
Distinguishing Incubation and Acute Disease Stages of Mild-to-Moderate COVID-19

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Flow Cytometry
2.3. Multipathway Posphoprotein Analyses
2.4. SARS-CoV-2 Specific Antibodies
2.5. Cytokine Analysis
2.6. Statistics
3. Results
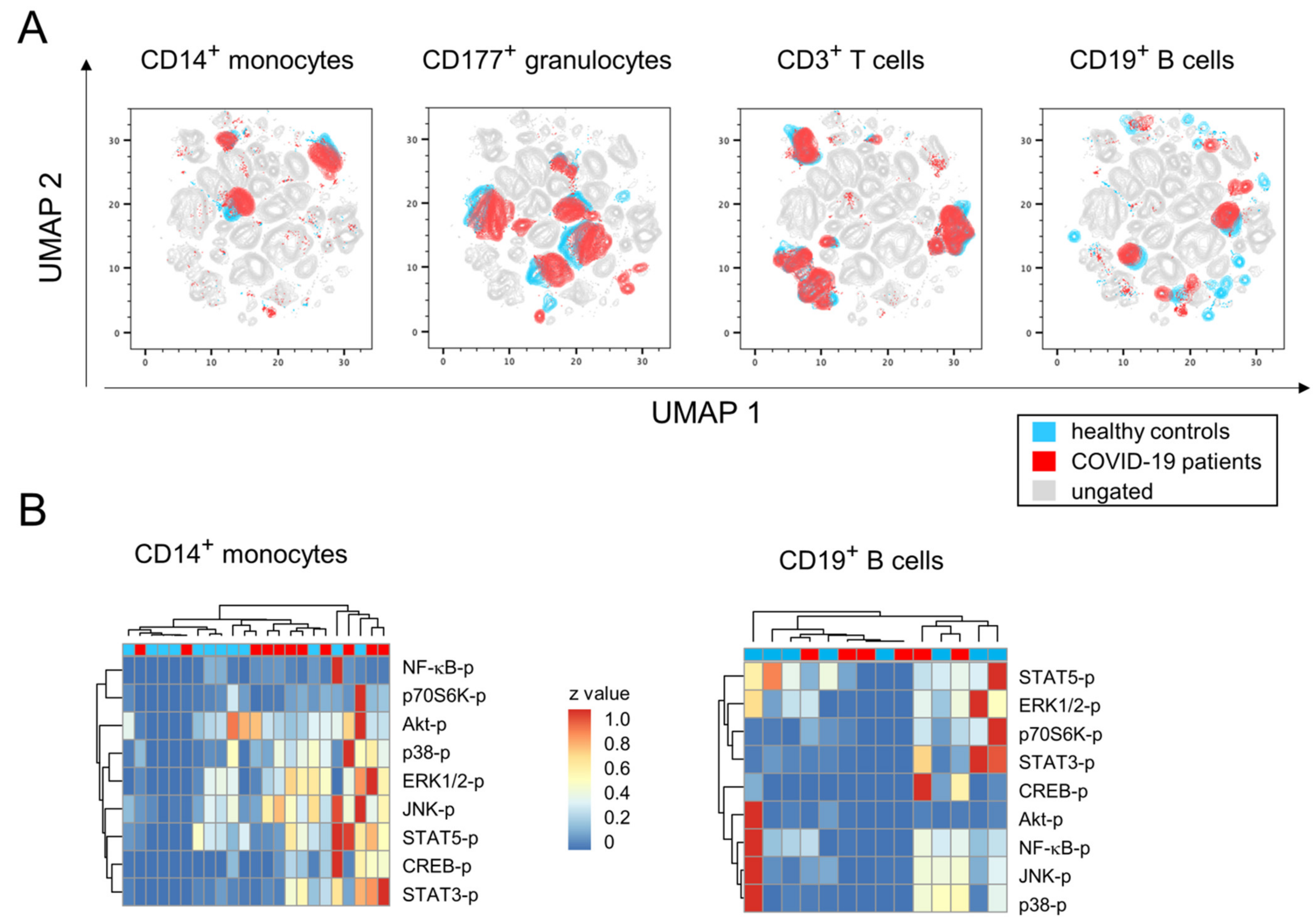
3.1. Mild-to-Moderate COVID-19 Induces Minor Changes in Immune Cell Subpopulation Distribution and Activation
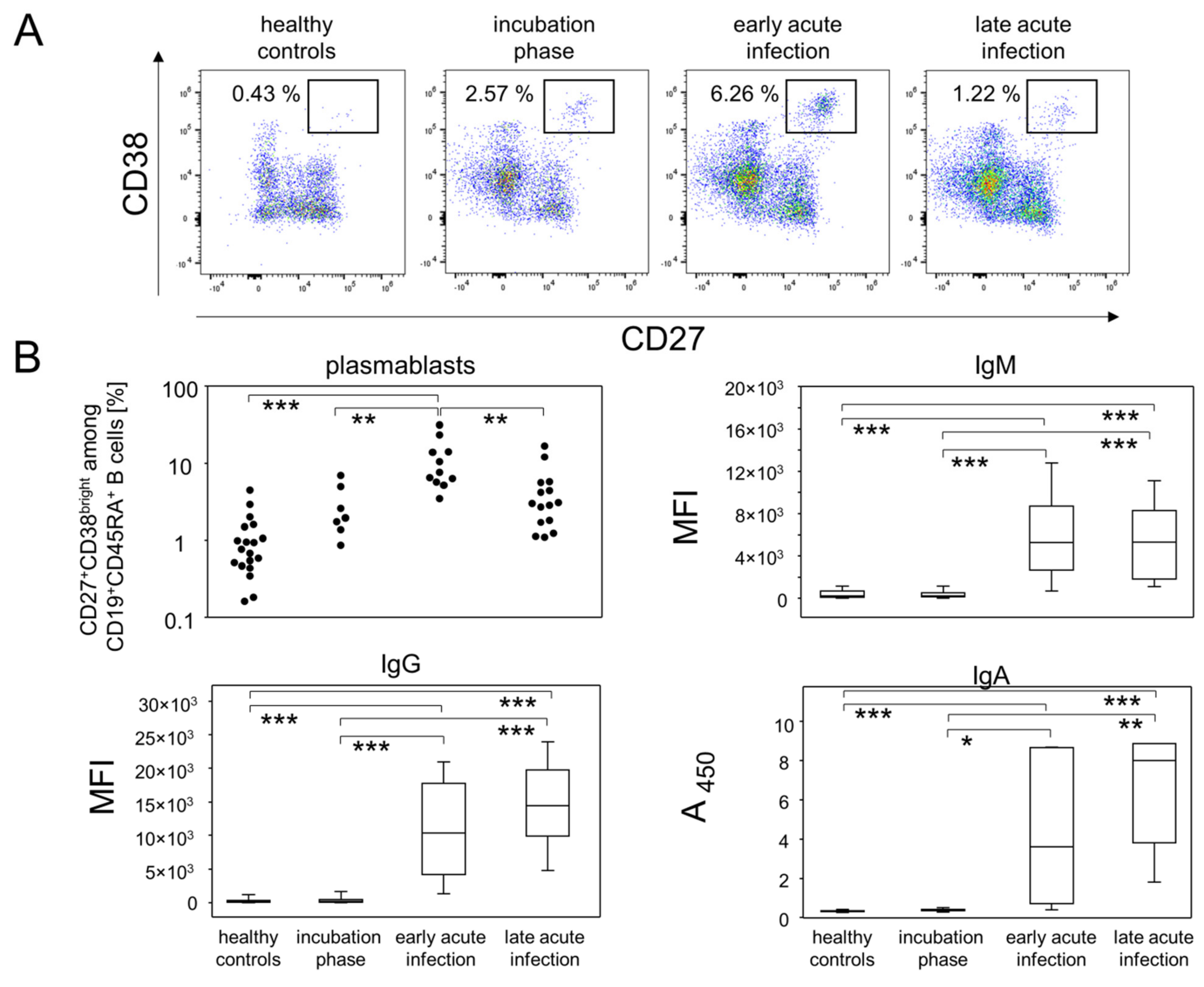
3.2. The Fraction of Plasmablasts in Combination with Antibody Titers Delineate Infection Phases
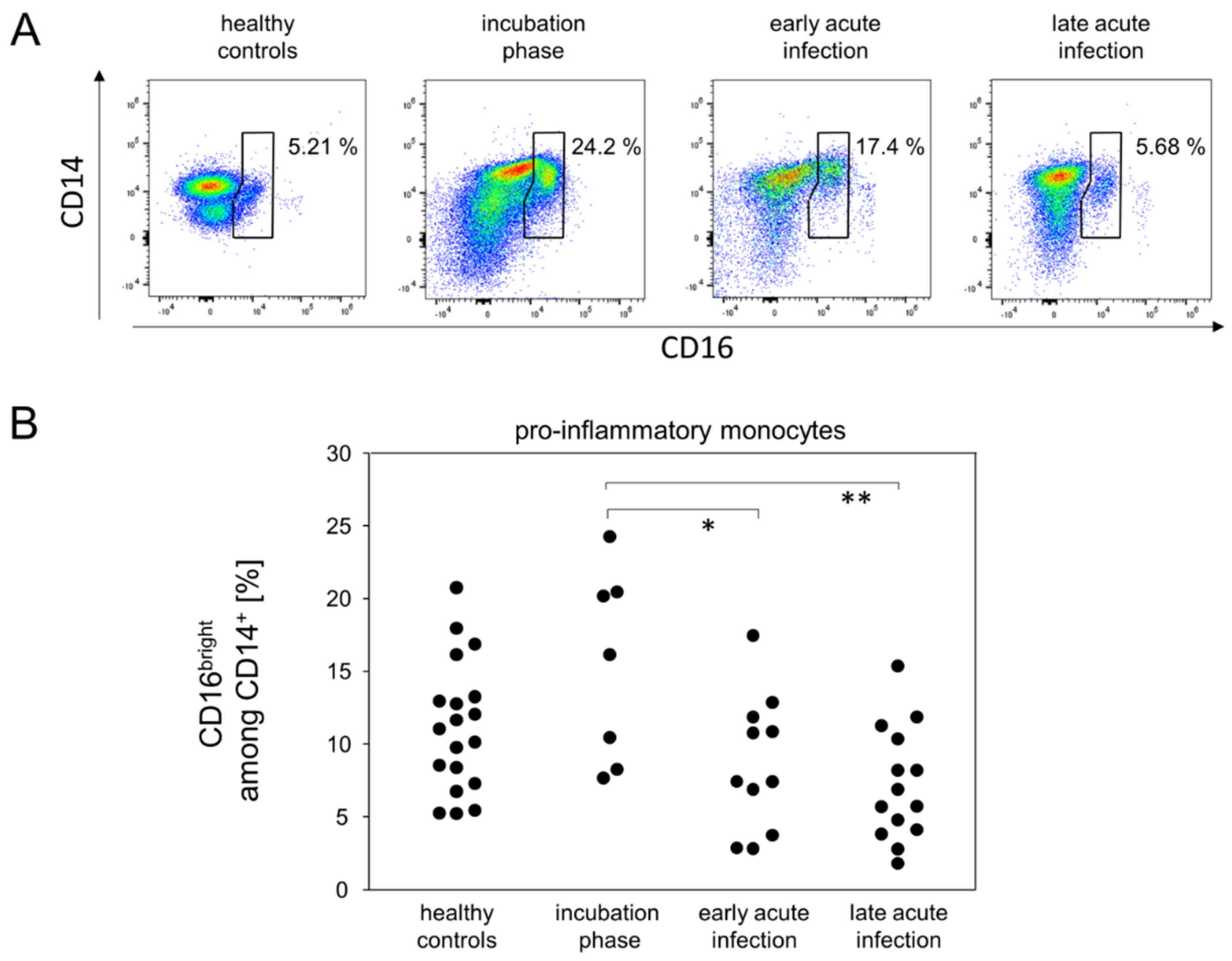
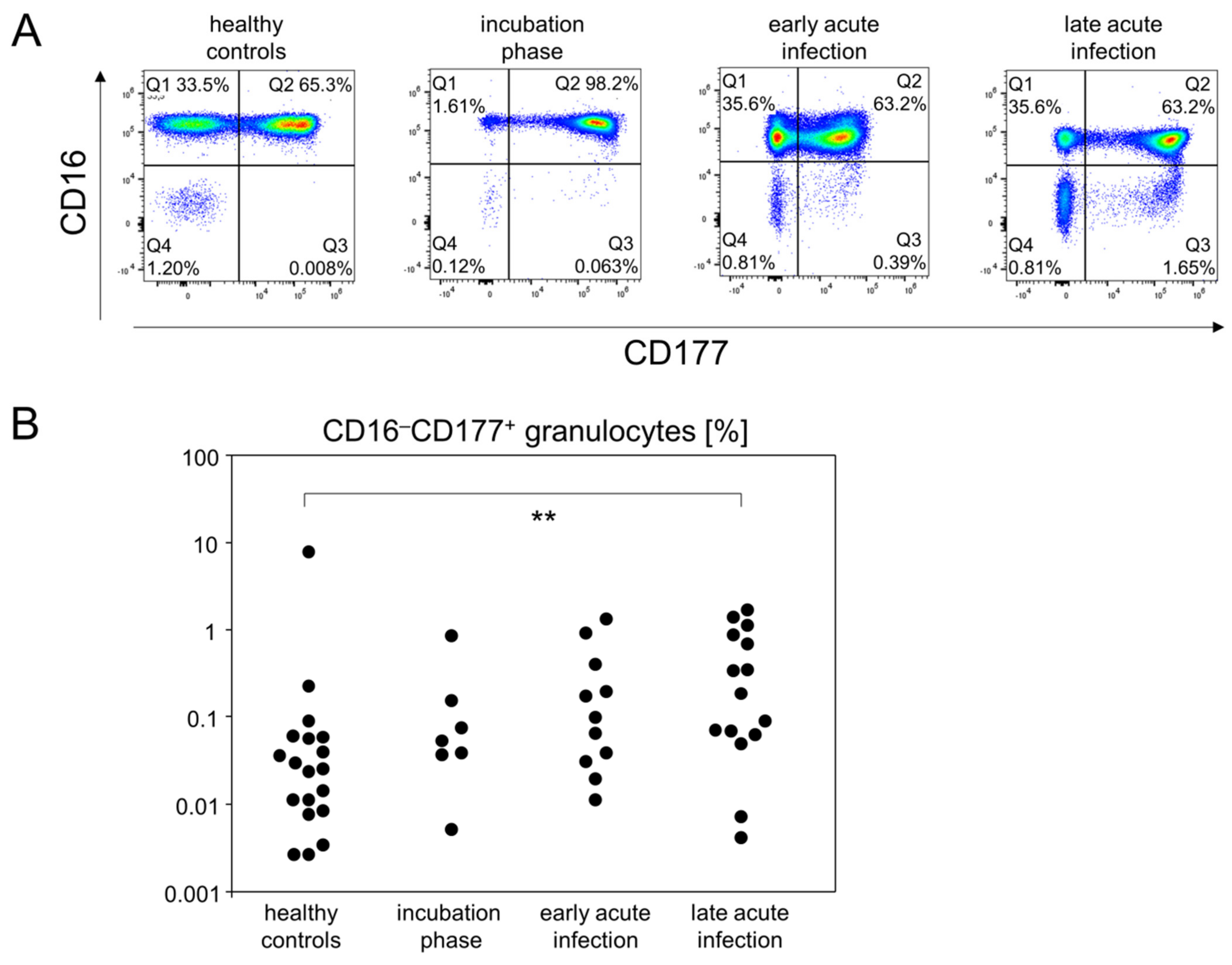
3.3. Pro-Inflammatory Monocytes and CD16−CD177+ Granulocytes Follow Different Kinetics during Mild-to-Moderate COVID-19
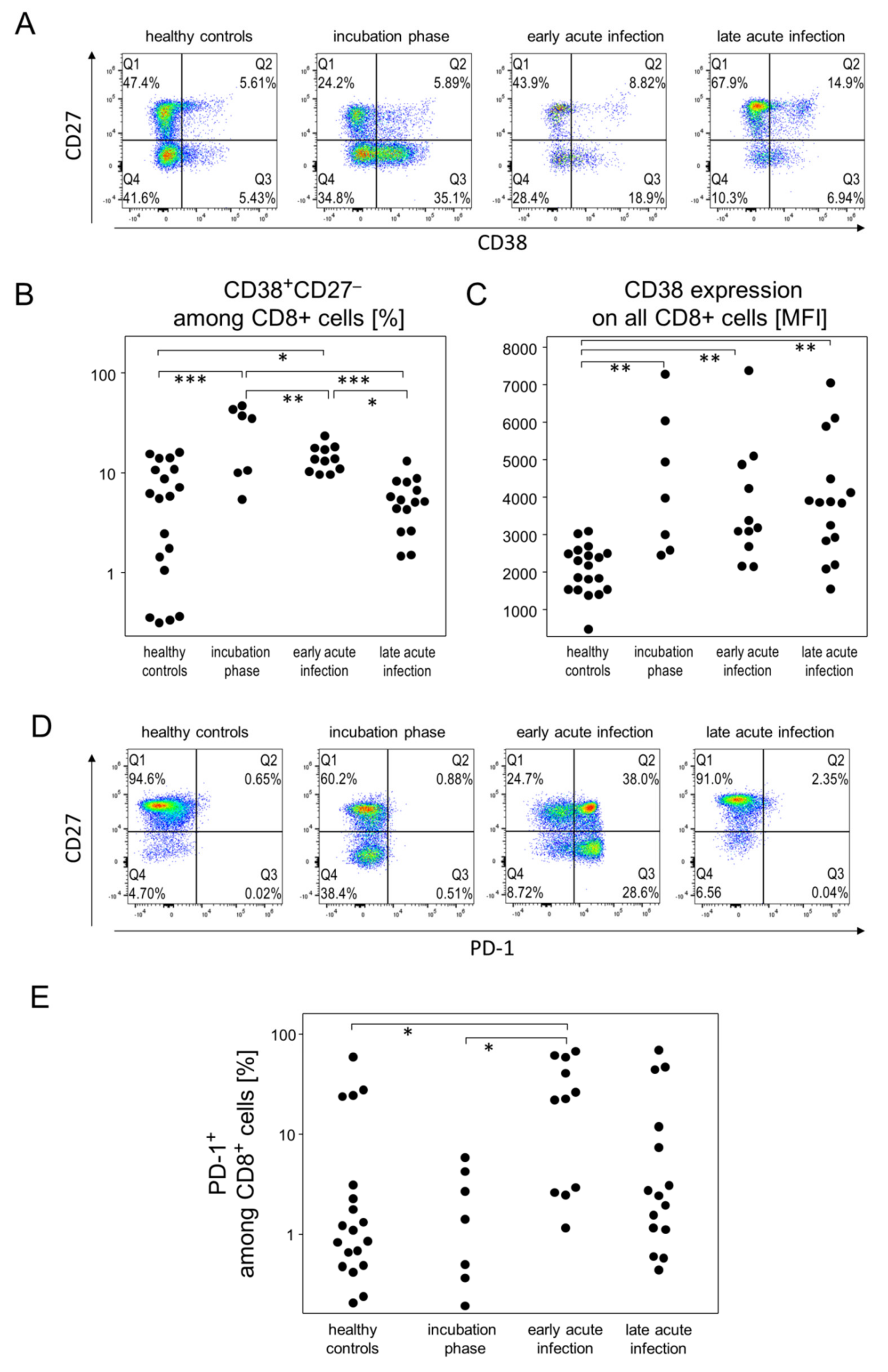
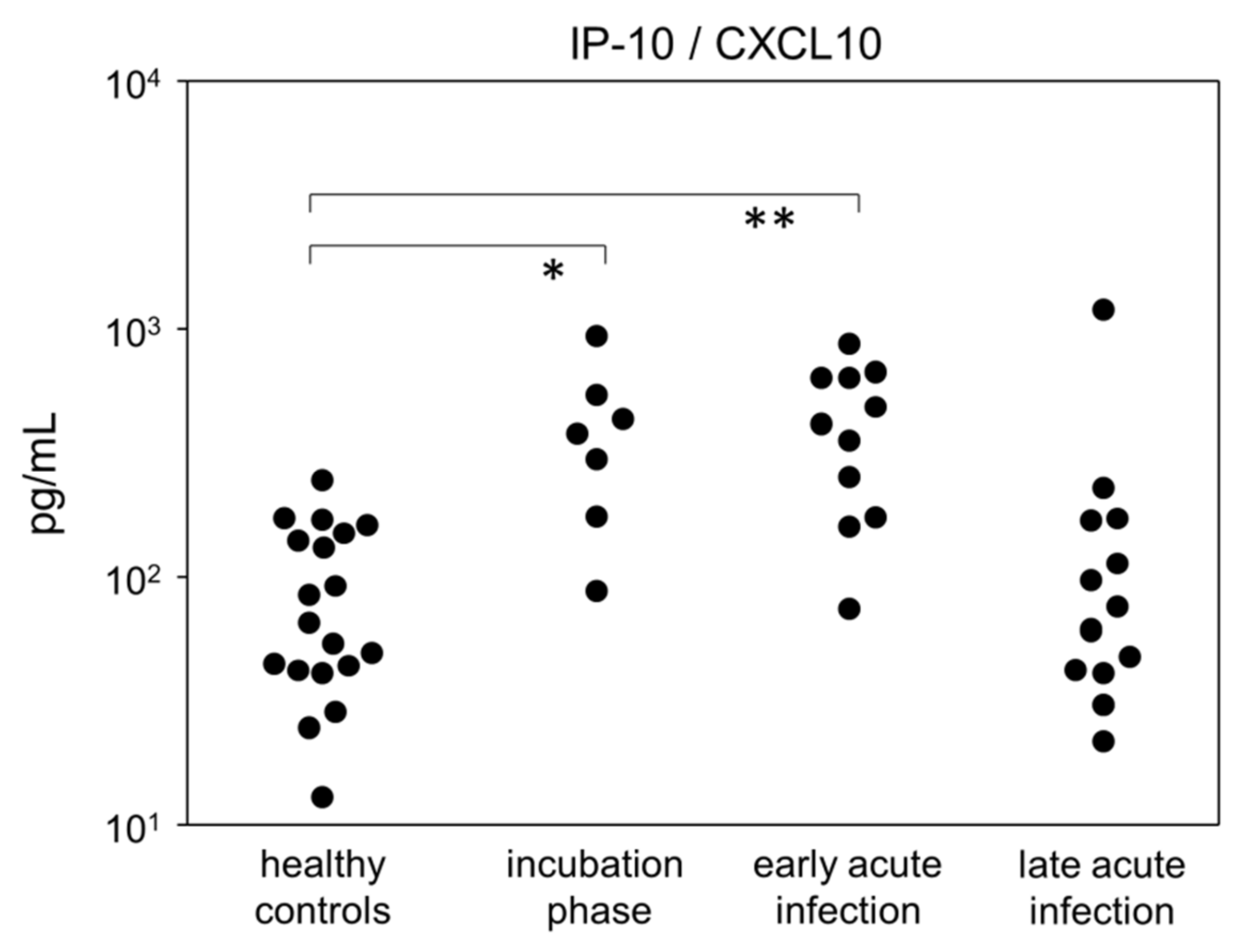
3.4. Incubation and Early Acute Infection Phases Are Characterized by an Increase in Activated Cytotoxic T Cells and IP-10
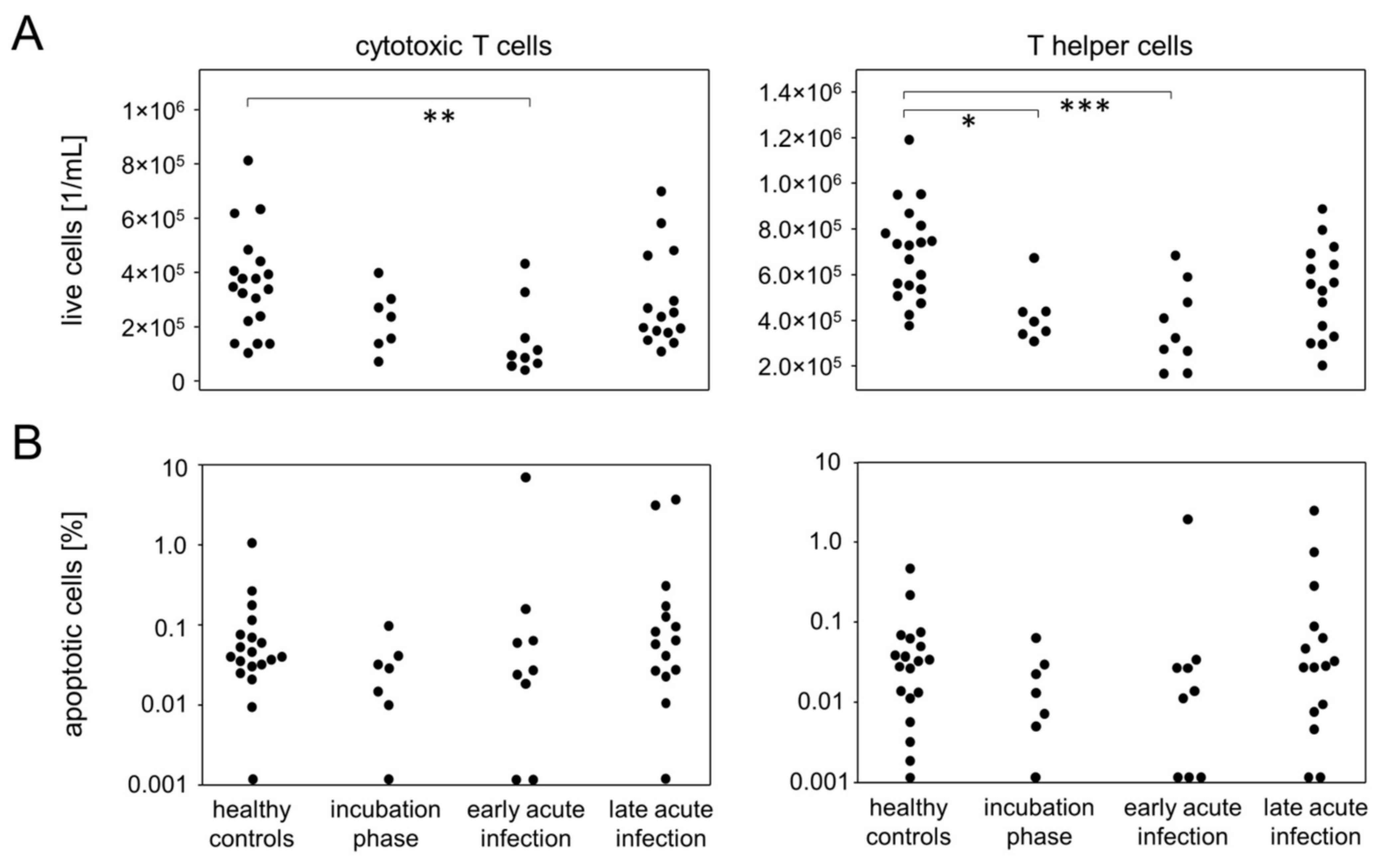
3.5. Incubation and Early Acute Infection Are Characterized by T Lymphopenia Unrelated to Apoptosis
3.6. IP-10 in Patients Correlates with Different Immune Parameters Than in Healthy Controls
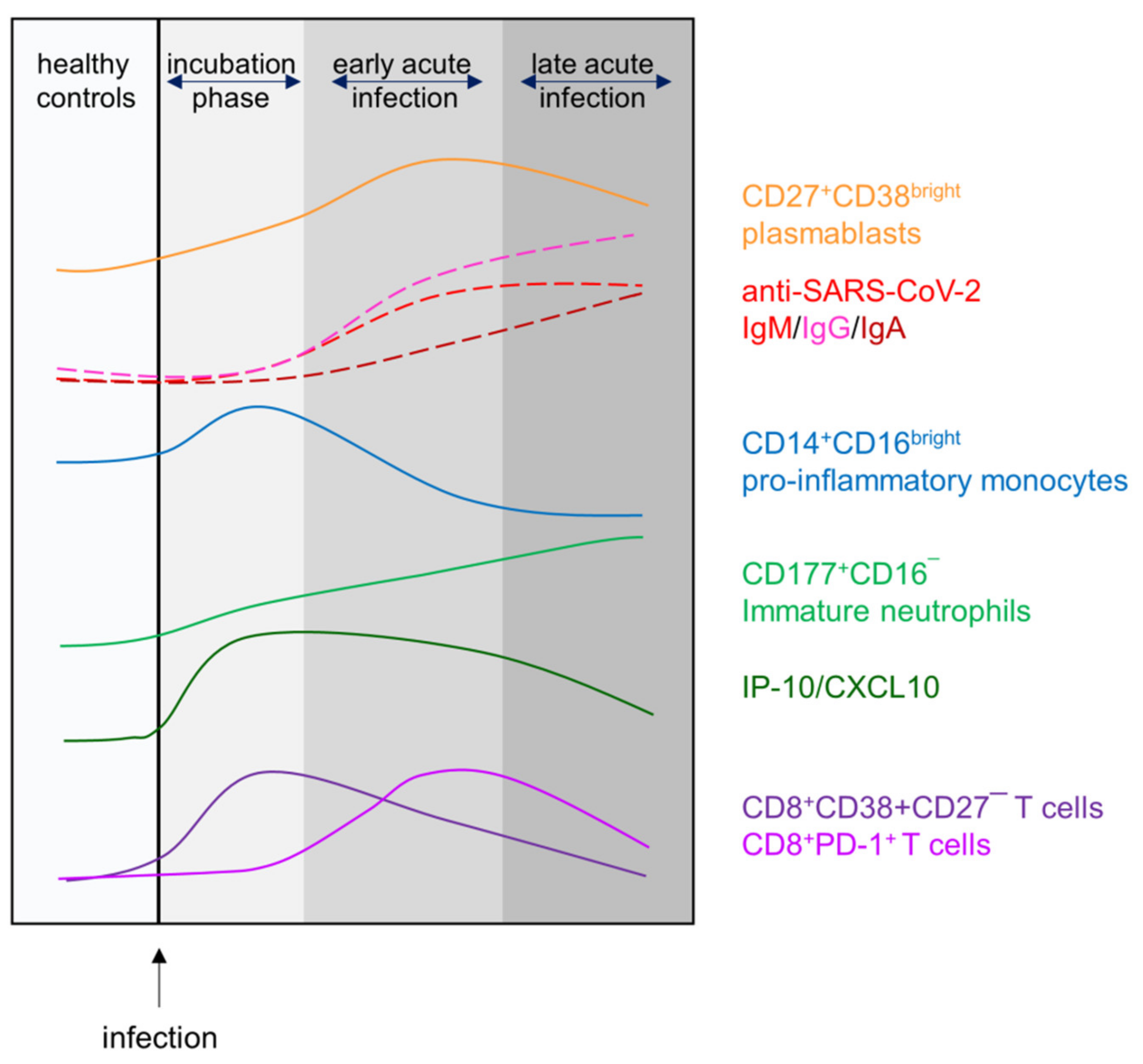
