
Variable In Vivo Hepatitis D Virus (HDV) RNA Editing Rates According to the HDV Genotype

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Non-Edited and Edited Clones
2.2. Quantification of the In Vivo Editing Rate in Plasma Samples
2.3. Determination of the Secondary Structures of the RNA Editing Region
2.4. Statistical Analyses
3. Results
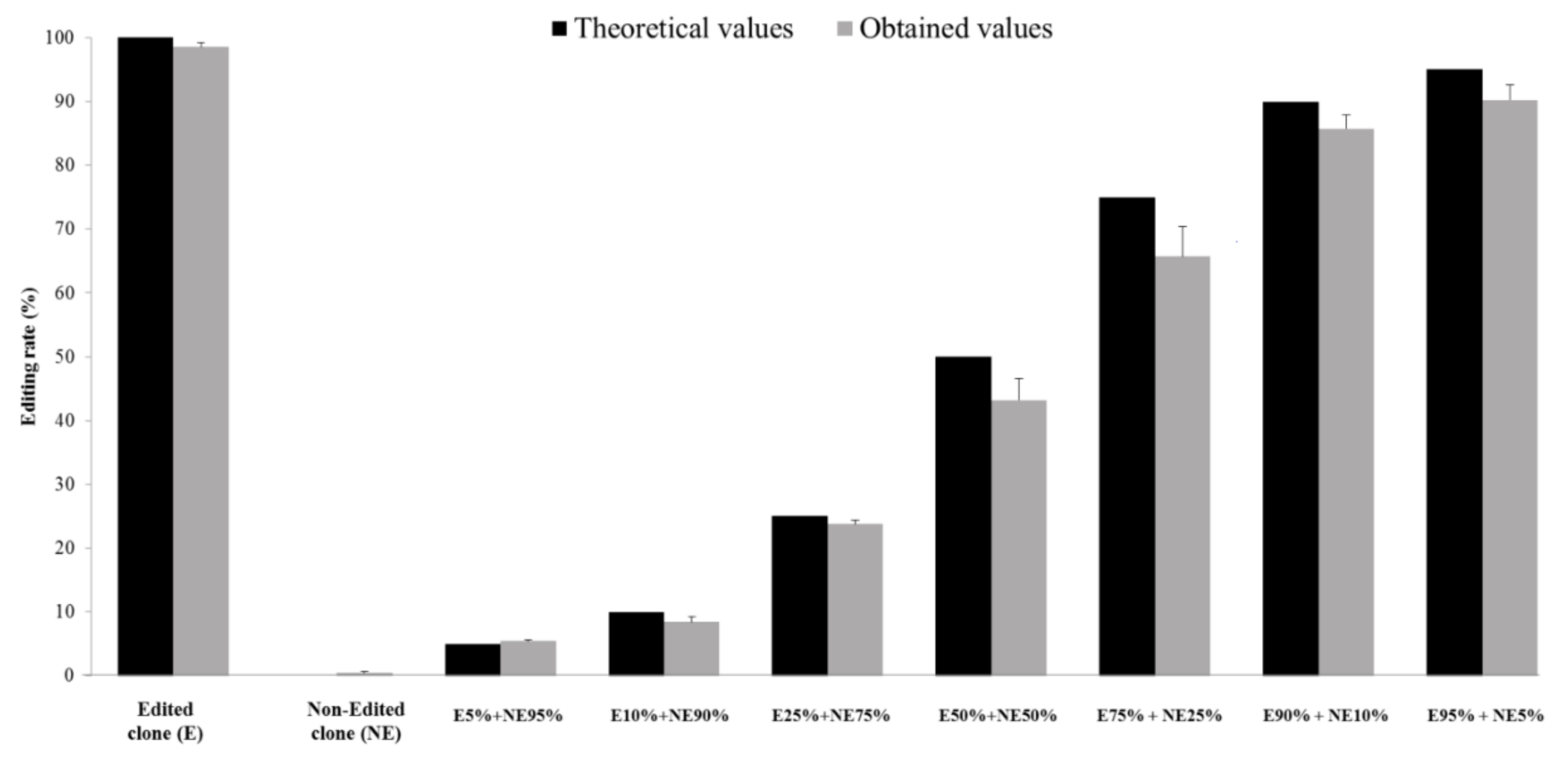
3.1. Provision of a New Tool to Quantify HDV RNA Editing Rate
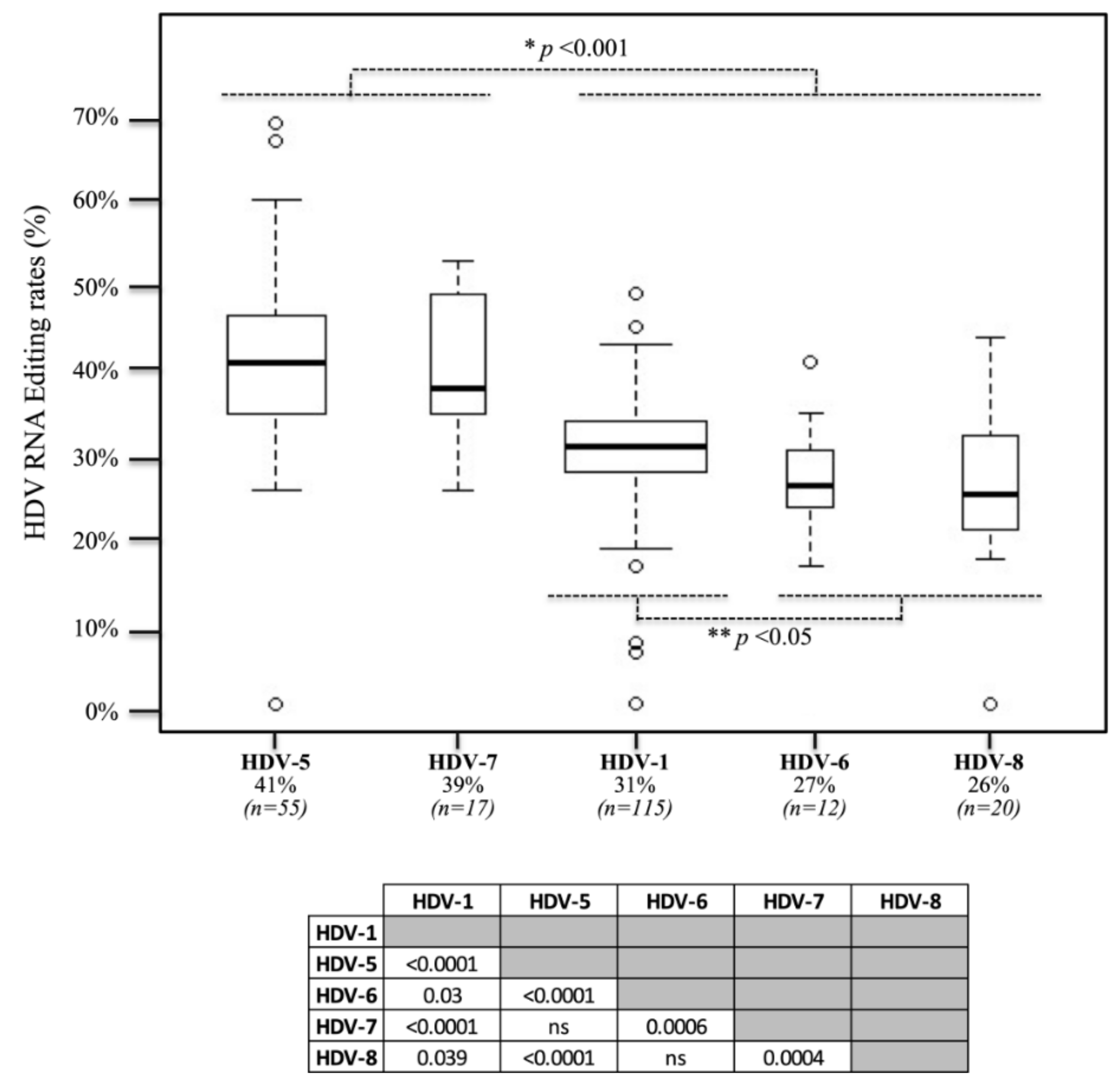
3.2. In Vivo Editing Rates in Clinical Samples
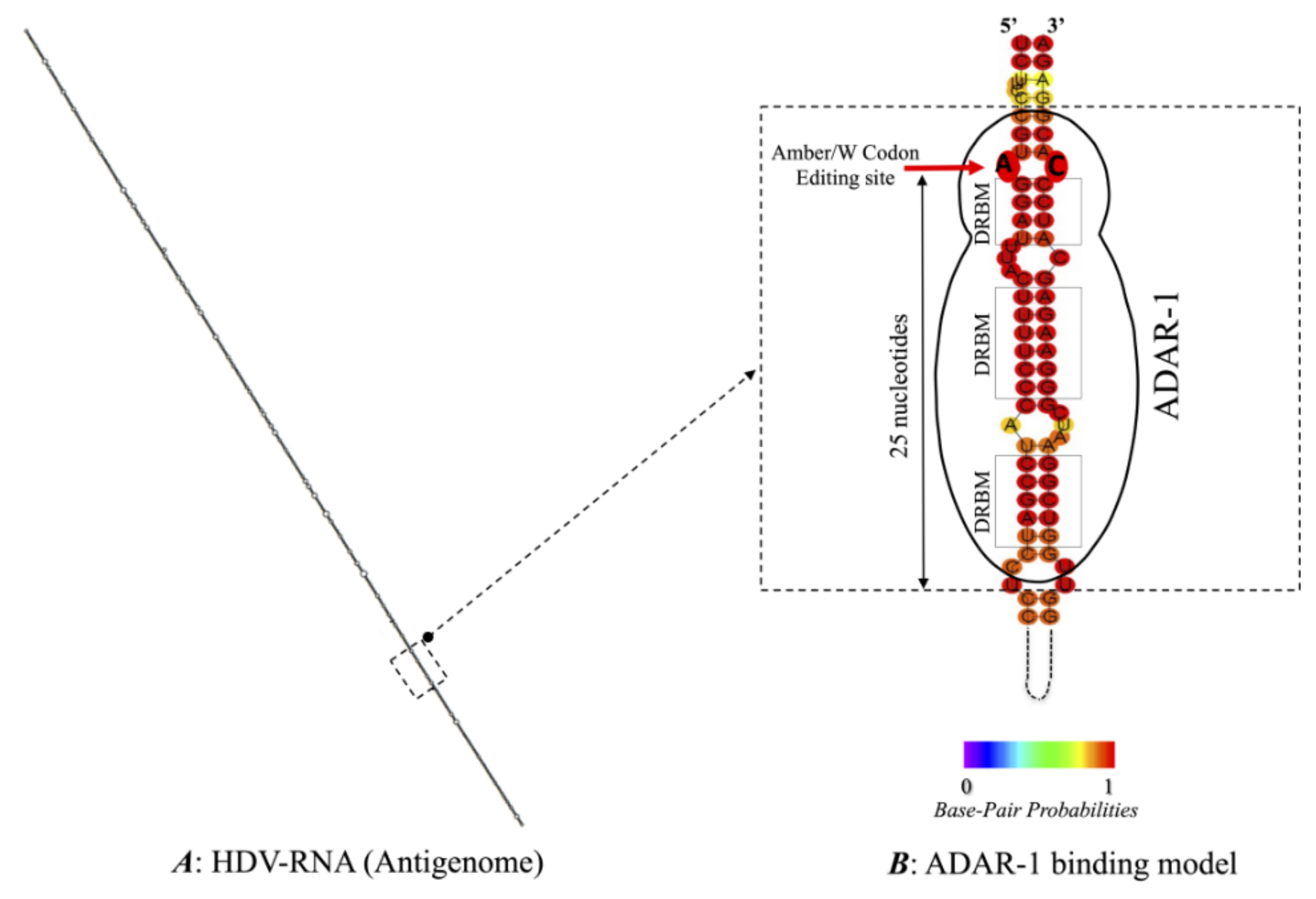
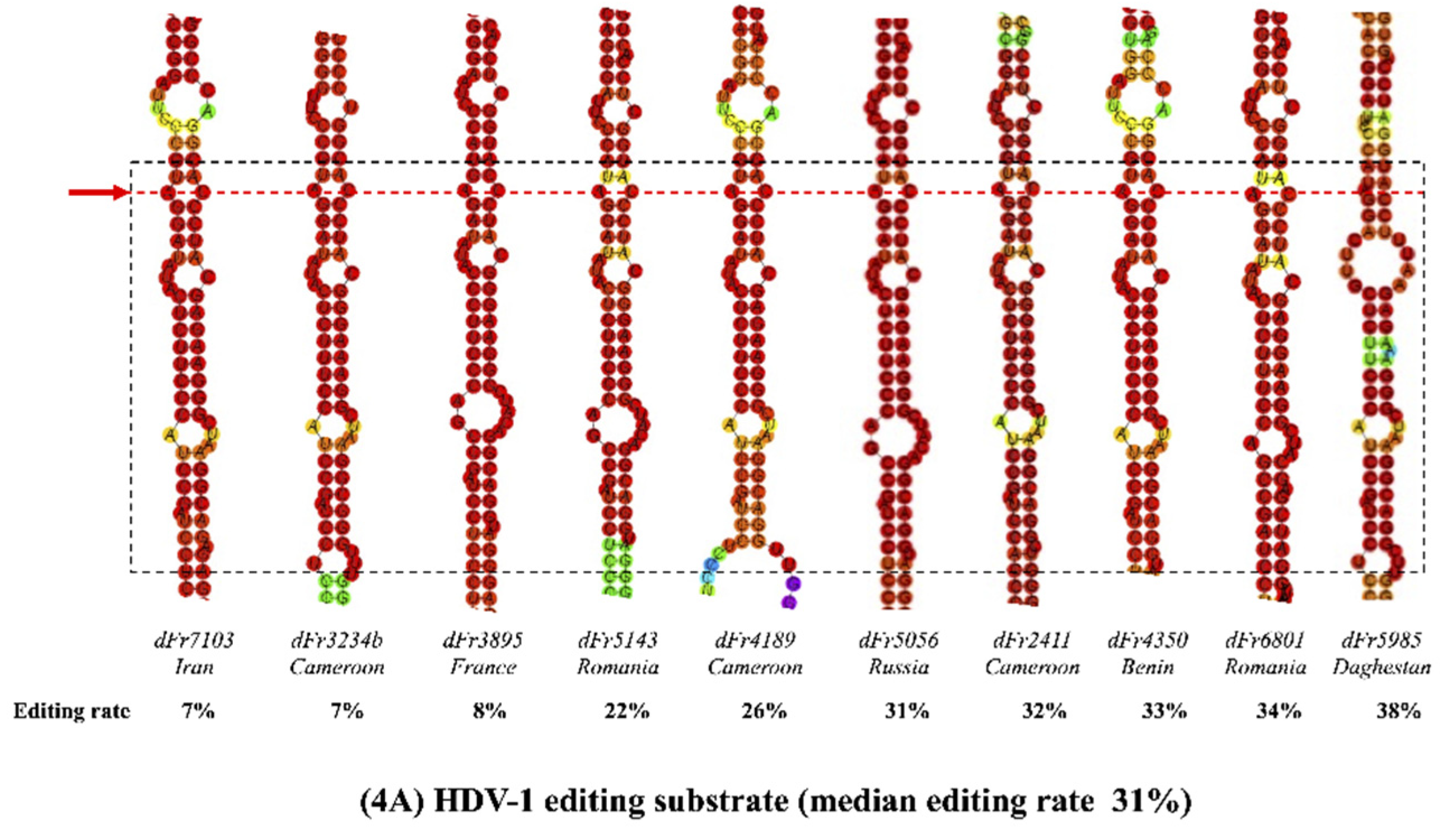
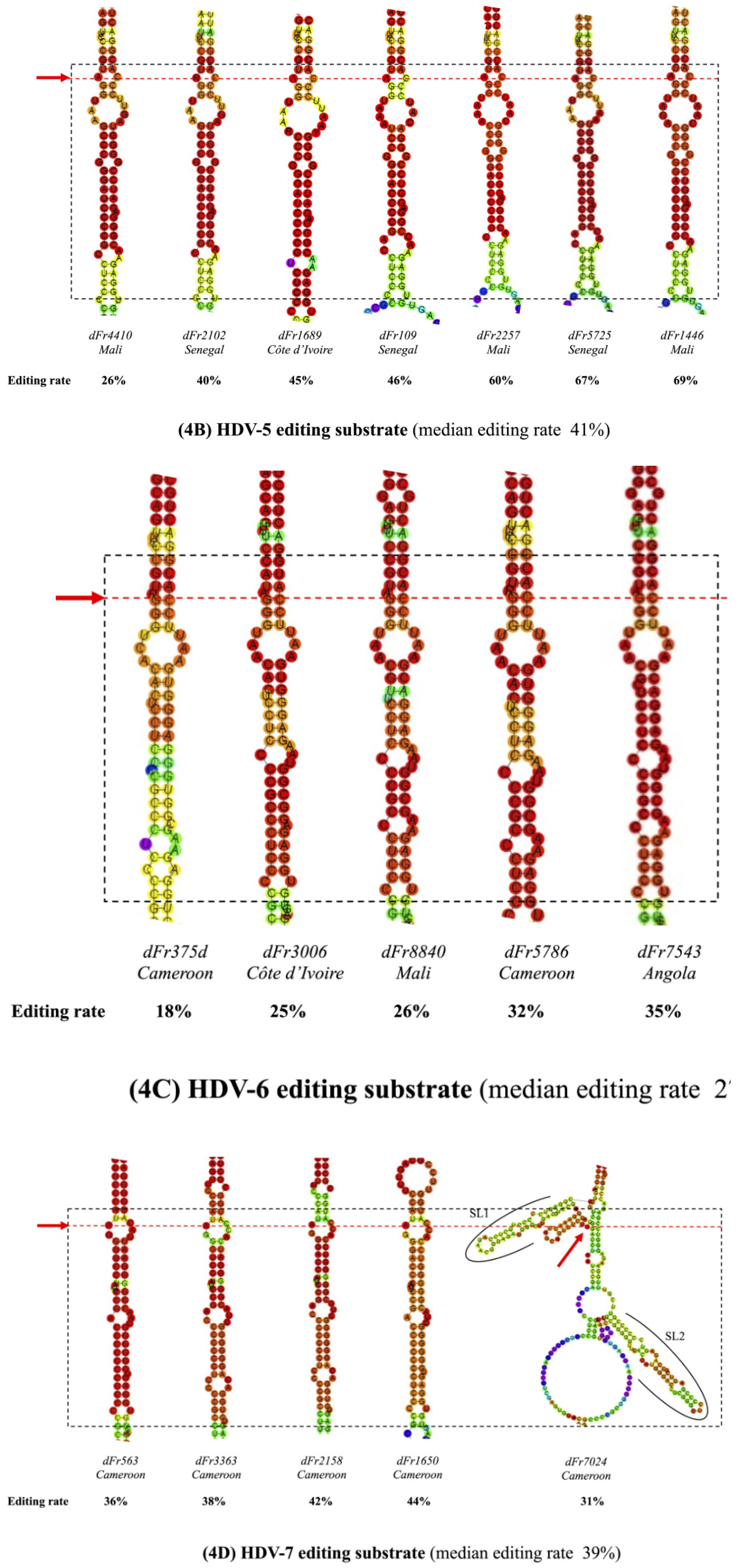
3.3. Predicted RNA Secondary Structures of the Editing Genome Region
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Primer | Sequence (5′-3′) |
|---|---|---|
| RT | Delta 920 fw | CATGCCGACCCGAAGAGGAAAG |
| Delta 1286 rv | GAAGGAAGGCCCTCGAGAACAAGA | |
| PCR | Delta 920 fw | TCGTCGGCAGCGTCAGATGTGTATAAGAGAAG |
| GCATGCCGACCCGAAGAGGAAAG | ||
| Delta N920 fw | TCGTCGGCAGCGTCAGATGTGTATAAGAGAAG | |
| NCATGCCGACCCGAAGAGGAAAG | ||
| Delta NN920 fw | TCGTCGGCAGCGTCAGATGTGTATAAGAGACA | |
| GNNCATGCCGACCCGAAGAGGAAAG | ||
| Delta NNN920 fw | TCGTCGGCAGCGTCAGATGTGTATAAGAGACA | |
| GNNNCATGCCGACCCGAAGAGGAAAG | ||
| Delta N1289 rv | GTCTCGTGGGCTCGGAGATGTGTATAAGAGAC | |
| AGNGAAGGAAGGCCCTCGAGAACAAGA |
References
- Gudima, S.; He, Y.; Meier, A.; Chang, J.; Chen, R.; Jarnik, M.; Nicolas, E.; Bruss, V.; Taylor, J. Assembly of Hepatitis Delta Virus: Particle Characterization, Including the Ability to Infect Primary Human Hepatocytes. J. Virol. 2007, 81, 3608–3617. [Google Scholar] [CrossRef] [Green Version]
