Heyman, P.; Jakab, F.; Krüger, D.H.; Maes, P.; Papa, A.; et al. Complex evolution and epidemiology of Dobrava-Belgrade hantavirus: Definition of genotypes and their characteristics. Arch. Virol. 2013, 158, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Song, J.-W.; Gu, S.H.; Bennett, S.N.; Arai, S.; Puorger, M.; Hilbe, M.; Yanagihara, R. Seewis virus, a genetically distinct hantavirus in the Eurasian common shrew (Sorex araneus). Virol. J. 2007, 4, 114. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.J.; Arai, S.; Hope, A.G.; Song, J.-W.; Cook, J.A.; Yanagihara, R. Genetic diversity and phylogeography of Seewis virus in the Eurasian common shrew in Finland and Hungary. Virol. J. 2009, 6, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, M.; Radosa, L.; Rosenfeld, U.M.; Schmidt, S.; Triebenbacher, C.; Löhr, P.W.; Fuchs, D.; Heroldová, M.; Jánová, E.; Stanko, M.; et al. Broad geographical distribution and high genetic diversity of shrew-borne Seewis hantavirus in Central Europe. Virus Genes 2012, 45, 48–55. [Google Scholar] [CrossRef]
- Yashina, L.; Abramov, S.; Gutorov, V.; Dupal, T.; Krivopalov, A.; Panov, V.; Danchinova, G.; Vinogradov, V.; Luchnikova, E.; Hay, J.; et al. Seewis virus: Phylogeography of a shrew-borne hantavirus in Siberia, Russia. Vector-Borne Zoonotic Dis. 2010, 10, 585–591. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.H.; Hejduk, J.; Markowski, J.; Kang, H.J.; Markowski, M.; Połatyńska, M.; Sikorska, B.; Liberski, P.P.; Yanagihara, R. Co-circulation of soricid- and talpid-borne hantaviruses in Poland. Infect. Genet. Evol. 2014, 28, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Kang, H.J.; Gu, S.H.; Ohdachi, S.D.; Cook, J.A.; Yashina, L.N.; Tanaka-Taya, K.; Abramov, S.A.; Morikawa, S.; Okabe, N.; et al. Genetic diversity of Artybash virus in the Laxmann’s shrew (Sorex caecutiens). Vector Borne Zoonotic Dis. 2016, 16, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Yashina, L.N.; Kartashov, M.Y.; Wang, W.; Li, K.; Zdanovskaya, N.I.; Ivanov, L.I.; Zhang, Y.Z. Co-circulation of distinct shrew-borne hantaviruses in the far east of Russia. Virus Res. 2019, 272, 197717. [Google Scholar] [CrossRef]
- Mills, J.N.; Childs, J.E.; Ksiazek, T.G.; Peters, C.J. Methods for Trapping and Sampling Small Mammals for Virologic Testing; US Department of Health and Human Services, Center for Disease Control and Prevention: Atlanta, GA, USA, 1995.
- Sikes, R.S. Animal Care and Use Committee of the American Society of Mammalogists. 2016 Guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. J. Mammal. 2016, 97, 663–688. [Google Scholar] [CrossRef]
- Cook, J.A.; Galbreath, K.E.; Bell, K.C.; Campbell, M.L.; Carrière, S.; Colella, J.P.; Dawson, N.G.; Dunnum, J.L.; Eckerlin, R.P.; Greiman, S.E.; et al. The Beringian Coevolution Project: Holistic collections of mammals and associated parasites reveal novel perspectives on evolutionary and environmental change in the North. Arctic Sci. 2017, 3, 585–617. [Google Scholar] [CrossRef]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Barrier, P.; Koivogue, L.; Meulen, J.; Krüger, D.H. Novel hantavirus sequences in shrew, Guinea. Emerg. Infect. Dis. 2007, 13, 520–522. [Google Scholar] [CrossRef]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Yashina, L.N.; Abramov, S.A.; Dupal, T.A.; Yakimenko, V.V.; Tantsev, A.K.; Malyshev, B.S.; Kartashev, M.Y. Hantaviruses in insectivore populations in Siberia. Probl. Part. Danger. Infect. 2018, 4, 89–93. (In Russian) [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [Green Version]
- Posada, D.; Crandall, K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Ling, J.; Vaheri, A.; Hepojiki, S.; Levanov, L.; Jaaskelainen, A.; Henttonen, H.; Vapalahti, O.; Sironen, T.; Hepojoki, S. Serological survey of Seewis virus (SWSV) antibodies in patient suspected for hantavirus infection in Finland. J. Gen. Virol. 2015, 96, 1664–1675. [Google Scholar] [CrossRef]
