
Antibody Generation and Immunogenicity Analysis of EBV gp42 N-Terminal Region

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction, Expression and Purification
2.2. Immunization
2.3. Antibody Screen and Preparation
2.4. Enzyme Linked Immunosorbent Assay (ELISA)
2.5. Surface Plasmon Resonance (SPR) Assays
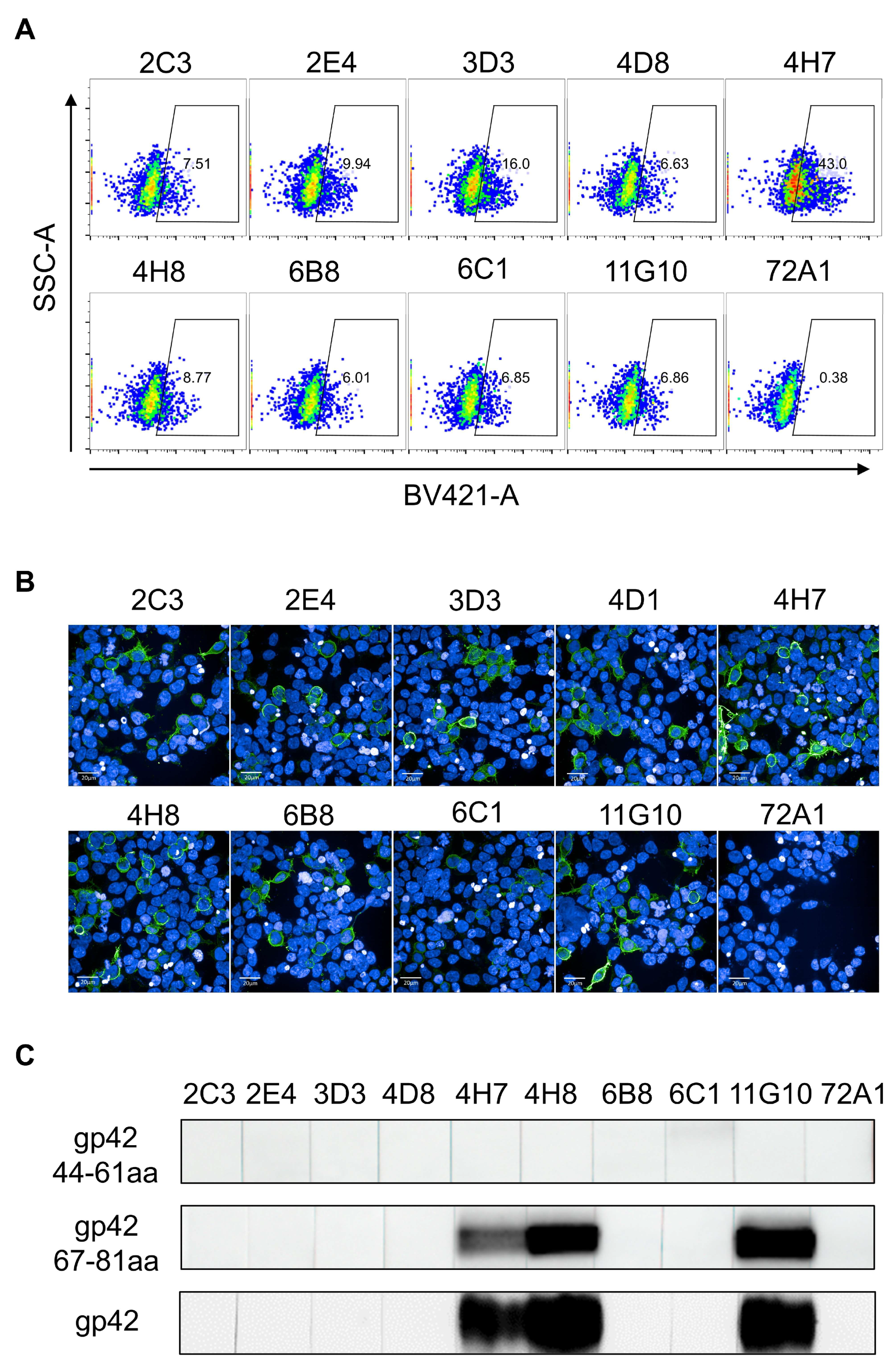
2.6. Surface Staining of gp42 by Flow Cytometry
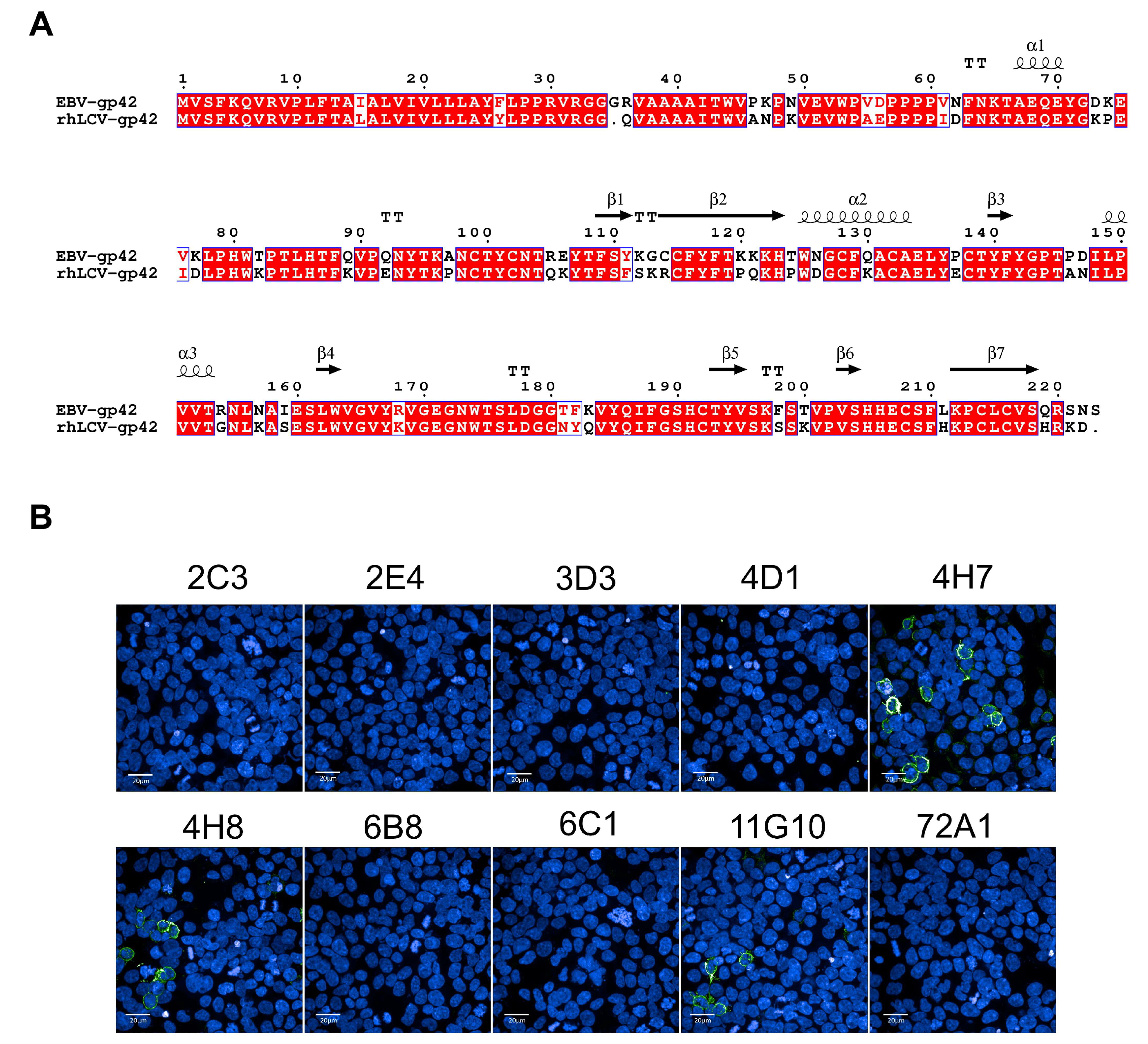
2.7. Surface Staining of gp42 by Immunofluorescence
2.8. SDS-PAGE and Western Blotting
2.9. Transmission Electron Microscopy (TEM)
2.10. High Performance Size Exclusion Chromatography (HPSEC)
2.11. Homology Comparison
2.12. Virus Production
2.13. Neutralization Assay on B Cells
2.14. Statistical Analysis
3. Results
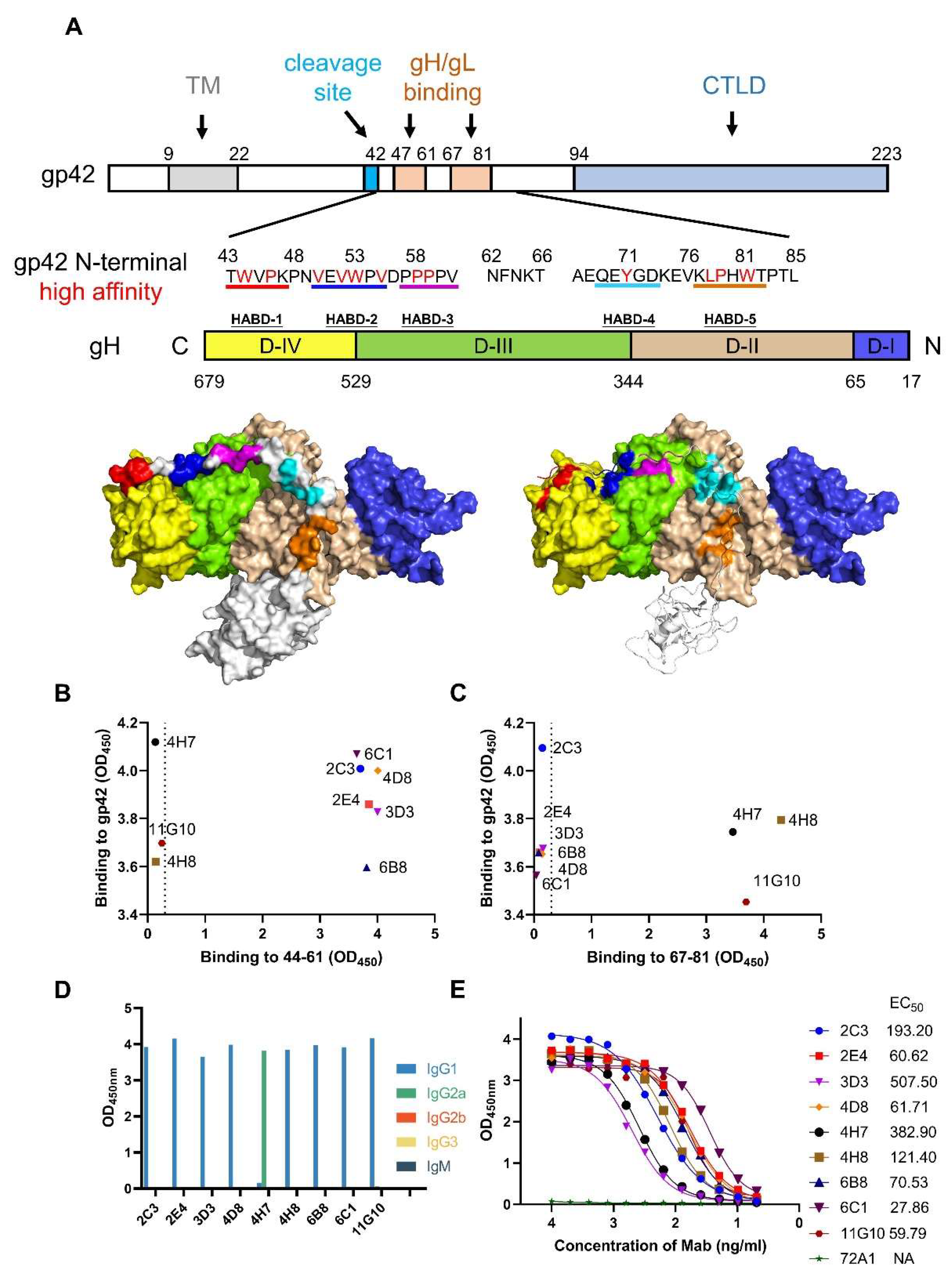
3.1. Generation and Characterization of gp42-44-61aa and gp42-67-81aa Specific mAbs
3.2. Cross-Reactivity Analysis of mAbs Targeting gp42 N-Terminal Region
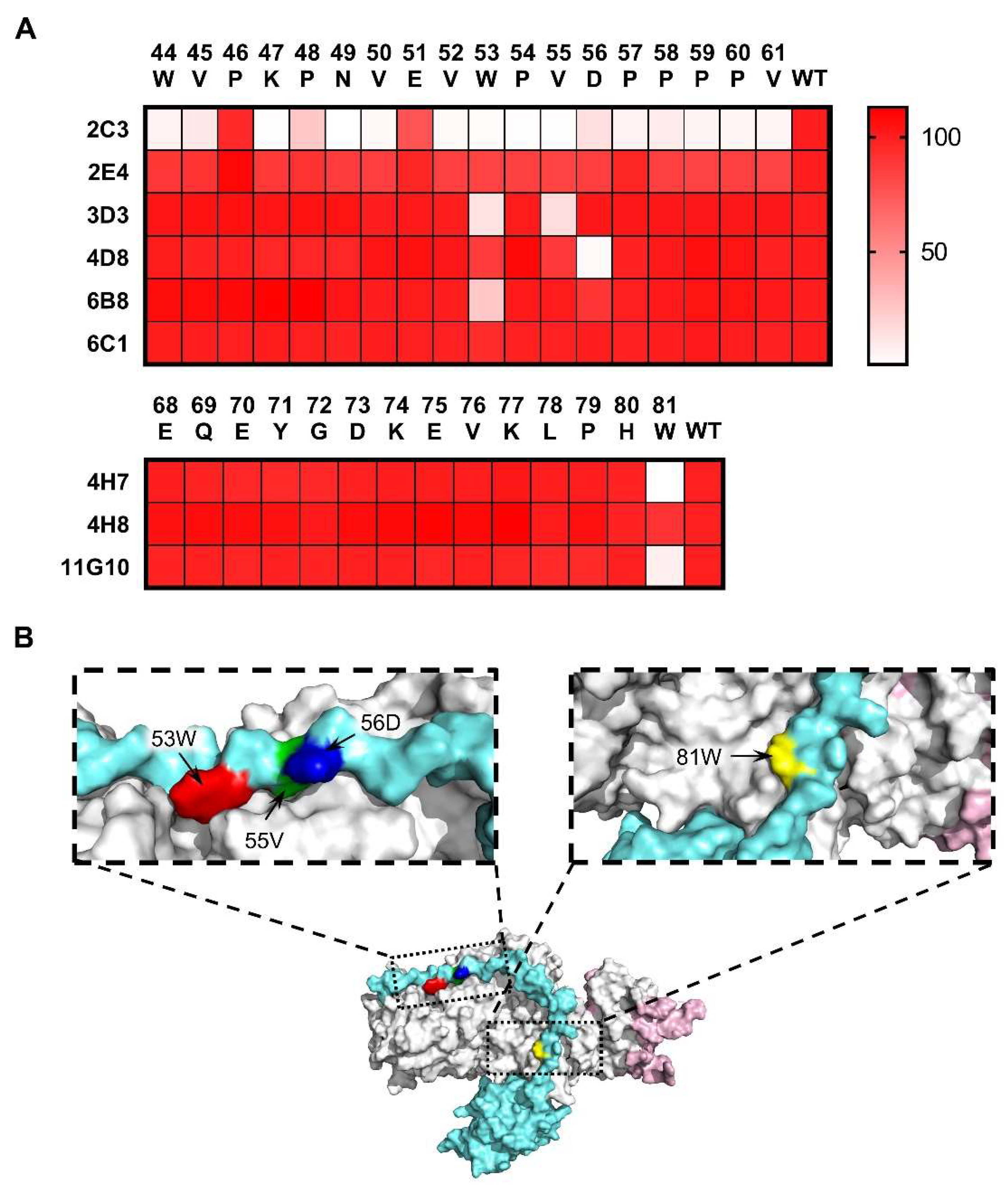
3.3. Epitope Analysis of mAbs by Indirect ELISA
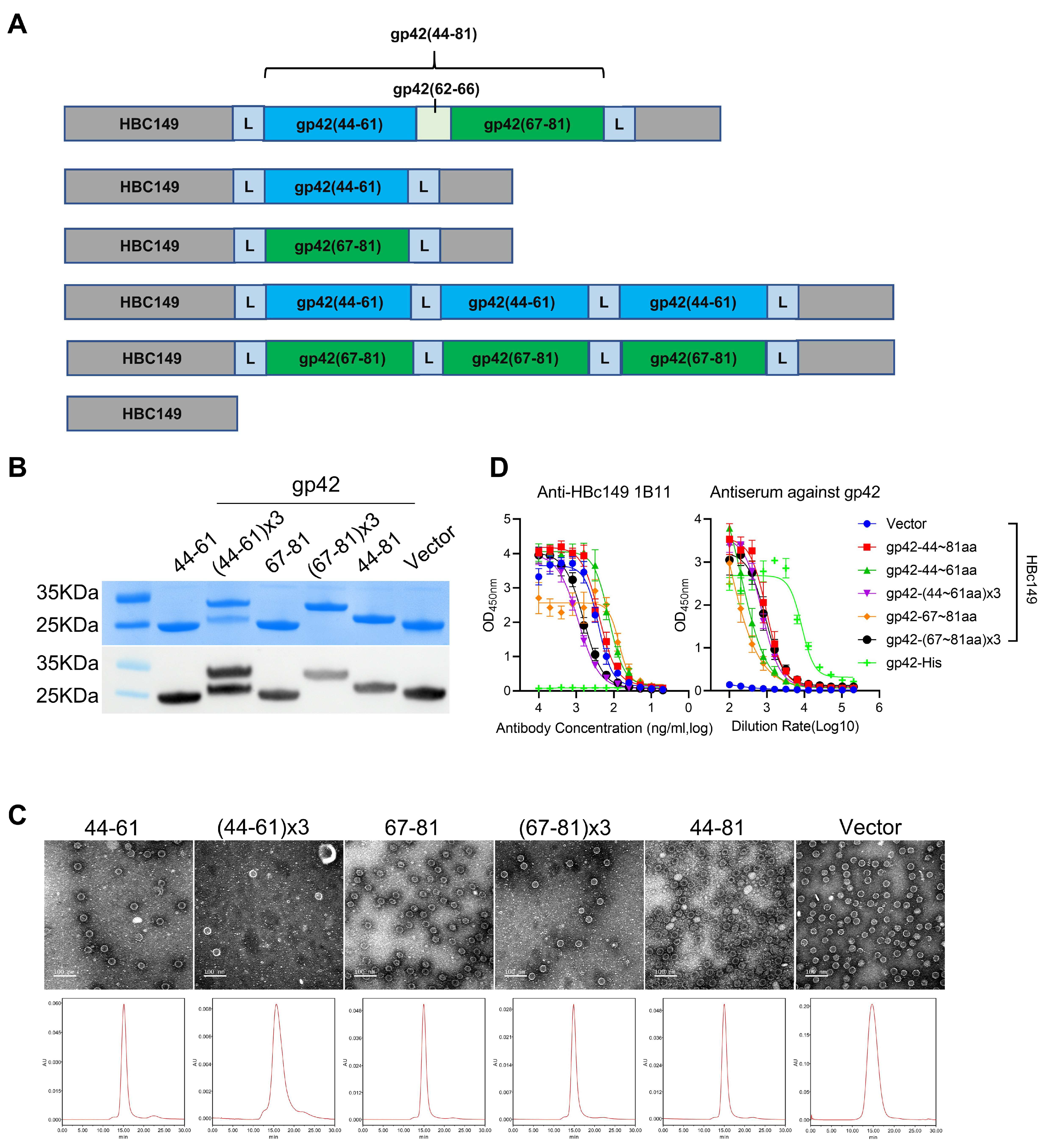
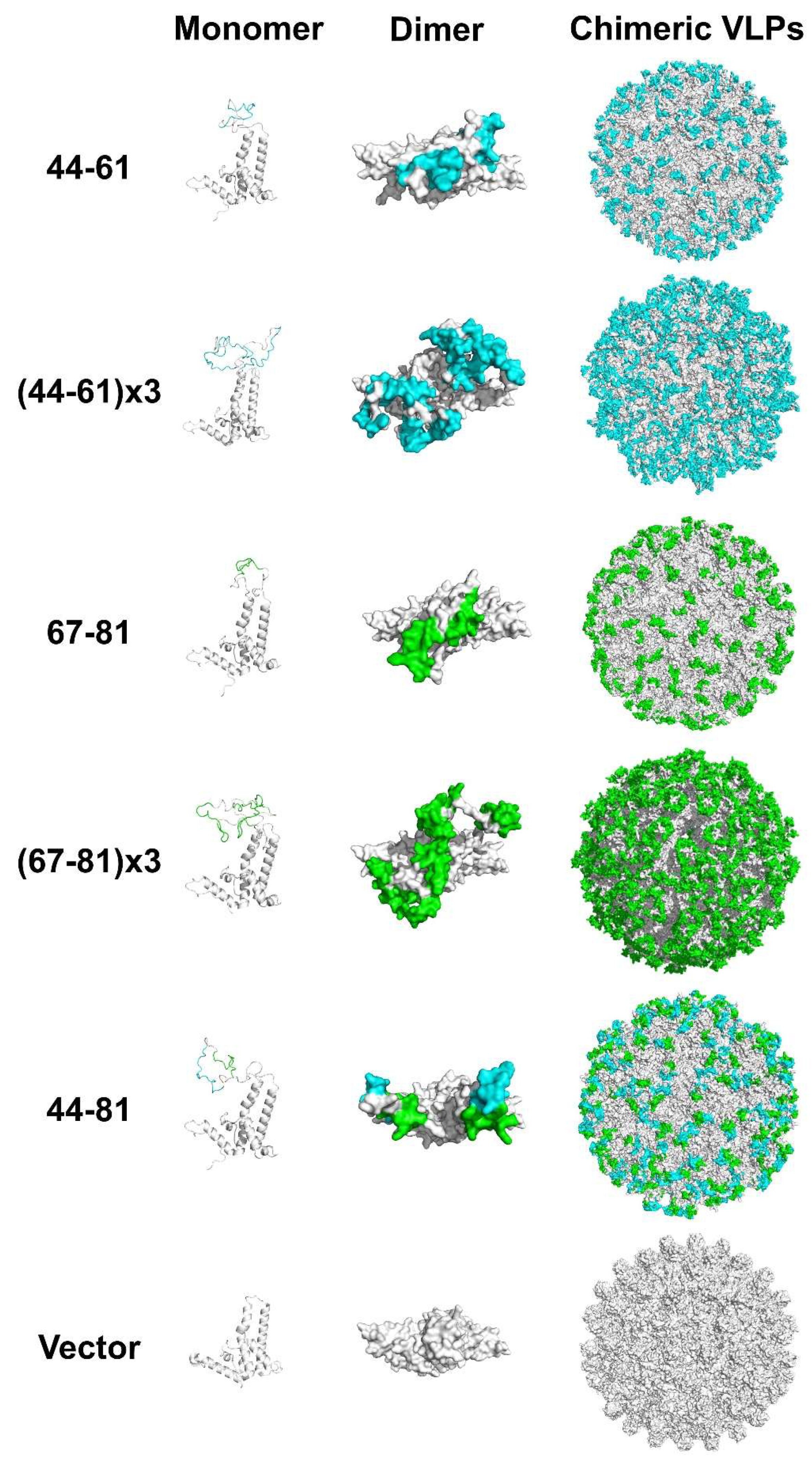
3.4. Construction and Characterization of Epitope-Displaying Chimeric VLPs
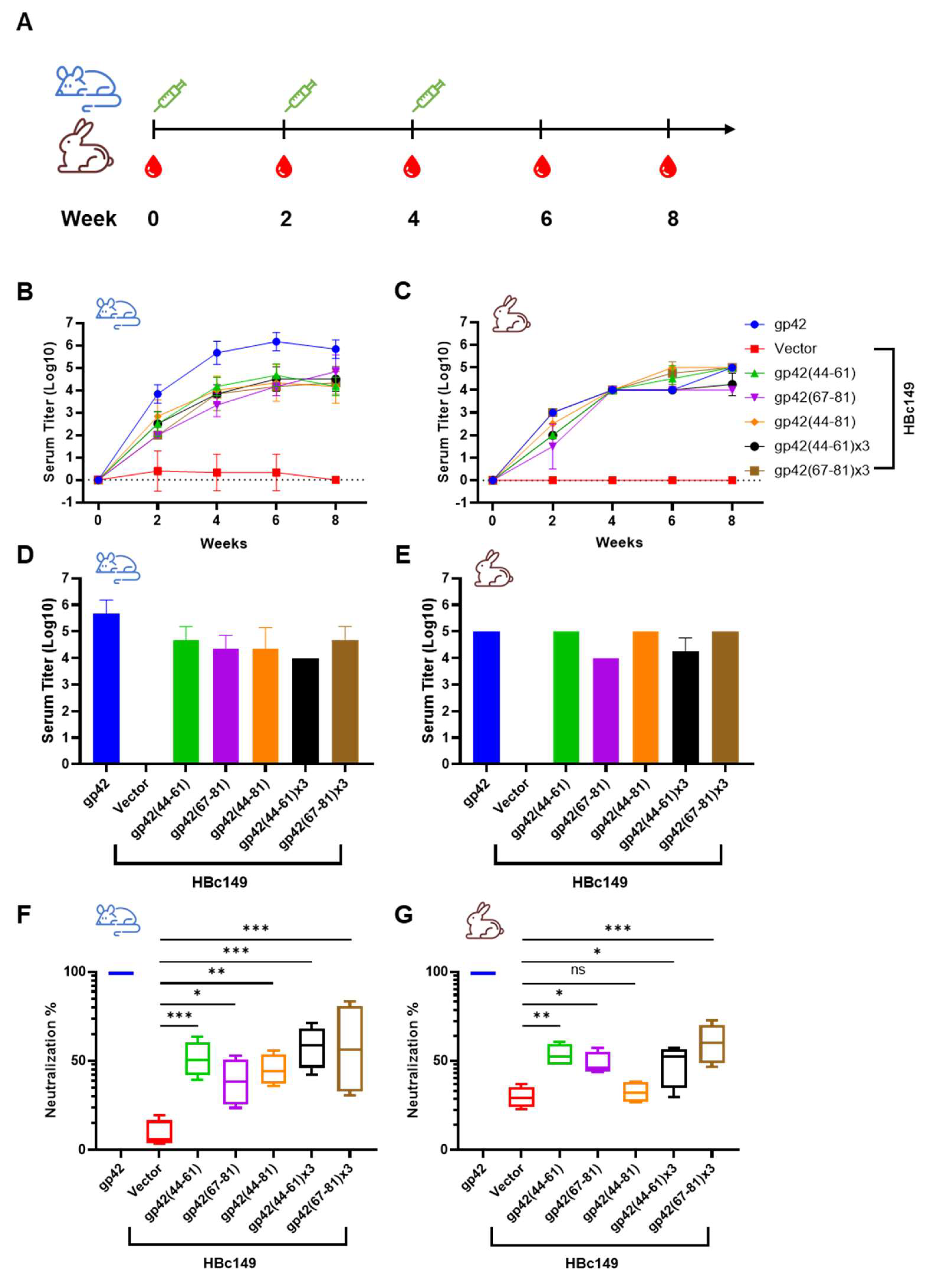
3.5. Immunogenicity of the Chimeric VLPs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Mui, U.N.; Haley, C.T.; Vangipuram, R.; Tyring, S.K. Human oncoviruses: Mucocutaneous manifestations, pathogenesis, therapeutics, and prevention: Hepatitis viruses, human T-cell leukemia viruses, herpesviruses, and Epstein-Barr virus. J. Am. Acad. Dermatol. 2019, 81, 23–41. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479-480, 498–507. [Google Scholar] [CrossRef] [Green Version]