- Wille, M.; Netter, H.J.; Littlejohn, M.; Yuen, L.; Shi, M.; Eden, J.-S.; Klaassen, M.; Holmes, E.C.; Hurt, A.C. A Divergent Hepatitis D-Like Agent in Birds. Viruses 2018, 10, 720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetzel, U.; Szirovicza, L.; Smura, T.; Prähauser, B.; Vapalahti, O.; Kipar, A.; Hepojoki, J. Identification of a Novel Deltavirus in Boa Con-strictors. mBio 2019. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.-S.; Pettersson, J.H.-O.; Le Lay, C.; Shi, M.; Lo, N.; Wille, M.; Eden, J.-S.; Holmes, E.C. Novel hepatitis D-like agents in vertebrates and inverte-brates. Virus Evol. 2019, 5, vez021. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, S.; Pirzer, F.; Goldmann, N.; Schmid, J.; Corman, V.M.; Gottula, L.T.; Schroeder, S.; Rasche, A.; Muth, D.; Drexler, J.F.; et al. Mammalian deltavirus without hepadnavirus coinfection in the neotropical rodentProechimys semispinosus. Proc. Natl. Acad. Sci. USA 2020, 117, 17977–17983. [Google Scholar] [CrossRef]
- Stockdale, A.J.; Kreuels, B.; Henrion, M.Y.; Giorgi, E.; Kyomuhangi, I.; de Martel, C.; Hutin, Y.; Geretti, A.M. The global prevalence of hepatitis D virus infection: Systematic review and meta-analysis. J. Hepatol. 2020, 73, 523–532. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Shen, D.-T.; Ji, D.-Z.; Han, P.-C.; Zhang, W.-M.; Ma, J.-F.; Chen, W.-S.; Goyal, H.; Pan, S.; Xu, H.-G. Prevalence and burden of hepatitis D virus infection in the global population: A systematic review and meta-analysis. Gut 2019, 68, 512–521. [Google Scholar] [CrossRef]
- Roulot, D.; Brichler, S.; Layese, R.; BenAbdesselam, Z.; Zoulim, F.; Thibault, V.; Scholtes, C.; Roche, B.; Castelnau, C.; Poynard, T.; et al. Origin, HDV genotype and persistent viremia determine outcome and treatment response in patients with chronic hepatitis delta. J. Hepatol. 2020, 73, 1046–1062. [Google Scholar] [CrossRef]
- Le Gal, F.; Brichler, S.; Drugan, T.; Alloui, C.; Roulot, D.; Pawlotsky, J.-M.; Dény, P.; Gordien, E. Genetic diversity and worldwide distribution of the deltavirus genus: A study of 2152 clinical strains. Hepatology 2017, 66, 1826–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzetto, M.; Smedile, A. Hepatitis D Virus clades and geno-types: A practical approach. In Hepatitis D: Virology, Management and Methodology; Il Pensiero Scientifico: Rome, Italy, 2019; Chapter 4. [Google Scholar]