- Lundkvist, A.; Meisel, H.; Koletzki, D.; Lankinen, H.; Cifire, F.; Geldmacher, A.; Sibold, C.; Gött, P.; Vaheri, A.; Krüger, D.H.; et al. Mapping of B-cell epitopes in the nucleocapsid protein of Puumala hantavirus. Viral Immunol. 2002, 15, 177–192. [Google Scholar] [CrossRef]
- Yoshimatsu, K.; Arikawa, J. Serological diagnosis with recombinant N antigen for hantavirus infection. Virus Res. 2014, 187, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Song, J.-W.; Sumibcay, L.; Bennett, S.N.; Nerurkar, V.R.; Parmenter, C.; Cook, J.A.; Yates, T.L.; Yanagihara, R. Hantavirus in northern short-tailed shrew, United States. Emerg. Infect. Dis. 2007, 13, 1420–1423. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Region | Collection Site | Species | Year | Hantavirus RNA Positive/Tested | Virus |
|---|---|---|---|---|---|
| Altai Republic | Teletskoye | Sorex araneus | 2007 | 7/10 | SWSV, ALTV |
| Sorex caecutiens | 2007 | 1/1 | ARTV | ||
| Sorex araneus | 2018 | 1/6 | SWSV | ||
| Sorex araneus | 2019 | 6/43 | SWSV | ||
| Sorex caecutiens | 2019 | 1/1 | ARTV | ||
| Sorex minutus | 2019 | 0/2 | - | ||
| Choya | Sorex araneus | 2019 | 8/23 | SWSV | |
| Sorex isodon | 2019 | 0/1 | - | ||
| Tomsk Oblast | Belyi Yar | Sorex araneus | 2019 | 2/10 | SWSV |
| Sorex caecutiens | 2019 | 0/3 | - | ||
| Volkovo | Sorex araneus | 2019 | 1/7 | SWSV | |
| Kargasok | Sorex araneus | 2019 | 15/46 | SWSV | |
| Sorex caecutiens | 2019 | 0/1 | - | ||
| Parabel | Sorex araneus | 2019 | 6/28 | SWSV, ALTV | |
| Sorex caecutiens | 2019 | 0/2 | - | ||
| Khabarovsk Krai | Khekhtsir | Sorex caecutiens | 2007 | 1/13 | ARTV |
| Sorex caecutiens | 2008 | 1/7 | LENV | ||
| Galkino | Sorex caecutiens | 2007 | 2/28 | ARTV | |
| Sorex tundrensis | 2007 | 10/32 | ARTV | ||
| Sorex caecutiens | 2008 | 0/11 | - | ||
| Sorex tundrensis | 2008 | 4/13 | ARTV | ||
| Krasnoyarsk Krai | Parnaya | Sorex araneus | 2008 | 5/17 | SWSV |
| Sorex caecutiens | 2008 | 2/2 | ARTV, LENV | ||
| Sorex tundrensis | 2008 | 0/3 | - | ||
| Irkutsk Oblast | Irkutsk City | Sorex araneus | 2007 | 0/2 | - |
| Sorex tundrensis | 2007 | 1/2 | SWSV | ||
| Sorex daphaenodon | 2007 | 1/2 | SWSV | ||
| Sakha Republic | Amga River | Sorex caecutiens | 2006 | 10/19 | ARTV, LENV |
| Sorex minutissimus | 2006 | 1/5 | ALTV-like | ||
| Kenkeme River | Sorex caecutiens | 2006 | 4/24 | LENV | |
| Sorex roboratus | 2006 | 4/12 | LENV | ||
| Sorex daphaenodon | 2006 | 0/4 | - | ||
| Lena River | Sorex caecutiens | 2006 | 1/6 | LENV | |
| Sorex tundrensis | 2006 | 0/5 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yashina, L.N.; Abramov, S.A.; Zhigalin, A.V.; Smetannikova, N.A.; Dupal, T.A.; Krivopalov, A.V.; Kikuchi, F.; Senoo, K.; Arai, S.; Mizutani, T.; et al. Geographic Distribution and Phylogeny of Soricine Shrew-Borne Seewis Virus and Altai Virus in Russia. Viruses 2021, 13, 1286. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071286
Yashina LN, Abramov SA, Zhigalin AV, Smetannikova NA, Dupal TA, Krivopalov AV, Kikuchi F, Senoo K, Arai S, Mizutani T, et al. Geographic Distribution and Phylogeny of Soricine Shrew-Borne Seewis Virus and Altai Virus in Russia. Viruses. 2021; 13(7):1286. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071286
Chicago/Turabian StyleYashina, Liudmila N., Sergey A. Abramov, Alexander V. Zhigalin, Natalia A. Smetannikova, Tamara A. Dupal, Anton V. Krivopalov, Fuka Kikuchi, Kae Senoo, Satoru Arai, Tetsuya Mizutani, and et al. 2021. "Geographic Distribution and Phylogeny of Soricine Shrew-Borne Seewis Virus and Altai Virus in Russia" Viruses 13, no. 7: 1286. https://0-doi-org.brum.beds.ac.uk/10.3390/v13071286