- Hutt-Fletcher, L.M. EBV glycoproteins: Where are we now? Future Virol. 2015, 10, 1155–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschner, A.N.; Sorem, J.; Longnecker, R.; Jardetzky, T.S. Structure of Epstein-Barr virus glycoprotein 42 suggests a mechanism for triggering receptor-activated virus entry. Structure 2009, 17, 223–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Turk, S.M.; Hutt-Fletcher, L.M. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J. Virol 1995, 69, 3987–3994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Hutt-Fletcher, L.M. Epstein-Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J. Virol 1998, 72, 158–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möhl, B.S.; Chen, J.; Sathiyamoorthy, K.; Jardetzky, T.S.; Longnecker, R. Structural and Mechanistic Insights into the Tropism of Epstein-Barr Virus. Mol. Cells 2016, 39, 286–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiels, B.; Stevenson, P.G.; Vanderplasschen, A.; Gillet, L. A gammaherpesvirus uses alternative splicing to regulate its tropism and its sensitivity to neutralization. PLoS Pathog. 2013, 9, e1003753. [Google Scholar] [CrossRef] [PubMed]
- Borza, C.M.; Hutt-Fletcher, L.M. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 2002, 8, 594–599. [Google Scholar] [CrossRef]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.; Keating, S.; Gomez, R.; Franken, K.L.; Ottenhoff, T.H.; Spriggs, M.; Schumacher, T.N.; Hutt-Fletcher, L.M.; et al. