
Genomic Epidemiology of Salmonid Alphavirus in Norwegian Aquaculture Reveals Recent Subtype-2 Transmission Dynamics and Novel Subtype-3 Lineages

 , , ,
, , ,
Abstract
:1. Introduction
2. Material and Methods
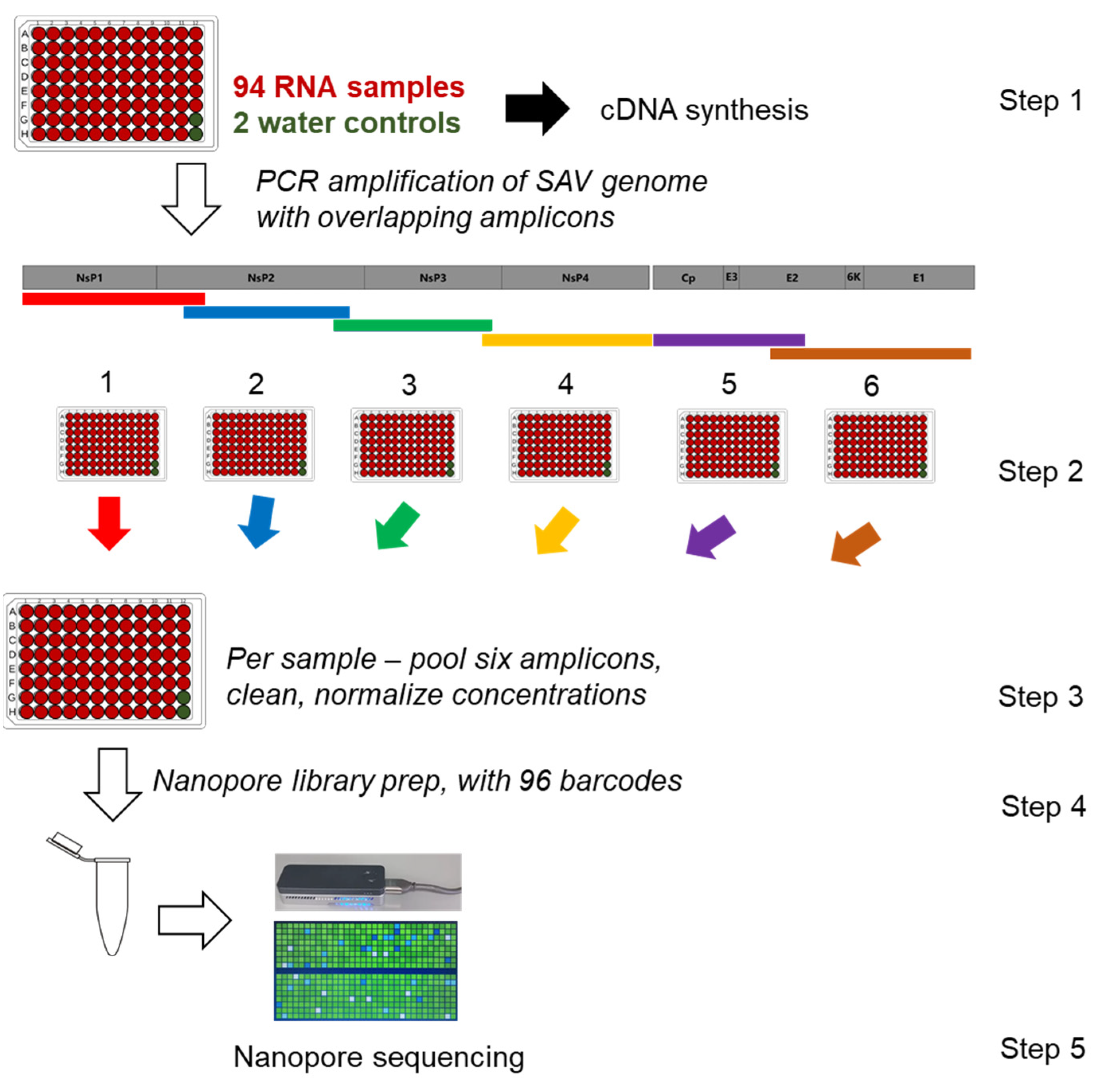
2.1. Overview of SAV Nanopore Sequencing Approach
2.2. Samples, Reverse Transcription and PCR
2.3. ONT Library Preparation, MinION Sequencing, Basecalling, and Trimming
2.4. Mapping, Consensus Sequence Generation, and Filtering
2.5. Phylogenetic and Phylogeographic Reconstructions
2.6. Sanger Sequencing of Additional SAV2 Genomes
3. Results
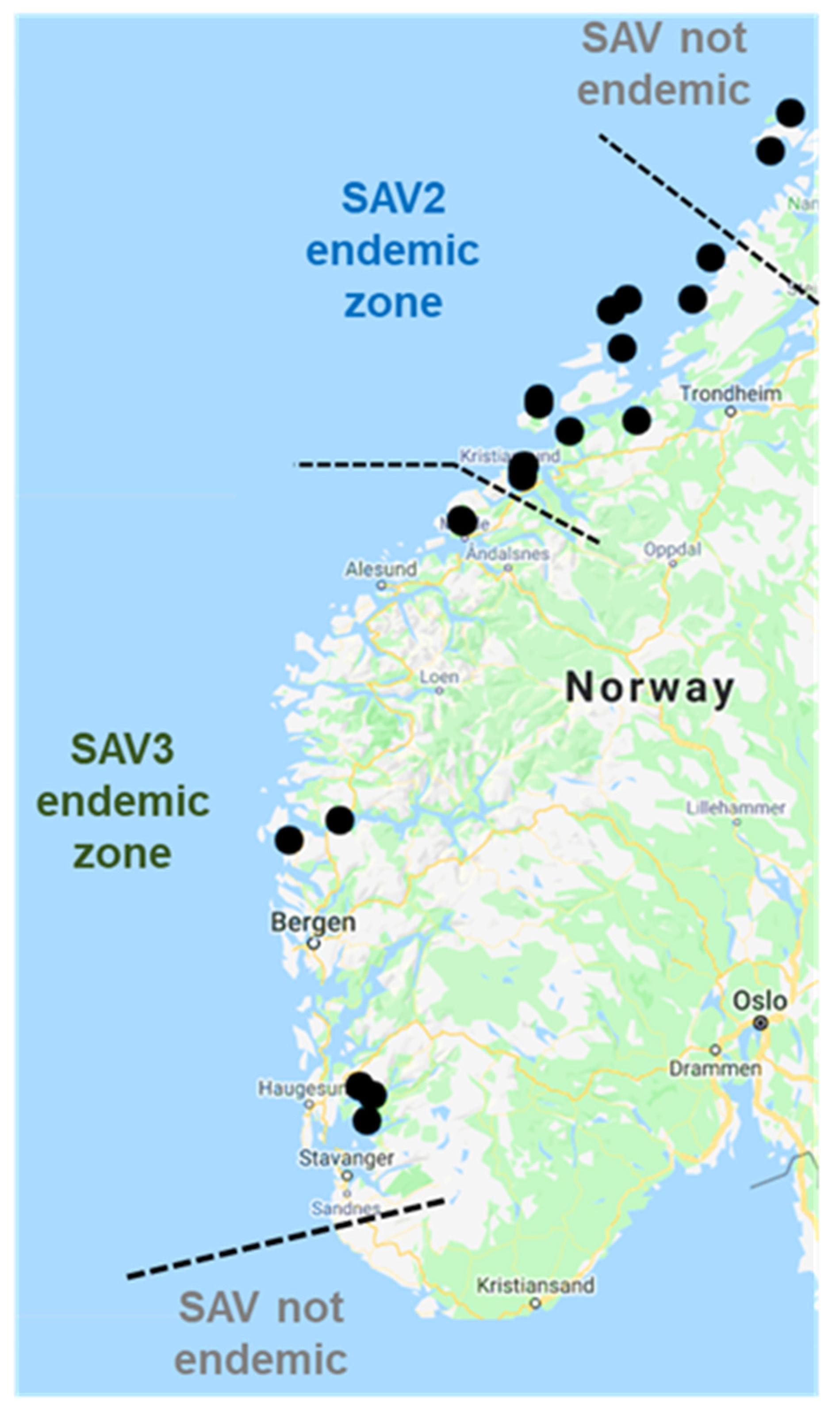
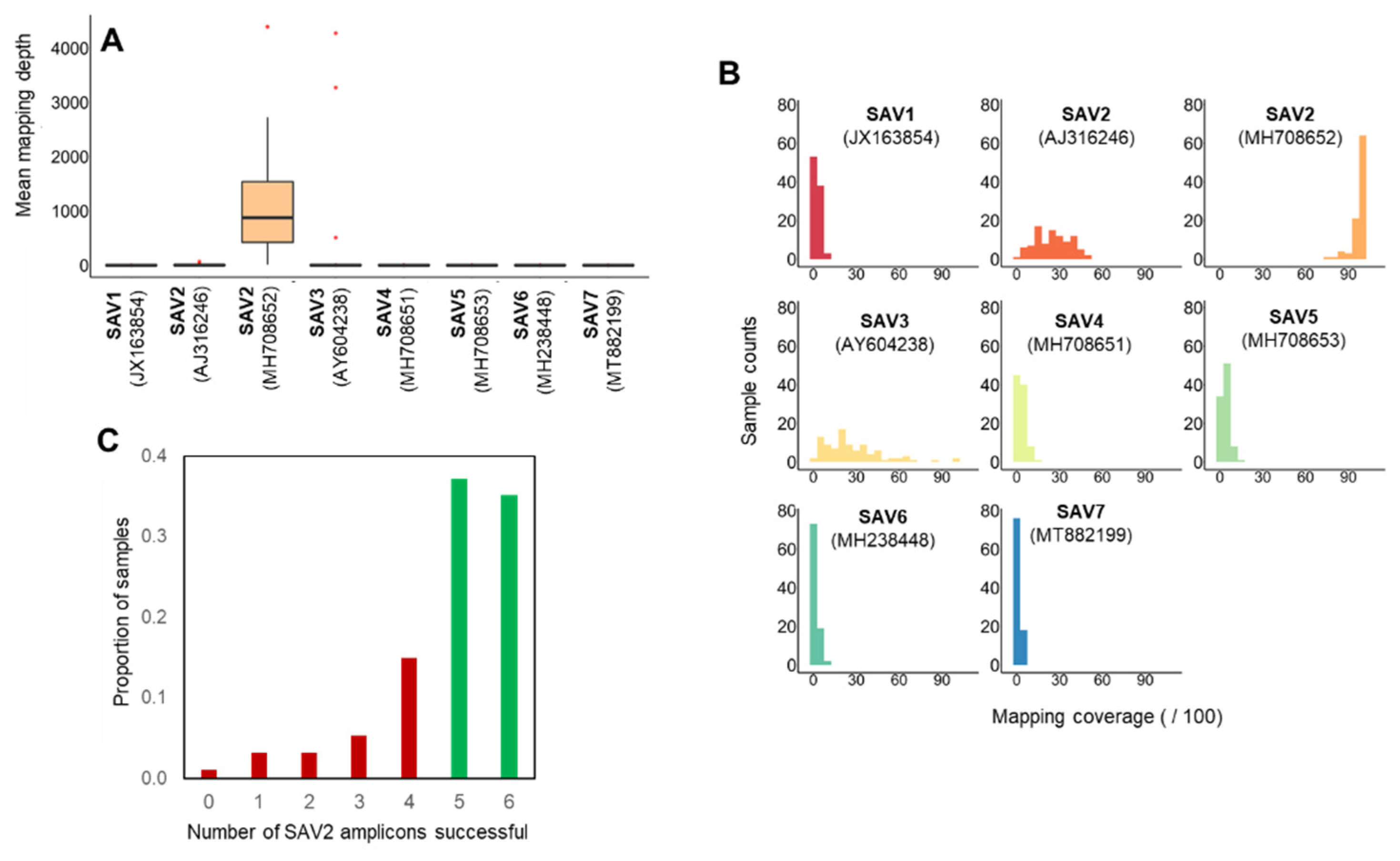
3.1. Nanopore Sequencing of SAV from Norwegian Field Samples
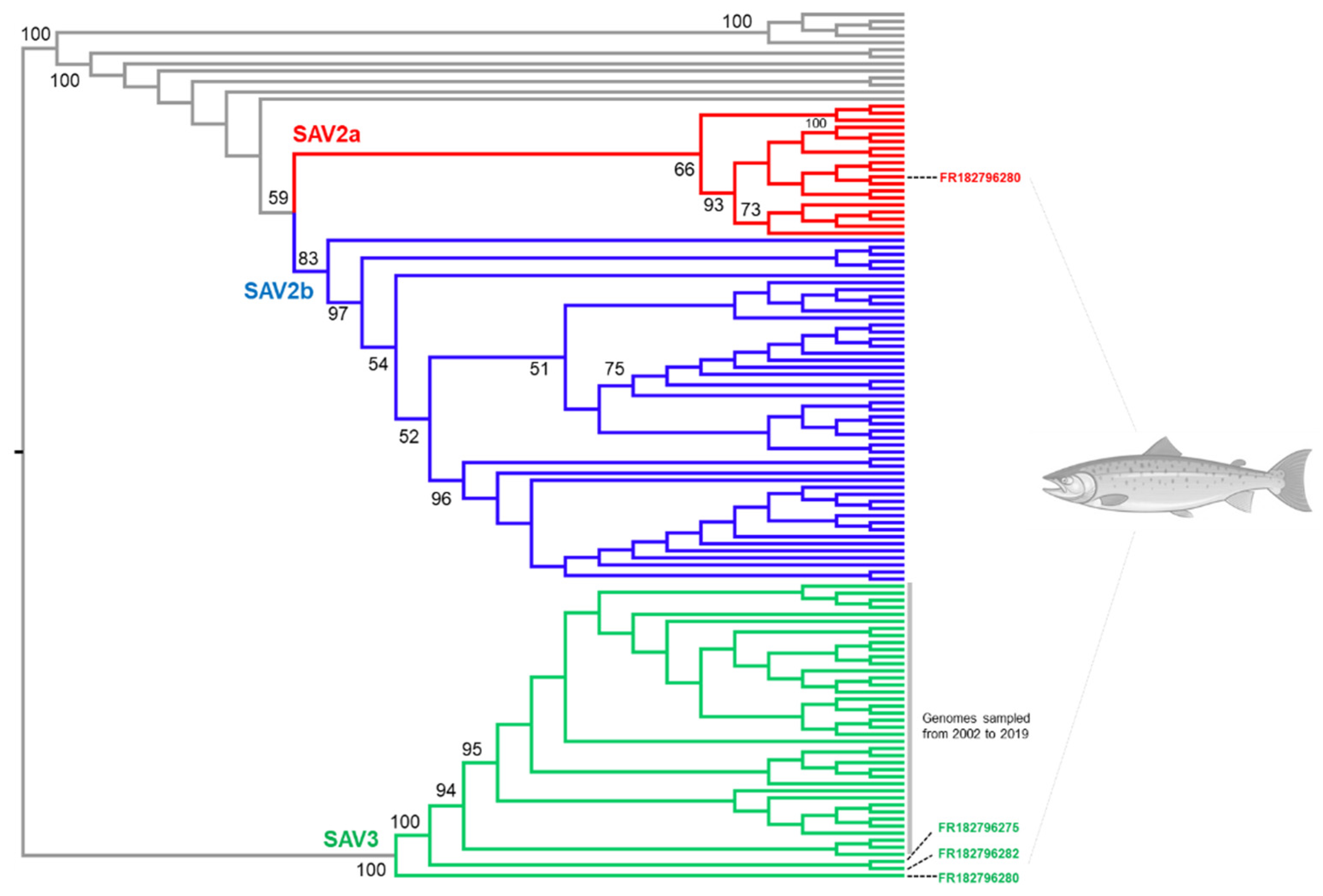
3.2. SAV2 Genetic Diversity in Recent Norwegian Aquaculture
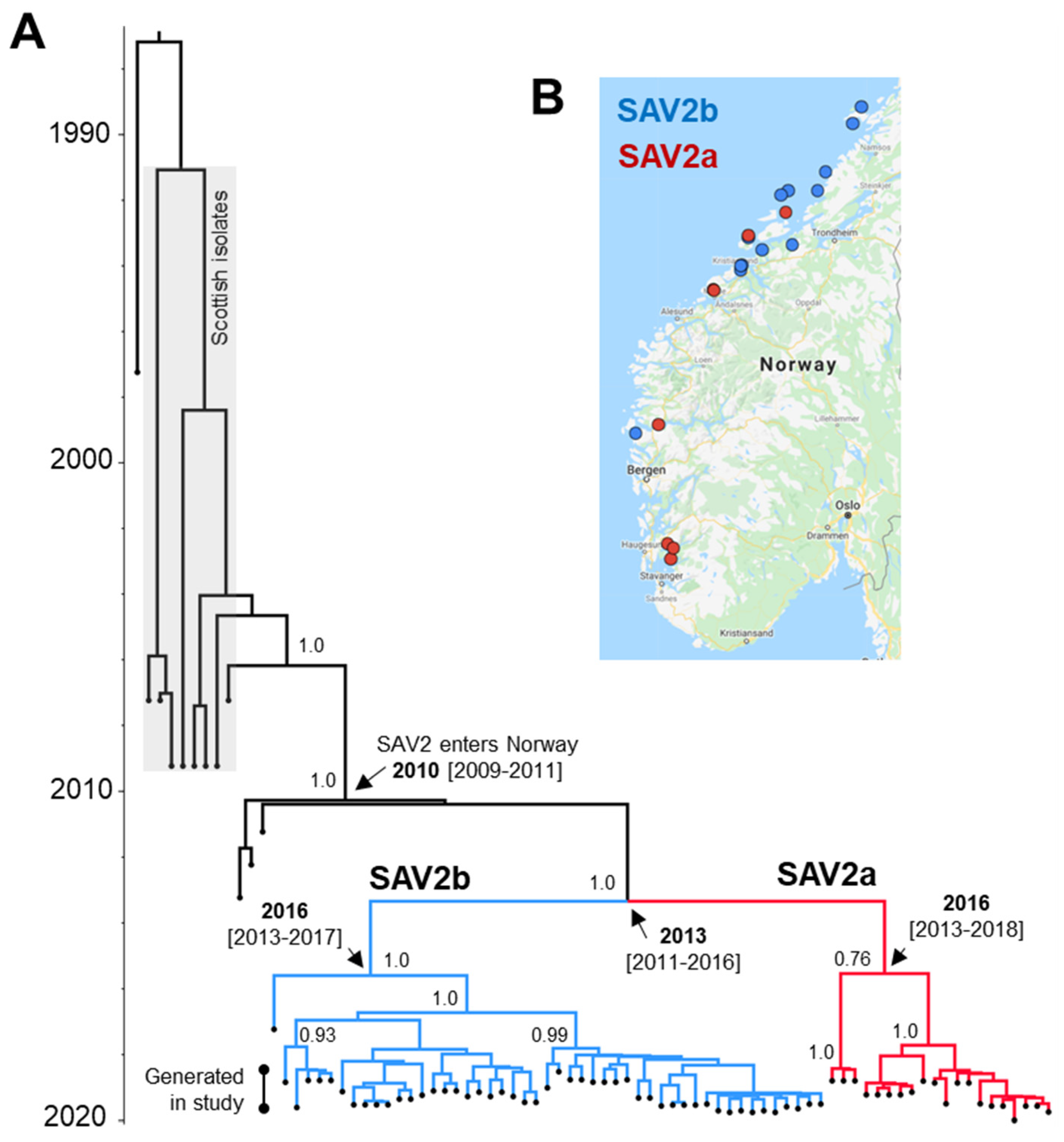
