
A Viral Long Non-Coding RNA Protects against Cell Death during Human Cytomegalovirus Infection of CD14+ Monocytes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Human Cytomegaloviruses
2.3. Fluorescence Microscopy
2.4. Sodium Dodecyl Sulphate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blotting
3. Results
3.1. β2.7 Protects against Mitochondrial Stress in Infected Monocytes
3.2. β2.7 Protects against Apoptosis in HCMV Infected Monocytes
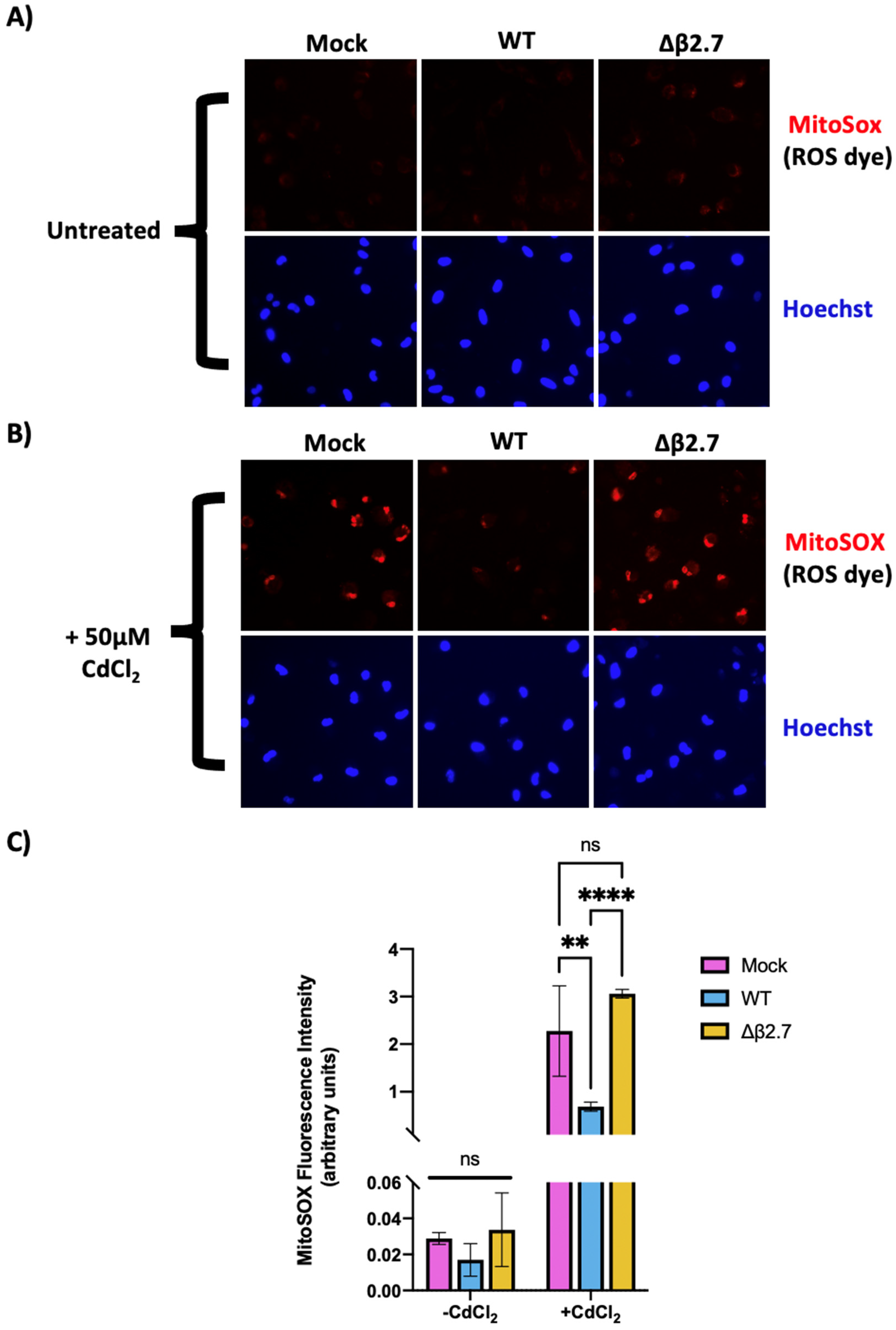
3.3. β2.7 Lowers ROS Levels in Infected Monocytes
3.4. Antioxidants or β2.7 Reduce ROS Levels Induced by Cadmium to Prevent Apoptosis
3.5. WT, but Not Δβ2.7, Virus Infection of Monocytes Upregulates the Antioxidant Enzyme, SOD2
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of Cytomegalovirus Seroprevalence and Demographic Characteristics Associated with Infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus Remains Latent in a Common Precursor of Dendritic and Myeloid Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of Endogenous Human Cytomegalovirus in CD34+ Bone Marrow Progenitors. J. Gen. Virol. 1996, 77 Pt 12, 3099–3102. [Google Scholar] [CrossRef] [PubMed]
- Söderberg-Nauclér, C.; Fish, K.N.; Nelson, J.A. Reactivation of Latent Human Cytomegalovirus by Allogeneic Stimulation of Blood Cells from Healthy Donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Wiedeman, J.; Sissons, J.G.; Borysiewicz, L.K.; Sinclair, J.H. Monocytes Are a Major Site of Persistence of Human Cytomegalovirus in Peripheral Blood Mononuclear Cells. J. Gen. Virol. 1991, 72 Pt 9, 2059–2064. [Google Scholar] [CrossRef]
- von Laer, D.; Meyer-Koenig, U.; Serr, A.; Finke, J.; Kanz, L.; Fauser, A.A.; Neumann-Haefelin, D.; Brugger, W.; Hufert, F.T. Detection of Cytomegalovirus DNA in CD34+ Cells from Blood and Bone Marrow. Blood 1995, 86, 4086–4090. [Google Scholar] [CrossRef] [Green Version]
- Collins-McMillen, D.; Chesnokova, L.; Lee, B.-J.; Fulkerson, H.L.; Brooks, R.; Mosher, B.S.; Yurochko, A.D. HCMV Infection and Apoptosis: How Do Monocytes Survive HCMV Infection? Viruses 2018, 10, 533. [Google Scholar] [CrossRef] [Green Version]
- Elder, E.; Sinclair, J. HCMV Latency: What Regulates the Regulators? Med. Microbiol. Immunol. 2019, 208, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Caviness, K.; Buehler, J.; Smithey, M.; Nikolich-Žugich, J.; Goodrum, F. Transcriptome-Wide Characterization of Human Cytomegalovirus in Natural Infection and Experimental Latency. Proc. Natl. Acad. Sci. USA 2017, 114, E10586–E10595. [Google Scholar] [CrossRef] [Green Version]
- Shnayder, M.; Nachshon, A.; Krishna, B.; Poole, E.; Boshkov, A.; Binyamin, A.; Maza, I.; Sinclair, J.; Schwartz, M.; Stern-Ginossar, N. Defining the Transcriptional Landscape during Cytomegalovirus Latency with Single-Cell RNA Sequencing. mBio 2018, 9, e00013-18. [Google Scholar] [CrossRef] [Green Version]