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Krishnan, U.M. The emergence of COVID-19 as a global pandemic: Understanding the epidemiology, immune response and potential therapeutic targets of SARS-CoV-2. Biochimie 2020, 179, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.R.; Fleming, T.R.; Longini, I.M.; Peto, R.; Briand, S.; Heymann, D.L.; Beral, V.; Snape, M.D.; Rees, H.; Ropero, A.-M.; et al. SARS-CoV-2 Variants and Vaccines. N. Engl. J. Med. 2021, 385, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Hall, V.J.; Foulkes, S.; Charlett, A.; Atti, A.; Monk, E.J.M.; Simmons, R.; Wellington, E.; Cole, M.J.; Saei, A.; Oguti, B.; et al. SARS-CoV-2 infection rates of antibody-positive compared with antibody-negative health-care workers in England: A large, multicentre, prospective cohort study (SIREN). Lancet 2021, 397, 1459–1469. [Google Scholar] [CrossRef]
- Da Rosa Mesquita, R.; Francelino Silva Junior, L.C.; Santos Santana, F.M.; Farias de Oliveira, T.; Campos Alcântara, R.; Monteiro Arnozo, G.; Rodrigues da Silva Filho, E.; Galdino dos Santos, A.G.; Oliveira da Cunha, E.J.; Salgueiro de Aquino, S.H.; et al. Clinical manifestations of COVID-19 in the general population: Systematic review. Wien. Klin. Wochenschr. 2021, 133, 377–382. [Google Scholar] [CrossRef]