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J. Virol. 2005, 79, 841–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathiyamoorthy, K.; Chen, J.; Longnecker, R.; Jardetzky, T.S. The COMPLEXity in herpesvirus entry. Curr. Opin. Virol. 2017, 24, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Dargan, D.J.; Stow, N.D. Fundamental and accessory systems in herpesviruses. Antivir. Res. 2002, 56, 1–11. [Google Scholar] [CrossRef]
- Ressing, M.E.; van Gent, M.; Gram, A.M.; Hooykaas, M.J.; Piersma, S.J.; Wiertz, E.J. Immune Evasion by Epstein-Barr Virus. Curr. Top Microbiol. Immunol. 2015, 391, 355–381. [Google Scholar] [CrossRef] [PubMed]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.; Gomez, R.; Heemskerk, B.; Toebes, M.; Mullen, M.M.; Jardetzky, T.S.; Longnecker, R.; Schilham, M.W.; et al. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc. Natl. Acad. Sci. USA 2003, 100, 11583–11588. [Google Scholar] [CrossRef] [Green Version]
- Rowe, C.L.; Matsuura, H.; Jardetzky, T.S.; Longnecker, R. Investigation of the function of the putative self-association site of Epstein-Barr virus (EBV) glycoprotein 42 (gp42). Virology 2011, 415, 122–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschner, A.N.; Omerovic, J.; Popov, B.; Longnecker, R.; Jardetzky, T.S. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J. Virol. 2006, 80, 9444–9454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschner, A.N.; Lowrey, A.S.; Longnecker, R.; Jardetzky, T.S. Binding-site interactions between Epstein-Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B-cell membrane fusion. J. Virol. 2007, 81, 9216–9229. [Google Scholar] [CrossRef] [Green Version]