- Radjef, N.; Gordien, E.; Ivaniushina, V.; Gault, E.; Anaïs, P.; Drugan, T.; Trinchet, J.-C.; Roulot, D.; Tamby, M.; Milinkovitch, M.C.; et al. Molecular phylogenetic analyses indicate a wide and ancient radiation of African hepatitis delta virus, suggesting a deltavirus genus of at least seven major clades. J. Virol. 2004, 78, 2537–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureau, C.; Negro, F. The hepatitis delta virus: Replication and pathogenesis. J. Hepatol. 2016, 64, S102–S116. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.J.; Kalpana, G.; Goldberg, J.; Mason, W.; Werner, B.; Gerin, J.; Taylor, J. Structure and replication of the genome of the hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1986, 83, 8774–8778. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.M. Infection by Hepatitis Delta Virus. Viruses 2020, 12, 648. [Google Scholar] [CrossRef] [PubMed]
- Greco-Stewart, V.; Pelchat, M. Interaction of Host Cellular Proteins with Components of the Hepatitis Delta Virus. Viruses 2010, 2, 189–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, R.; Grubb, D.; Elleuch, A.; Nohales, M.A.; Delgado, S.; Gago, S. Rolling-circle replication of viroids, viroid-like satellite RNAs and hepatitis delta virus: Variations on a theme. RNA Biol. 2011, 8, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macnaughton, T.B.; Shi, S.T.; Modahl, L.E.; Lai, M.M.C. Rolling Circle Replication of Hepatitis Delta Virus RNA Is Carried Out by Two Different Cellular RNA Polymerases. J. Virol. 2002, 76, 3920–3927. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.K.; Lazinski, D.W. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. USA 2002, 99, 15118–15123. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.L. RNA Editing in Hepatitis Delta Virus. Curr. Top. Microbiol. Immunol. 2006, 307, 67–89. [Google Scholar] [CrossRef]