3.3. Phylogeographical Reconstructions of SAV2


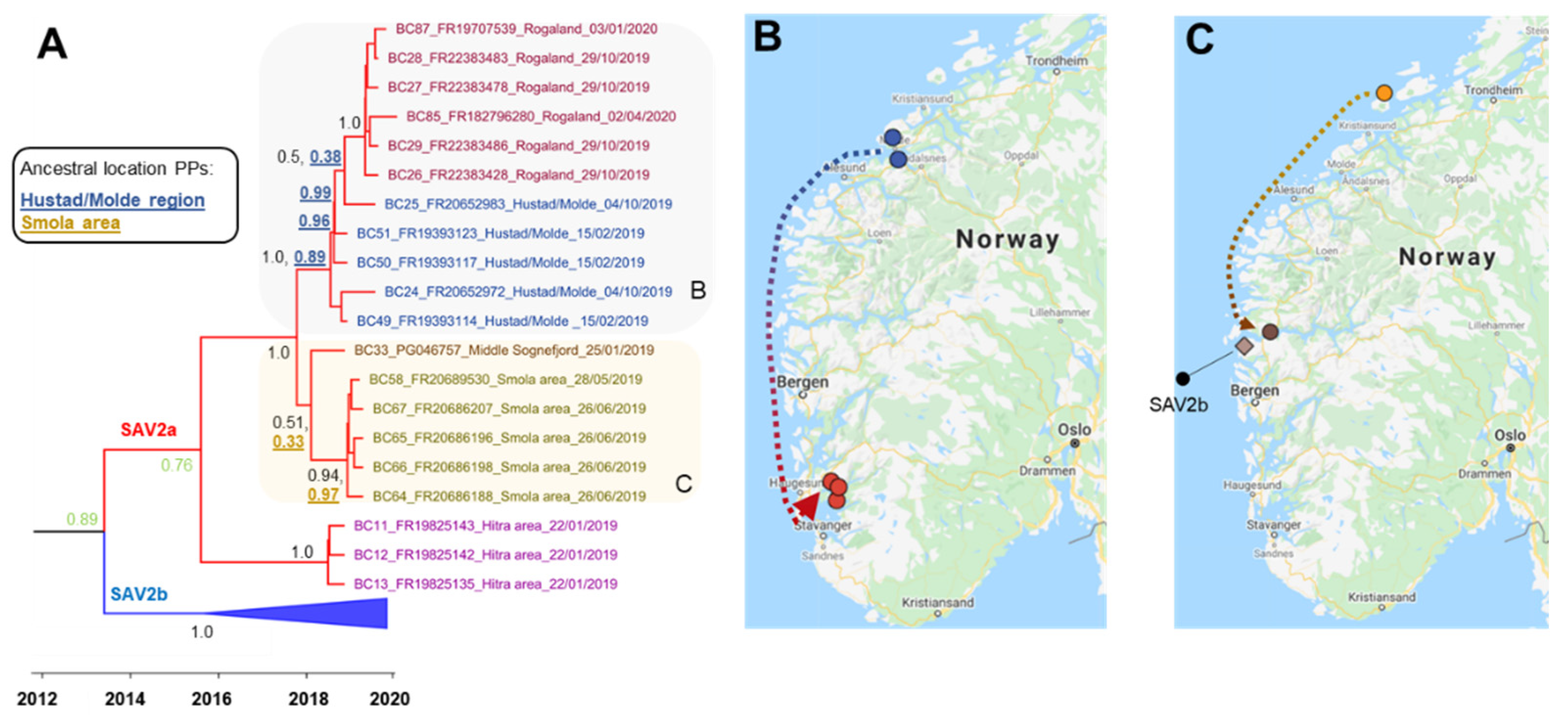
3.3.1. Phylogeographical Reconstructions of SAV2a
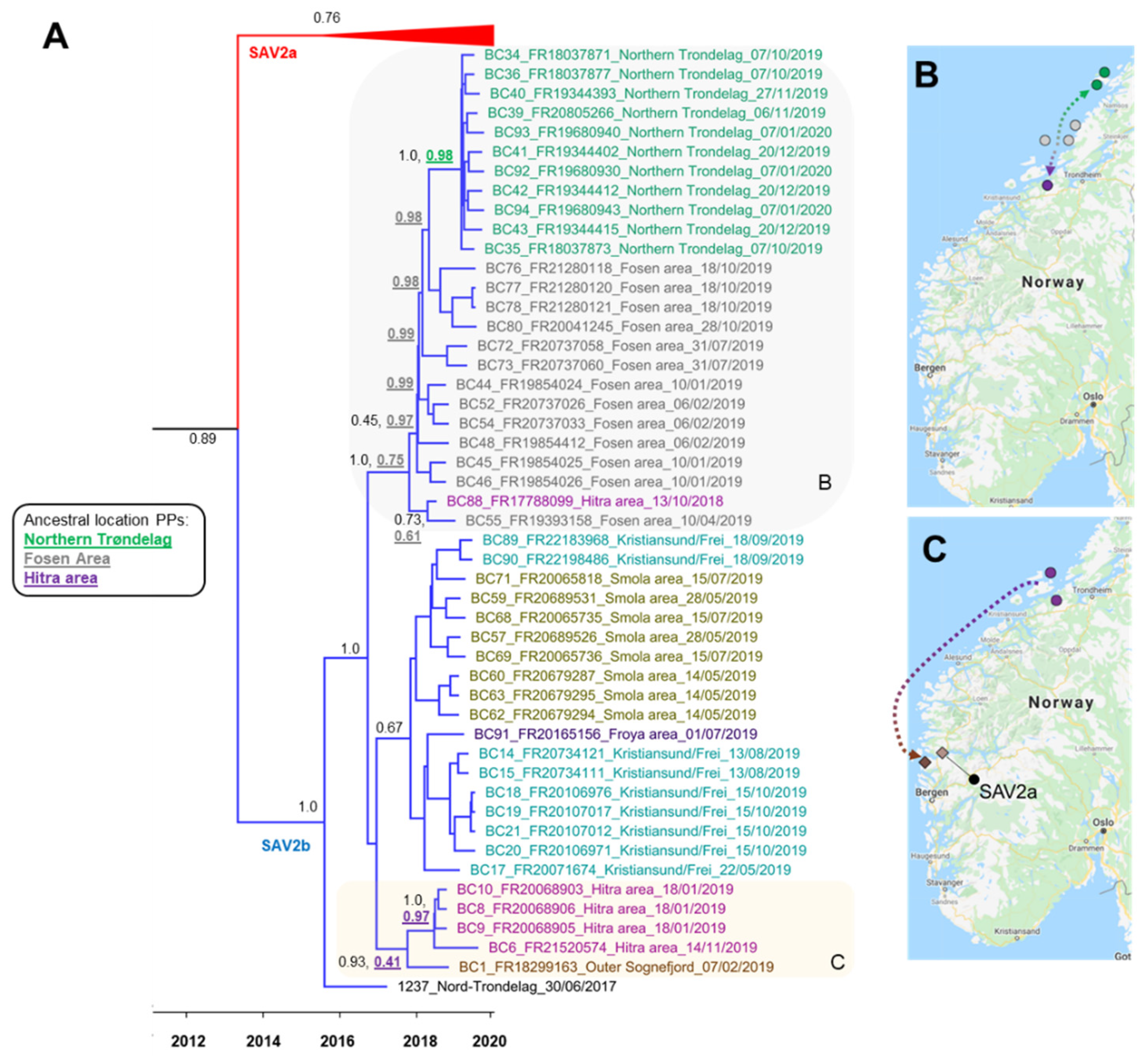
3.3.2. Phylogeographical Reconstructions of SAV2b
3.4. Novel SAV3 Strains Detected in Fish Co-Infected with SAV2 and SAV3
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef]
- Gire, S.K.; Goba, A.; Andersen, K.G.; Sealfon, R.S.; Park, D.J.; Kanneh, L.; Jalloh, S.; Momoh, M.; Fullah, M.; Dudas, G.; et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014, 345, 1369–1372. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Quick, J.; Claro, I.M.; Thézé, J.; de Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.G.; Hill, S.C.; Black, A.; da Costa, A.C.; et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef]
- Lo, S.W.; Jamrozy, D. Genomics and epidemiological surveillance. Nat. Rev. Microbiol. 2020, 18, 478. [Google Scholar] [CrossRef]