- Sathiyamoorthy, K.; Jiang, J.; Hu, Y.X.; Rowe, C.L.; Möhl, B.S.; Chen, J.; Jiang, W.; Mellins, E.D.; Longnecker, R.; Zhou, Z.H.; et al. Assembly and architecture of the EBV B cell entry triggering complex. PLoS Pathog. 2014, 10, e1004309. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Marquardt, G.; Kirschner, A.N.; Longnecker, R.; Jardetzky, T.S. Mapping the N-terminal residues of Epstein-Barr virus gp42 that bind gH/gL by using fluorescence polarization and cell-based fusion assays. J. Virol. 2010, 84, 10375–10385. [Google Scholar] [CrossRef] [Green Version]
- Snijder, J.; Ortego, M.S.; Weidle, C.; Stuart, A.B.; Gray, M.D.; McElrath, M.J.; Pancera, M.; Veesler, D.; McGuire, A.T. An Antibody Targeting the Fusion Machinery Neutralizes Dual-Tropic Infection and Defines a Site of Vulnerability on Epstein-Barr Virus. Immunity 2018, 48, 799–811.e799. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhao, B.; Ding, M.; Song, S.; Kang, Y.; Yu, Y.; Xu, M.; Xiang, T.; Gao, L.; Feng, Q.; et al. A novel vaccine candidate based on chimeric virus-like particle displaying multiple conserved epitope peptides induced neutralizing antibodies against EBV infection. Theranostics 2020, 10, 5704–5718. [Google Scholar] [CrossRef]