- Cheng, Q.; Jayan, G.C.; Casey, J.L. Differential Inhibition of RNA Editing in Hepatitis Delta Virus Genotype III by the Short and Long Forms of Hepatitis Delta Antigen. J. Virol. 2003, 77, 7786–7795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, J.L. Control of ADAR1 Editing of Hepatitis Delta Virus RNAs. Curr. Top. Microbiol. Immunol. 2011, 353, 123–143. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-W.; Juang, H.-H.; Kuo, C.-Y.; Li, H.-P.; Iang, S.-B.; Lin, S.-H.; Yeh, C.-T.; Chao, M. Structural Pattern Differences in Unbranched Rod-like RNA of Hepatitis Delta Virus affect RNA Editing. Viruses 2019, 11, 934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeywickrama-Samarakoon, N.; Cortay, J.-C.; Sureau, C.; Müller, S.; Alfaiate, D.; Guerrieri, F.; Chaikuad, A.; Schröder, M.; Merle, P.; Levrero, M.; et al. Hepatitis Delta Virus histone mimicry drives the recruitment of chromatin remodelers for viral RNA replication. Nat. Commun. 2020, 11, 419. [Google Scholar] [CrossRef]
- Wei, Y.; Ganem, D. Activation of heterologous gene expression by the large isoform of hepatitis delta antigen. J. Virol. 1998, 72, 2089–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-H.; Chang, S.C.; Wu, C.H.H.; Chang, M.-F. A Novel Chromosome Region Maintenance 1-independent Nuclear Export Signal of the Large Form of Hepatitis Delta Antigen That Is Required for the Viral Assembly. J. Biol. Chem. 2001, 276, 8142–8148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.-H.; Jeong, S.-H.; Hwang, S.B. Large hepatitis delta antigen modulates transforming growth factor-beta signaling cas-cades: Implication of hepatitis delta virus-induced liver fibrosis. Gastroenterology 2007, 132, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-Y.; Oh, S.-H.; Kang, S.M.; Lim, Y.-S.; Hwang, S.B. Hepatitis delta virus large antigen sensitizes to TNF-alpha-induced NF-kappaB signaling. Mol. Cells 2009, 28, 49–55. [Google Scholar] [CrossRef]