- Rossetto, C.C.; Tarrant-Elorza, M.; Pari, G.S. Cis and Trans Acting Factors Involved in Human Cytomegalovirus Experimental and Natural Latent Infection of CD14 (+) Monocytes and CD34 (+) Cells. PLoS Pathog. 2013, 9, e1003366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatherer, D.; Seirafian, S.; Cunningham, C.; Holton, M.; Dargan, D.J.; Baluchova, K.; Hector, R.D.; Galbraith, J.; Herzyk, P.; Wilkinson, G.W.G.; et al. High-Resolution Human Cytomegalovirus Transcriptome. Proc. Natl. Acad. Sci. USA 2011, 108, 19755–19760. [Google Scholar] [CrossRef] [Green Version]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I Binding by a Virally Encoded RNA Regulates Mitochondria-Induced Cell Death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef] [Green Version]
- Poole, E.; Reeves, M.; Sinclair, J.H. The Use of Primary Human Cells (Fibroblasts, Monocytes, and Others) to Assess Human Cytomegalovirus Function. In Methods in Molecular Biology (Clifton, N.J.); Humana Press: Totowa, NJ, USA, 2014; Volume 1119, pp. 81–98. [Google Scholar]
- McSharry, B.P.; Tomasec, P.; Neale, M.L.; Wilkinson, G.W.G. The Most Abundantly Transcribed Human Cytomegalovirus Gene (Beta 2.7) Is Non-Essential for Growth in Vitro. J. Gen. Virol. 2003, 84, 2511–2516. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Murphy, E.A. A Myeloid Progenitor Cell Line Capable of Supporting Human Cytomegalovirus Latency and Reactivation, Resulting in Infectious Progeny. J. Virol. 2012, 86, 9854–9865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.; Vaníček, J.; Robins, H.; Shenk, T.; Levine, A.J. Suppression of Immediate-Early Viral Gene Expression by Herpesvirus-Coded MicroRNAs: Implications for Latency. Proc. Natl. Acad. Sci. USA 2008, 105, 5453–5458. [Google Scholar] [CrossRef] [Green Version]
- Warming, S.; Costantino, N.; Court, D.L.; Jenkins, N.A.; Copeland, N.G. Simple and Highly Efficient BAC Recombineering Using GalK Selection. Nucleic Acids Res. 2005, 33, e36. [Google Scholar] [CrossRef]
- Lau, B.; Poole, E.; Van Damme, E.; Bunkens, L.; Sowash, M.; King, H.; Murphy, E.; Wills, M.; Van Loock, M.; Sinclair, J. Human Cytomegalovirus MiR-UL112-1 Promotes the down-Regulation of Viral Immediate Early-Gene Expression during Latency to Prevent T-Cell Recognition of Latently Infected Cells. J. Gen. Virol. 2016, 97, 2387–2398. [Google Scholar] [CrossRef]
- Elder, E.G.; Krishna, B.A.; Poole, E.; Perera, M.; Sinclair, J. Regulation of Host and Viral Promoters during Human Cytomegalovirus Latency via US28 and CTCF. J. Gen. Virol. 2021, 102, 001609. [Google Scholar] [CrossRef]
- Alkharashi, N.A.O.; Periasamy, V.S.; Athinarayanan, J.; Alshatwi, A.A. Cadmium Triggers Mitochondrial Oxidative Stress in Human Peripheral Blood Lymphocytes and Monocytes: Analysis Using in Vitro and System Toxicology Approaches. J. Trace Elem. Med. Biol. Organ. Soc. Miner. Trace Elem. GMS 2017, 42, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Fuente, H.D.L.; Portales-Perez, D.; Baranda, L.; Diaz-Barriga, F.; Saavedra-Alanis, V.; Layseca, E.; González-Amaro, R. Effect of Arsenic, Cadmium and Lead on the Induction of Apoptosis of Normal Human Mononuclear Cells. Clin. Exp. Immunol. 