- Napoli, C.; Benincasa, G.; Criscuolo, C.; Faenza, M.; Liberato, C.; Rusciano, M. Immune reactivity during COVID-19: Implications for treatment. Immunol. Lett. 2021, 231, 28–34. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef]
- Wilk, A.J.; Lee, M.J.; Wei, B.; Parks, B.; Pi, R.; Martínez-Colón, G.J.; Ranganath, T.; Zhao, N.Q.; Taylor, S.; Becker, W.; et al. Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19. J. Exp. Med. 2021, 218, e20210582. [Google Scholar] [CrossRef]
- Boscolo-Rizzo, P.; Guida, F.; Polesel, J.; Marcuzzo, A.V.; Capriotti, V.; D’Alessandro, A.; Zanelli, E.; Marzolino, R.; Lazzarin, C.; Antonucci, P.; et al. Sequelae in adults at 12 months after mild-to-moderate coronavirus disease 2019 (COVID-19). Int. Forum Allergy Rhinol. 2021, 1–4. [Google Scholar] [CrossRef]
- Logue, J.K.; Franko, N.M.; McCulloch, D.J.; McDonald, D.; Magedson, A.; Wolf, C.R.; Chu, H.Y. Sequelae in Adults at 6 Months After COVID-19 Infection. JAMA Netw. Open 2021, 4, e210830. [Google Scholar] [CrossRef]
- Seeßle, J.; Waterboer, T.; Hippchen, T.; Simon, J.; Kirchner, M.; Lim, A.; Müller, B.; Merle, U. Persistent Symptoms in Adult Patients 1 Year After Coronavirus Disease 2019 (COVID-19): A Prospective Cohort Study. Clin. Infect. Dis. 2021, ciab611. [Google Scholar] [CrossRef]