- Williams, V.; Brichler, S.; Khan, E.; Chami, M.; Dény, P.; Kremsdorf, D.; Gordien, E. Large hepatitis delta antigen activates STAT-3 and NF-κB via oxidative stress. J. Viral Hepat. 2012, 19, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Alfaiate, D.; Lucifora, J.; Abeywickrama-Samarakoon, N.; Michelet, M.; Testoni, B.; Cortay, J.-C.; Sureau, C.; Zoulim, F.; Dény, P.; Durantel, D. HDV RNA replication is asso-ciated with HBV repression and interferon-stimulated genes induction in super-infected hepatocytes. Antivir. Res. 2016, 136, 19–31. [Google Scholar] [CrossRef]
- D’Souza, S.; Lau, K.C.; Coffin, C.S.; Patel, T.R. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J. Gastroenterol. 2020, 26, 5759–5783. [Google Scholar] [CrossRef]
- Wang, K.S.; Choo, Q.L.; Weiner, A.J.; Ou, J.H.; Najarian, R.C.; Thayer, R.M.; Mullenbach, G.T.; Denniston, K.J.; Gerin, J.L.; Houghton, M. Structure, sequence and expression of the hepatitis delta (delta) viral genome. Nature 1986, 323, 508–514. [Google Scholar] [CrossRef]
- Dény, P.; Zignego, A.L.; Rascalou, N.; Ponzetto, A.; Tiollais, P.; Bréchot, C. Nucleotide sequence analysis of three different hepatitis delta viruses isolated from a woodchuck and humans. J. Gen. Virol. 1991, 72, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Karayiannis, P.; Thomas, H.; Monjardino, J. Editing efficiency of hepatitis delta virus RNA is related to the course of infection in woodchucks. J. Gen. Virol. 1995, 76, 3071–3078. [Google Scholar] [CrossRef]
- Homs, M.; Rodriguez-Frias, F.; Gregori, J.; Ruiz, A.; Reimundo, P.; Casillas, R.; Tabernero, D.; Godoy, C.; Barakat, S.; Quer, J.; et al. Evidence of an Exponential Decay Pattern of the Hepatitis Delta Virus Evolution Rate and Fluctuations in Quasispecies Complexity in Long-Term Studies of Chronic Delta Infection. PLoS ONE 2016, 11, e0158557. [Google Scholar] [CrossRef] [PubMed]