- Wang, F. Nonhuman primate models for Epstein-Barr virus infection. Curr. Opin. Virol. 2013, 3, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, E. Prospects for genetically modified non-human primate models, including the common marmoset. Neurosci. Res. 2015, 93, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Gujer, C.; Chatterjee, B.; Landtwing, V.; Raykova, A.; McHugh, D.; Münz, C. Animal models of Epstein Barr virus infection. Curr. Opin. Virol. 2015, 13, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Orlova, N.; Quink, C.; Wang, F. Cloning of the Epstein-Barr virus-related rhesus lymphocryptovirus as a bacterial artificial chromosome: A loss-of-function mutation of the rhBARF1 immune evasion gene. J. Virol. 2011, 85, 1330–1339. [Google Scholar] [CrossRef] [Green Version]
- Herrman, M.; Mühe, J.; Quink, C.; Wang, F. Epstein-Barr Virus gp350 Can Functionally Replace the Rhesus Lymphocryptovirus Major Membrane Glycoprotein and Does Not Restrict Infection of Rhesus Macaques. J. Virol. 2016, 90, 1222–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlova, N.; Fogg, M.H.; Carville, A.; Wang, F. Antibodies to lytic infection proteins in lymphocryptovirus-infected rhesus macaques: A model for humoral immune responses to epstein-barr virus infection. Clin. Vaccine Immunol. 2011, 18, 1427–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühe, J.; Wang, F. Non-human Primate Lymphocryptoviruses: Past, Present, and Future. Curr. Top Microbiol. Immunol. 2015, 391, 385–405. [Google Scholar] [CrossRef]
- Rivailler, P.; Jiang, H.; Cho, Y.G.; Quink, C.; Wang, F. Complete nucleotide sequence of the rhesus lymphocryptovirus: Genetic validation for an Epstein-Barr virus animal model. J. Virol. 2002, 76, 421–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carville, A.; Mansfield, K.G. Comparative pathobiology of macaque lymphocryptoviruses. Comp. Med. 2008, 58, 57–67. [Google Scholar] [PubMed]