- Roussel, M.; Ferrant, J.; Reizine, F.; Le Gallou, S.; Dulong, J.; Carl, S.; Lesouhaitier, M.; Gregoire, M.; Bescher, N.; Verdy, C.; et al. Comparative immune profiling of acute respiratory distress syndrome patients with or without SARS-CoV-2 infection. Cell Rep. Med. 2021, 2, 100291. [Google Scholar] [CrossRef]
- Iannetta, M.; Buccisano, F.; Fraboni, D.; Malagnino, V.; Campogiani, L.; Teti, E.; Spalliera, I.; Rossi, B.; Di Lorenzo, A.; Palmieri, R.; et al. Baseline T-lymphocyte subset absolute counts can predict both outcome and severity in SARS-CoV-2 infected patients: A single center study. Sci. Rep. 2021, 11, 12762. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Moratto, D.; Chiarini, M.; Giustini, V.; Serana, F.; Magro, P.; Roccaro, A.M.; Imberti, L.; Castelli, F.; Notarangelo, L.D.; Quiros-Roldan, E. Flow Cytometry Identifies Risk Factors and Dynamic Changes in Patients with COVID-19. J. Clin. Immunol. 2020, 40, 970–973. [Google Scholar] [CrossRef]
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020, 5, eabd7114. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, eabc8511. [Google Scholar] [CrossRef]
- Long, Q.-X.; Liu, B.-Z.; Deng, H.-J.; Wu, G.-C.; Deng, K.; Chen, Y.-K.; Liao, P.; Qiu, J.-F.; Lin, Y.; Cai, X.-F.; et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat. Med. 2020, 26, 845–848. [Google Scholar] [CrossRef]