- Sopena, S.; Godoy, C.; Tabernero, D.; Homs, M.; Gregori, J.; Riveiro-Barciela, M.; Ruiz, A.; Esteban, R.; Buti, M.; Rodríguez-Frías, F. Quantitative characterization of hepatitis delta virus genome edition by next-generation sequencing. Virus Res. 2018, 243, 52–59. [Google Scholar] [CrossRef]
- Le Gal, F.; Dziri, S.; Gerber, A.; Alloui, C.; Ben Abdesselam, Z.; Roulot, D.; Brichler, S.; Gordien, E. Performance Characteristics of a New Consensus Commercial Kit for Hepatitis D Virus RNA Viral Load Quantification. J. Clin. Microbiol. 2017, 55, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivaniushina, V.; Radjef, N.; Alexeeva, M.; Gault, E.; Semenov, S.; Salhi, M.; Kiselev, O.; Dény, P. Hepatitis delta virus genotypes I and II cocirculate in an endemic area of Yakutia, Russia. J. Gen. Virol. 2001, 82, 2709–2718. [Google Scholar] [CrossRef]
- Sato, S.; Wong, S.K.; Lazinski, D.W. Hepatitis Delta Virus Minimal Substrates Competent for Editing by ADAR1 and ADAR2. J. Virol. 2001, 75, 8547–8555. [Google Scholar] [CrossRef] [Green Version]
- Linnstaedt, S.D.; Kasprzak, W.K.; Shapiro, B.A.; Casey, J.L. The role of a metastable RNA secondary structure in hepatitis delta virus genotype III RNA editing. RNA 2006, 12, 1521–1533. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Filipovska, J.; Yano, K.; Furuya, A.; Inukai, N.; Narita, T.; Wada, T.; Sugimoto, S.; Konarska, M.M.; Handa, H. Stimulation of RNA Polymerase II Elongation by Hepatitis Delta Antigen. Science 2001, 293, 124–127. [Google Scholar] [CrossRef]
- Chao, M.; Hsieh, S.Y.; Taylor, J. Role of two forms of hepatitis delta virus antigen: Evidence for a mechanism of self-limiting ge-nome replication. J. Virol. 1990, 64, 5066–5069. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.P.; Lai, M.M. Oligomerization of hepatitis delta antigen is required for both the trans-activating and trans-dominant in-hibitory activities of the delta antigen. J. Virol. 