2002, 129, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kondo, T.; Zhao, Q.-L.; Li, F.-J.; Tanabe, K.; Arai, Y.; Zhou, Z.-C.; Kasuya, M. Apoptosis Induced by Cadmium in Human Lymphoma U937 Cells through Ca2+-Calpain and Caspase-Mitochondria- Dependent Pathways *. J. Biol. Chem. 2000, 275, 39702–39709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olszowski, T.; Baranowska-Bosiacka, I.; Gutowska, I.; Piotrowska, K.; Mierzejewska, K.; Korbecki, J.; Kurzawski, M.; Tarnowski, M.; Chlubek, D. The Effects of Cadmium at Low Environmental Concentrations on THP-1 Macrophage Apoptosis. Int. J. Mol. Sci. 2015, 16, 21410–21427. [Google Scholar] [CrossRef] [Green Version]
- Hossein-Khannazer, N.; Azizi, G.; Eslami, S.; Mohammed, H.A.; Fayyaz, F.; Hosseinzadeh, R.; Usman, A.B.; Kamali, A.N.; Mohammadi, H.; Jadidi-Niaragh, F.; et al. The Effects of Cadmium Exposure in the Induction of Inflammation. Immunopharmacol. Immunotoxicol. 2020, 42, 1–8. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial Membrane Potential Probes and the Proton Gradient: A Practical Usage Guide. BioTechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Elder, E.; Krishna, B.; Williamson, J.; Aslam, Y.; Farahi, N.; Wood, A.; Romashova, V.; Roche, K.; Murphy, E.; Chilvers, E.; et al. Monocytes Latently Infected with Human Cytomegalovirus Evade Neutrophil Killing. iScience 2019, 12, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, J.; Sissons, P. Latency and Reactivation of Human Cytomegalovirus. J. Gen. Virol. 2006, 87, 1763–1779. [Google Scholar] [CrossRef]
- Kew, V.G.; Wills, M.R.; Reeves, M.B. LPS Promotes a Monocyte Phenotype Permissive for Human Cytomegalovirus Immediate-Early Gene Expression upon Infection but Not Reactivation from Latency. Sci. Rep. 2017, 7, 810. [Google Scholar] [CrossRef]
- Hargett, D.; Shenk, T.E. Experimental Human Cytomegalovirus Latency in CD14+ Monocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 20039–20044. [Google Scholar] [CrossRef] [Green Version]
- Goodrum, F.D.; Jordan, C.T.; High, K.; Shenk, T. Human Cytomegalovirus Gene Expression during Infection of Primary Hematopoietic Progenitor Cells: A Model for Latency. Proc. Natl. Acad. Sci. USA 2002, 99, 16255–16260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, J.L.; Slobedman, B. Human Cytomegalovirus Latent Infection of Myeloid Cells Directs Monocyte Migration by Up-Regulating Monocyte Chemotactic Protein-1. J. Immunol. 2008, 180, 6577–6585. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.; Reeves, M.; Sinclair, J.; High, K.; Shenk, T. Human Cytomegalovirus Sequences Expressed in Latently Infected Individuals Promote a Latent Infection in Vitro. Blood 2007, 110, 937–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Gao, Y.; Zhou, Q.; Zhang, Q.; Peng, Y.; Tian, K.; Wang, J.; Zheng, X. Human Cytomegalovirus Latent Infection Alters the Expression of Cellular and Viral MicroRNA. Gene 2014, 536, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Song, X.; Ma, P.; Lv, L.; Zhang, Y.; Deng, J.; Zhang, Y. Human Cytomegalovirus MiR-US33as-5p Targets IFNAR1 to Achieve Immune Evasion During Both Lytic and Latent Infection. Front. Immunol. 2021, 12, 461. [Google Scholar] [CrossRef]