- Takeda, S.; Shiosaki, K.; Kaneda, Y.; Nakasatomi, T.; Yoshizaki, H.; Someya, K.; Konno, Y.; Eda, Y.; Kino, Y.; Yamamoto, N.; et al. Hemagglutinating virus of Japan protein is efficient for induction of CD4+ T-cell response by a hepatitis B core particle-based HIV vaccine. Clin. Immunol. 2004, 112, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Huo, C.; Yang, J.; Lei, L.; Qiao, L.; Xin, J.; Pan, Z. Hepatitis B virus core particles containing multiple epitopes confer protection against enterovirus 71 and coxsackievirus A16 infection in mice. Vaccine 2017, 35, 7322–7330. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.L.; Kirschner, A.N.; Jardetzky, T.S.; Longnecker, R. Characteristics of Epstein-Barr virus envelope protein gp42. Virus Genes 2010, 40, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Mullen, M.M.; Haan, K.M.; Longnecker, R.; Jardetzky, T.S. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 2002, 9, 375–385. [Google Scholar] [CrossRef]
- Rowe, C.L.; Chen, J.; Jardetzky, T.S.; Longnecker, R. Membrane anchoring of Epstein-Barr virus gp42 inhibits fusion with B cells even with increased flexibility allowed by engineered spacers. mBio 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Homad, L.J.; Akins, N.R.; Stoffers, C.M.; Lackhar, S.; Malhi, H.; Wan, Y.H.; Rawlings, D.J.; McGuire, A.T. Neutralizing Antibodies Protect against Oral Transmission of Lymphocryptovirus. Cell Rep. Med. 2020, 1, 100033. [Google Scholar] [CrossRef]