- Guo, L.; Ren, L.; Yang, S.; Xiao, M.; Chang, D.; Yang, F.; Dela Cruz, C.S.; Wang, Y.; Wu, C.; Xiao, Y.; et al. Profiling early humoral response to diagnose novel coronavirus disease (COVID-19). Clin. Infect. Dis. 2020, 71, 778–785. [Google Scholar] [CrossRef] [Green Version]
- Sterlin, D.; Mathian, A.; Miyara, M.; Mohr, A.; Anna, F.; Claër, L.; Quentric, P.; Fadlallah, J.; Devilliers, H.; Ghillani, P.; et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci. Transl. Med. 2021, 13, eabd2223. [Google Scholar] [CrossRef]
- Appay, V.; Dunbar, P.R.; Callan, M.; Klenerman, P.; Gillespie, G.M.A.; Papagno, L.; Ogg, G.S.; King, A.; Lechner, F.; Spina, C.A.; et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 2002, 8, 379–385. [Google Scholar] [CrossRef]
- Cooper, D.A.; Imrie, A.A.; Penny, R. Antibody Response to Human Immunodeficiency Virus After Primary Infection. J. Infect. Dis. 1987, 155, 1113–1118. [Google Scholar] [CrossRef]
- Hampl, H.; Kapprell, H.P.; Sawitzky, D.; Wilske, W.; Gürtler, L. Detection of specific human immunodeficiency virus IgM antibodies. Med. Microbiol. Immunol. 1995, 184, 69–71. [Google Scholar] [CrossRef]
- Green, A.M.; Harris, E. Monocyte-Plasmablast Crosstalk during Dengue. Cell Host Microbe 2014, 16, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Kazer, S.W.; Walker, B.D.; Shalek, A.K. Evolution and Diversity of Immune Responses during Acute HIV Infection. Immunity 2020, 53, 908–924. [Google Scholar] [CrossRef]