- COVID-19 Genomics UK (COG-UK). An integrated national scale SARS-CoV-2 genomic surveillance network. Lancet Microbe 2020, 1, e99–e100. [Google Scholar] [CrossRef]
- Béné, C.; Barange, M.; Subasinghe, R.; Pinstrup-Andersen, P.; Merino, G.; Hemre, G.-I.; Williams, M. Feeding 9 billion by 2050—Putting fish back on the menu. Food Secur. 2015, 7, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Stentiford, G.D.; Sritunyalucksana, K.; Flegel, T.W.; Williams, B.A.; Withyachumnarnkul, B.; Itsathitphaisarn, O.; Bass, D. New paradigms to help solve the global aquaculture disease crisis. PLoS Pathog. 2017, 13, e1006160. [Google Scholar] [CrossRef] [PubMed]
- Mugimba, K.K.; Byarugaba, D.K.; Mutoloki, S.; Evensen, Ø.; Munang’andu, H.M. Challenges and solutions to viral diseases of finfish in marine aquaculture. Pathogens 2021, 10, 673. [Google Scholar] [CrossRef] [PubMed]
- Houston, R.D.; Bean, T.P.; Macqueen, D.J.; Gundappa, M.K.; Jin, Y.H.; Jenkins, T.L.; Selly, S.L.C.; Martin, S.A.M.; Stevens, J.R.; Santos, E.M.; et al. Harnessing genomics to fast-track genetic improvement in aquaculture. Nat. Rev. Genet. 2020, 21, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Miccoli, A.; Manni, M.; Picchietti, S.; Scapigliati, G. State-of-the-art vaccine research for aquaculture use: The case of three economically relevant fish species. Vaccines 2021, 9, 140. [Google Scholar] [CrossRef]
- Bayliss, S.C.; Verner-Jeffreys, D.W.; Bartie, K.L.; Aanensen, D.M.; Sheppard, S.K.; Adams, A.; Feil, E.J. The promise of whole genome pathogen sequencing for the molecular epidemiology of emerging aquaculture pathogens. Front. Microbiol. 2017, 8, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, R.R.; Haydon, D.T.; Lycett, S.J.; Murcia, P.R. Supersize me: How whole-genome sequencing and big data are transforming epidemiology. Trends Microbiol. 2014, 22, 282–291. [Google Scholar] [CrossRef]