1992, 66, 6641–6648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, K.-N.; Chuang, Y.-T.; Liu, H.; Lo, S.J. Hepatitis D virus RNA editing is inhibited by a GFP fusion protein containing a C-terminally deleted delta antigen. J. Gen. Virol. 2004, 85, 947–957. [Google Scholar] [CrossRef]
- Niro, G.A.; Smedile, A. Current Concept in the Pathophysiology of Hepatitis Delta Infection. Curr. Infect. Dis. Rep. 2011, 14, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, S.; Cassidy, W.M.; Valinluck, B.; Redeker, A.G. Interactions of HDV, HBV and HIV in chronic B and D infections and in reactivation of chronic D infection. Prog. Clin. Boil. Res. 1991, 364, 207–210. [Google Scholar]
- Cole, S.M.; Macnaughton, T.B.; Gowans, E.J. Differential roles for HDAg-p24 and -p27 in HDV pathogenesis. Prog. Clin. Boil. Res. 1993, 382, 131–138. [Google Scholar]
- Govindarajan, S. Inhibition of HBV replication during coinfection with HBV and HDV: Inhibition of the inhibition by coinfection with HIV. Hepatology 1990, 11, 703–704. [Google Scholar] [CrossRef] [PubMed]
- Baltes, A.; Akpinar, F.; Inankur, B.; Yin, J. Inhibition of infection spread by co-transmitted defective interfering particles. PLoS ONE 2017, 12, e0184029. [Google Scholar] [CrossRef] [PubMed] [Green Version]






| Parameters | p Value |
|---|---|
| % Editing/HDV genotype | 0.0038 |
| % Editing/HBV viral load | 0.01 |
| % Editing/HDV viral load | ns |
| Strain | HDV Genotype | Editing (%) | A-C Mismatch * | DRMB † | Length # |
|---|---|---|---|---|---|
| dFr7103 | 1 | 7 | Yes | 3 | 4-8-12 |
| dFr3234b | 1 | 7 | Yes | 4 | 4-8-7-4 |
| dFr3895 | 1 | 8 | Yes | 3 | 3-8-21 |
| dFr5143 | 1 | 22 | Yes | 3 | 4-8-12 |
| dFr4189 | 1 | 26 | Yes | 4 | 4-8-7-9 |
| dFr5056 | 1 | 31 | Yes | 3 | 4-8-21 |
| dFr2411 | 1 | 32 | Yes | 3 | 4-8-11 |
| dFr4350 | 1 | 33 | Yes | 3 | 4-8-21 |