- Kwissa, M.; Nakaya, H.I.; Onlamoon, N.; Wrammert, J.; Villinger, F.; Perng, G.C.; Yoksan, S.; Pattanapanyasat, K.; Chokephaibulkit, K.; Ahmed, R.; et al. Dengue Virus Infection Induces Expansion of a CD14+CD16+ Monocyte Population that Stimulates Plasmablast Differentiation. Cell Host Microbe 2014, 16, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zhao, Y.-D.; Wang, X.-M. CXCL10 an important chemokine associated with cytokine storm in COVID-19 infected patients. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7497–7505. [Google Scholar] [CrossRef]
- Pasrija, R.; Naime, M. The deregulated immune reaction and cytokines release storm (CRS) in COVID-19 disease. Int. Immunopharmacol. 2021, 90, 107225. [Google Scholar] [CrossRef]
- Lei, R.; Mohan, C. Immunological Biomarkers of COVID-19. Crit. Rev. Immunol. 2020, 40, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, J.; Lord, J.M. Neutrophils and COVID-19: Active Participants and Rational Therapeutic Targets. Front. Immunol. 2021, 12, 2097. [Google Scholar] [CrossRef] [PubMed]
- Ferbas, J.; Kaplan, A.H.; Hausner, M.A.; Hultin, L.E.; Matud, J.L.; Liu, Z.; Panicali, D.L.; Nerng-Ho, H.; Detels, R.; Giorgi, J.V. Virus Burden in Long-Term Survivors of Human Immunodeficiency Virus (HIV) Infection Is a Determinant of Anti-HIV