- Ulrich, K.; Wehner, S.; Bekaert, M.; Di Paola, N.; Dilcher, M.; Muir, K.F.; Taggart, J.B.; Matejusova, I.; Weidmann, M. Molecular epidemiological study on Infectious Pancreatic Necrosis Virus isolates from aquafarms in Scotland over three decades. J. Gen. Virol. 2018, 99, 1567–1581. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.D.; Karlsen, M.; Petterson, E.; Haugland, Ø.; Matejusova, I.; Macqueen, D.J. Genome sequencing of SAV3 reveals repeated seeding events of viral strains in Norwegian aquaculture. Front. Microbiol. 2020, 11, 740. [Google Scholar] [CrossRef]
- Gallagher, M.D.; Matejusova, I.; Ruane, N.M.; Macqueen, D.J. Genome-wide target enriched viral sequencing reveals extensive ‘hidden’ salmonid alphavirus diversity in farmed and wild fish populations. Aquaculture 2020, 522, 735117. [Google Scholar] [CrossRef]
- Mordecai, G.J.; Miller, K.M.; Bass, A.L.; Bateman, A.W.; Teffer, A.K.; Caleta, J.M.; Di Cicco, E.; Schulze, A.D.; Kaukinen, K.H.; Li, S.; et al. Aquaculture mediates global transmission of a viral pathogen to wild salmon. Sci. Adv. 2021, 7, eabe2592. [Google Scholar] [CrossRef]
- Benkaroun, J.; Muir, K.F.; Allshire, R.; Tamer, C.; Weidmann, M. Isolation of a new infectious pancreatic necrosis virus (IPNV) variant from a fish farm in Scotland. Viruses 2021, 13, 385. [Google Scholar] [CrossRef]
- Houston, R.D.; Macqueen, D.J. Atlantic salmon (Salmo salar L.) genetics in the 21st century: Taking leaps forward in aquaculture and biological understanding. Anim. Genet. 2019, 50, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Statistics Norway, Aquaculture. Available online: https://www.ssb.no/en/fiskeoppdrett/ (accessed on 5 November 2021).