- Moghaddam, A.; Rosenzweig, M.; Lee-Parritz, D.; Annis, B.; Johnson, R.P.; Wang, F. An animal model for acute and persistent Epstein-Barr virus infection. Science 1997, 276, 2030–2033. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Hutt-Fletcher, L.M. Compatibility of the gH homologues of Epstein-Barr virus and related lymphocryptoviruses. J. Gen. Virol. 2007, 88, 2129–2136. [Google Scholar] [CrossRef]
- Strnad, B.C.; Schuster, T.; Klein, R.; Hopkins, R.F., 3rd; Witmer, T.; Neubauer, R.H.; Rabin, H. Production and characterization of monoclonal antibodies against the Epstein-Barr virus membrane antigen. J. Virol. 1982, 41, 258–264. [Google Scholar] [CrossRef] [Green Version]
- Molesworth, S.J.; Lake, C.M.; Borza, C.M.; Turk, S.M.; Hutt-Fletcher, L.M. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J. Virol. 2000, 74, 6324–6332. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Spriggs, M.K.; Kovats, S.; Turk, S.M.; Comeau, M.R.; Nepom, B.; Hutt-Fletcher, L.M. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 1997, 71, 4657–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, M.F.; Rohrer, U.H.; Kündig, T.M.; Bürki, K.; Hengartner, H.; Zinkernagel, R.M. The influence of antigen organization on B cell responsiveness. Science 1993, 262, 1448–1451. [Google Scholar] [CrossRef] [PubMed]
- Dintzis, R.Z.; Vogelstein, B.; Dintzis, H.M. Specific cellular stimulation in the primary immune response: Experimental test of a quantized model. Proc. Natl. Acad. Sci. USA 1982, 79, 884–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, J.; Kumar, A.; Kaur, J. Strategies for optimization of heterologous protein expression in E. coli: Roadblocks and reinforcements. Int. J. Biol. Macromol. 