CD8+ Lymphocyte Activity. J. Infect. Dis. 1995, 172, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Eller, M.A.; Goonetilleke, N.; Tassaneetrithep, B.; Eller, L.A.; Costanzo, M.C.; Johnson, S.; Betts, M.R.; Krebs, S.J.; Slike, B.M.; Nitayaphan, S.; et al. Expansion of Inefficient HIV-Specific CD8 T Cells during Acute Infection. J. Virol. 2016, 90, 4005–4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Rha, M.-S.; Jeong, H.W.; Ko, J.-H.; Choi, S.J.; Seo, I.-H.; Lee, J.S.; Sa, M.; Kim, A.R.; Joo, E.-J.; Ahn, J.Y.; et al. PD-1-Expressing SARS-CoV-2-Specific CD8+ T Cells Are Not Exhausted, but Functional in Patients with COVID-19. Immunity 2021, 54, 44–52.e3. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [Green Version]
- Carsetti, R.; Quinti, I.; Locatelli, F. COVID-19—Pathogenesis and immunological findings across the clinical manifestation spectrum. Curr. Opin. Pulm. Med. 2021, 27, 193–198. [Google Scholar] [CrossRef]
- Humphries, F.; Shmuel-Galia, L.; Jiang, Z.; Wilson, R.; Landis, P.; Ng, S.L.; Parsi, K.M.; Maehr, R.; Cruz, J.; Morales-Ramos, A.; et al. A diamidobenzimidazole STING agonist protects against SARS-CoV-2 infection. Sci. Immunol. 2021, 6, eabi9002. [Google Scholar] [CrossRef]