- Crane, M.; Hyatt, A. Viruses of fish: An overview of significant pathogens. Viruses 2011, 3, 2025–2046. [Google Scholar] [CrossRef]
- Nelson, R.T.; McLoughlin, M.F.; Rowley, H.M.; Platten, M.A.; McCormick, J.I. Isolation of a toga-like virus from farmed Atlantic salmon Salmo salar with pancreas disease. Dis. Aquat. Org. 1995, 22, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Rowley, H.M.; Doherty, C.E.; McLoughlin, M.F.; Welsh, M.D. Isolation of salmon pancreas disease virus (SPDV) from farmed Atlantic salmon, Salmo salar L., in Scotland. J. Fish Dis. 1990, 21, 469–471. [Google Scholar] [CrossRef]
- Christie, K.E.; Fyrand, K.; Holtet, L.; Rowley, H.M. Isolation of pancreas disease virus from farmed Atlantic salmon, Salmo salar L. in Norway. J. Fish Dis. 1998, 21, 391–394. [Google Scholar] [CrossRef]
- Weston, J.H.; Welsh, M.D.; McLoughlin, M.F.; Todd, D. Salmon pancreas disease virus, an alphavirus infecting farmed Atlantic salmon, Salmo salar L. Virology 1999, 256, 188–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, J.H.; Villoing, S.; Bremont, M.; Castric, J.; Pfeffer, M.; Jewhurst, V.; McLoughlin, M.; Rødseth, O.; Christie, K.E.; Koumans, J.; et al. Comparison of 2 aquatic alphaviruses, salmon pancreas disease virus and sleeping disease virus, by using genome sequence analysis, monoclonal reactivity, and cross-infection. J. Virol. 2002, 76, 6155–6163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLoughlin, M.F.; Graham, D.A. Alphavirus infections in salmonids—A review. J. Fish Dis. 2007, 30, 511–531. [Google Scholar] [CrossRef]
- Deperasińska, I.; Schulz, P.; Siwicki, A.K. Salmonid Alphavirus (SAV). J. Vet. Res. 2018, 62, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fringuelli, E.; Rowley, H.M.; Wilson, J.C.; Hunter, R.; Rodger, H.; Graham, D.A. Phylogenetic analyses and molecular epidemiology of European salmonid alphaviruses (SAV) based on partial E2 and nsP3 gene nucleotide sequences. J. Fish Dis. 2008, 31, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Tighe, A.J.; Gallagher, M.D.; Carlsson, J.; Matejusova, I.; Swords, F.; Macqueen, D.J.; Ruane, N.M. Nanopore whole genome sequencing and partitioned phylogenetic analysis supports a new salmonid alphavirus genotype (SAV7). Dis. Aquat. Org. 2020, 142, 203–211. [Google Scholar] [CrossRef]
- Karlsen, M.; Gjerset, B.; Hansen, T.; Rambaut, A. Multiple introductions of salmonid alphavirus from a wild reservoir have caused independent and self-sustainable epizootics in aquaculture. J. Gen. Virol. 2014, 95, 52–59. [Google Scholar] [CrossRef]
- Bruno, D.W.; Noguera, P.A.; Black, J.; Murray, W.; Macqueen, D.J.; Matejusova, I. Identification of a wild reservoir of salmonid alphavirus in common dab Limanda limanda, with emphasis on virus culture and sequencing. Aquacult. Environ. Interact. 2014, 5, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Hodneland, K.; Bratland, A.; Christie, K.E.; Endresen, C.; Nylund, A. New subtype of salmonid alphavirus (SAV), Togaviridae, from Atlantic salmon Salmo salar and rainbow trout Oncorhynchus mykiss in Norway. Dis. Aquat. Org. 2005, 66, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Hjortaas, M.J.; Skjelstad, H.R.; Taksdal, T.; Olsen, A.B.; Johansen, R.; Bang-Jensen, B.; Orpetveit, I.; Sindre, H. The first detections of subtype 2-related salmonid alphavirus (SAV2) in Atlantic salmon, Salmo salar L., in Norway. J. Fish Dis. 2013, 36, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Hjortaas, M.J.; Fringuelli, E.; Monjane, A.L.; Mikalsen, A.B.; Jonassen, C.M.; Savage, P.; Sindre, H. Emergence of Salmonid Alphavirus genotype 2 in Norway-molecular characterization of viral strains circulating in Norway and Scotland. Viruses 2021, 13, 1556. [Google Scholar] [CrossRef] [PubMed]