2018, 106, 803–822. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Cossart, P. Post-translational modifications in host cells during bacterial infection. FEBS Lett. 2010, 584, 2748–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mAbs | Ka (105/Ms) | Kd (10−4/s) | KD (nM) |
|---|---|---|---|
| 2C3 | 0.79 | 10.90 | 13.80 |
| 2E4 | 1.35 | 2.47 | 1.82 |
| 3D3 | 0.47 | 9.70 | 20.70 |
| 4D8 | 1.52 | 5.99 | 3.93 |
| 4H7 | 0.36 | 0.62 | 1.73 |
| 4H8 | 0.30 | 1.27 | 4.32 |
| 6B8 | 2.16 | 5.86 | 2.72 |
| 6C1 | 1.24 | 3.60 | 2.90 |
| 11G10 | 0.36 | 0.33 | 0.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, J.; Wei, D.; Wu, Q.; Zhong, L.; Chen, K.; Huang, Y.; Zhang, W.; Chen, J.; Xia, N.; Zhang, X.; et al. Antibody Generation and Immunogenicity Analysis of EBV gp42 N-Terminal Region. Viruses 2021, 13, 2380. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122380
Hong J, Wei D, Wu Q, Zhong L, Chen K, Huang Y, Zhang W, Chen J, Xia N, Zhang X, et al. Antibody Generation and Immunogenicity Analysis of EBV gp42 N-Terminal Region. Viruses. 2021; 13(12):2380. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122380
Chicago/Turabian StyleHong, Junping, Dongmei Wei, Qian Wu, Ling Zhong, Kaiyun Chen, Yang Huang, Wanlin Zhang, Junyu Chen, Ningshao Xia, Xiao Zhang, and et al. 2021. "Antibody Generation and Immunogenicity Analysis of EBV gp42 N-Terminal Region" Viruses 13, no. 12: 2380. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122380