| dFr6801 | 1 | 34 | Yes | 3 | 4-8-22 |
| dFr5985 | 1 | 38 | No | 4 | 3-8-7-4 |
| dFr4410 | 5 | 26 | Yes | 4 | 2-4-9-4 |
| dFr2102 | 5 | 40 | Yes | 5 | 2-4-9-4-2 |
| dFr1689 | 5 | 45 | Yes | 4 | 2-3-9-4 |
| dFr109 | 5 | 46 | Yes | 4 | 2-3-8-4 |
| dFr2257 | 5 | 60 | Yes | 4 | 2-3-9-4 |
| dFr5725 | 5 | 67 | Yes | 5 | 2-4-8-4-1 |
| dFr1446e | 5 | 69 | Yes | 5 | 2-3-9-4-1 |
| dFr375d | 6 | 18 | No | 4 | 2-13-1-4 |
| dFr3006 | 6 | 25 | No | 4 | 2-7-9-15 |
| dFr8840 | 6 | 26 | No | 5 | 2-7-4-4-15 |
| dFr5786 | 6 | 32 | No | 5 | 2-7-4-4-16 |
| dFr7543 | 6 | 35 | No | 5 | 2-7-4-4-15 |
| dFr563 | 7 | 36 | Yes | 3 | 9-10-16 |
| dFr3363 | 7 | 38 | Yes | 3 | 9-7-8 |
| dFr2158 | 7 | 42 | Yes | 3 | 9-7-8 |
| dFr1650 | 7 | 44 | Yes | 4 | 9-10-2-4 |
| dFr2072 | 8 | 23 | No | 5 | 2-2-9-4-11 |
| dFr1011e | 8 | 25 | Yes | 3 | 15-4-9 |
| dFr6493 | 8 | 28 | Yes | 3 | 12-4-5 |
| dFr1274 | 8 | 31 | Yes | 4 | 4-7-4-8 |
| dFr367e | 8 | 44 | No | 5 | 2-2-8-4-15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziri, S.; Rodriguez, C.; Gerber, A.; Brichler, S.; Alloui, C.; Roulot, D.; Dény, P.; Pawlotsky, J.M.; Gordien, E.; Le Gal, F. Variable In Vivo Hepatitis D Virus (HDV) RNA Editing Rates According to the HDV Genotype. Viruses 2021, 13, 1572. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081572
Dziri S, Rodriguez C, Gerber A, Brichler S, Alloui C, Roulot D, Dény P, Pawlotsky JM, Gordien E, Le Gal F. Variable In Vivo Hepatitis D Virus (HDV) RNA Editing Rates According to the HDV Genotype. Viruses. 2021; 13(8):1572. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081572
Chicago/Turabian StyleDziri, Samira, Christophe Rodriguez, Athenaïs Gerber, Ségolène Brichler, Chakib Alloui, Dominique Roulot, Paul Dény, Jean Michel Pawlotsky, Emmanuel Gordien, and Frédéric Le Gal. 2021. "Variable In Vivo Hepatitis D Virus (HDV) RNA Editing Rates According to the HDV Genotype" Viruses 13, no. 8: 1572. https://0-doi-org.brum.beds.ac.uk/10.3390/v13081572