- Stien, L.; Tørud, B.; Gismervik, S.; Lien, M.; Medaas, C.; Osmundsen, T.; Kristiansen, T.; Størkersen, K. Governing the welfare of Norwegian farmed salmon: Three conflict cases. Mar. Policy 2020, 117. [Google Scholar] [CrossRef]
- Sommerset, I.; Bang Jensen, B.; Bornø, B.; Haukaas, A.; Brun, E. (Eds.) The Health Situation in Norwegian Aquaculture 2020; Norwegian Veterinary Institute: Oslo, Norway, 2021. [Google Scholar]
- Gallagher, M.D.; Matejusova, I.; Nguyen, L.; Ruane, N.M.; Falk, K.; Macqueen, D.J. Nanopore sequencing for rapid diagnostics of salmonid RNA viruses. Sci. Rep. 2018, 8, 16307. [Google Scholar] [CrossRef] [Green Version]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 5 November 2021).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouckaert, R.R.; Drummond, A.J. bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol. Biol. 2017, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Høgåsen, H.R.; Lyngstad, T.M.; Jansen, M.D.; Nilsen, A. Risk of Disease Transfer with Wellboats in Norway—Technical Report; Veterinærinstituttets rapportserie Veterinærinstituttet: Oslo, Norway, 2015. [Google Scholar]
- Jansen, M.D.; Bang Jensen, B.; McLoughlin, M.F.; Rodger, H.D.; Taksdal, T.; Sindre, H.; Graham, D.A.; Lillehaug, A. The epidemiology of pancreas disease in salmonid aquaculture: A summary of the current state of knowledge. J. Fish Dis. 2017, 40, 141–155. [Google Scholar] [CrossRef]
- Røsaeg, M.V.; Thorarinsson, R.; Aunsmo, A. Effect of vaccines against pancreas disease in farmed Atlantic salmon. J. Fish Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hillestad, B.; Johannessen, S.; Melingen, G.O.; Moghadam, H.K. Identification of a new infectious pancreatic necrosis virus (IPNV) variant in atlantic salmon (Salmo salar L.) that can cause high mortality even in genetically resistant fish. Front. Genet. 2021, 12. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amplicon | Forward Primer (5’–3’) | Reverse Primer (5’–3’) | Product Length 1 | Genomic Location 1 |
|---|---|---|---|---|
| 1 | AGACTGCGTTTCCAGGGTTC | CCCGTAGATGCCAATCGTGT | 2162 bp | 89–2251 bp |
| 2 | GAATACGTTTACGAATTGTCCTCC | ACCGAGACGGACTTGAAATACC | 1967 bp | 2083–4049 bp |
| 3 | GACCTGGTGTTTTGTGACGC | TCCCGTGTTAGCCCTCTAGG | 1895 bp | 3838–5732 bp |
| 4 | GCAGCGTCCACRGCCATAGT | CATCAGGCGTTTTACAGGGTC | 2015 bp | 5531–7545 bp |
| 5 | ATGTTTCCCATGCAATTCACCAAC | GGTGCGGCTTGCCCTGGGTGAT | 1816 bp | 7818–9633 bp |
| 6 | AGAGAACGCAGCAAGGGC | GGCACTTCTTCACCACGCA | 2431 bp | 9318–11,748 bp |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macqueen, D.J.; Eve, O.; Gundappa, M.K.; Daniels, R.R.; Gallagher, M.D.; Alexandersen, S.; Karlsen, M. Genomic Epidemiology of Salmonid Alphavirus in Norwegian Aquaculture Reveals Recent Subtype-2 Transmission Dynamics and Novel Subtype-3 Lineages. Viruses 2021, 13, 2549. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122549
Macqueen DJ, Eve O, Gundappa MK, Daniels RR, Gallagher MD, Alexandersen S, Karlsen M. Genomic Epidemiology of Salmonid Alphavirus in Norwegian Aquaculture Reveals Recent Subtype-2 Transmission Dynamics and Novel Subtype-3 Lineages. Viruses. 2021; 13(12):2549. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122549
Chicago/Turabian StyleMacqueen, Daniel J., Oliver Eve, Manu Kumar Gundappa, Rose Ruiz Daniels, Michael D. Gallagher, Svein Alexandersen, and Marius Karlsen. 2021. "Genomic Epidemiology of Salmonid Alphavirus in Norwegian Aquaculture Reveals Recent Subtype-2 Transmission Dynamics and Novel Subtype-3 Lineages" Viruses 13, no. 12: 2549. https://0-doi-org.brum.beds.ac.uk/10.3390/v13122549