- Bessière, P.; Wasniewski, M.; Picard-Meyer, E.; Servat, A.; Figueroa, T.; Foret-Lucas, C.; Coggon, A.; Lesellier, S.; Boué, F.; Cebron, N.; et al. Intranasal type I interferon treatment is beneficial only when administered before clinical signs onset in the SARS-CoV-2 hamster model. PLoS Pathog. 2021, 17, e1009427. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Controls (n = 19) | COVID-19 Patients (n = 22) | p-Value | |
|---|---|---|---|
| sex (male/female) | 9/10 | 11/11 | 1 * |
| median age (min–max) | 68 (22–89) | 57 (22–85) | 0.574 # |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, M.; Volzke, J.; Subin, B.; Schmidt, C.J.; Geerdes-Fenge, H.; Reisinger, E.C.; Müller-Hilke, B. Distinguishing Incubation and Acute Disease Stages of Mild-to-Moderate COVID-19. Viruses 2022, 14, 203. https://0-doi-org.brum.beds.ac.uk/10.3390/v14020203
Müller M, Volzke J, Subin B, Schmidt CJ, Geerdes-Fenge H, Reisinger EC, Müller-Hilke B. Distinguishing Incubation and Acute Disease Stages of Mild-to-Moderate COVID-19. Viruses. 2022; 14(2):203. https://0-doi-org.brum.beds.ac.uk/10.3390/v14020203
Chicago/Turabian StyleMüller, Michael, Johann Volzke, Behnam Subin, Christian Johann Schmidt, Hilte Geerdes-Fenge, Emil Christian Reisinger, and Brigitte Müller-Hilke. 2022. "Distinguishing Incubation and Acute Disease Stages of Mild-to-Moderate COVID-19" Viruses 14, no. 2: 203. https://0-doi-org.brum.beds.ac.uk/10.3390/v14020203