- Jenkins, C.; Abendroth, A.; Slobedman, B. A Novel Viral Transcript with Homology to Human Interleukin-10 Is Expressed during Latent Human Cytomegalovirus Infection. J. Virol. 2004, 78, 1440–1447. [Google Scholar] [CrossRef] [Green Version]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The Mitochondrial Membrane Potential (Δψm) in Apoptosis; an Update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef]
- Zhao, J.; Sinclair, J.; Houghton, J.; Bolton, E.; Bradley, A.; Lever, A. Cytomegalovirus Β2.7 RNA Transcript Protects Endothelial Cells against Apoptosis during Ischemia/Reperfusion Injury. J. Heart Lung Transplant. 2010, 29, 342–345. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015; ISBN 978-0-19-871748-5. [Google Scholar]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Dupré-Crochet, S.; Erard, M.; Nüβe, O. ROS Production in Phagocytes: Why, When, and Where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.S.; Gupta Vallur, P.; Phaëton, R.; Mythreye, K.; Hempel, N. Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxid. Basel Switz. 2017, 6, 86. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.; Nogalski, M.T.; Bentz, G.L.; Smith, M.S.; Parmater, A.; Yurochko, A.D. PI3K-Dependent Upregulation of Mcl-1 by Human Cytomegalovirus Is Mediated by Epidermal Growth Factor Receptor and Inhibits Apoptosis in Short-Lived Monocytes. J. Immunol. 2010, 184, 3213–3222. [Google Scholar] [CrossRef] [Green Version]
- Krishna, B.A.; Spiess, K.; Poole, E.L.; Lau, B.; Voigt, S.; Kledal, T.N.; Rosenkilde, M.M.; Sinclair, J.H. Targeting the Latent Cytomegalovirus Reservoir with an Antiviral Fusion Toxin Protein. Nat. Commun. 2017, 8, 14321. [Google Scholar] [CrossRef] [Green Version]
- Poole, E.; Lau, J.C.H.; Sinclair, J. Latent Infection of Myeloid Progenitors by Human Cytomegalovirus Protects Cells from FAS-Mediated Apoptosis through the Cellular IL-10/PEA-15 Pathway. J. Gen. Virol. 2015, 96, 2355–2359. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Elner, S.G.; Bian, Z.-M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-Inflammatory Cytokines Increase Reactive Oxygen Species through Mitochondria and NADPH Oxidase in Cultured RPE Cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.-Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial Reactive Oxygen Species Promote Production of Proinflammatory Cytokines and Are Elevated in TNFR1-Associated Periodic Syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell. Longev. 2016, 2016, e2795090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.-G. ROS Play a Critical Role in the Differentiation of Alternatively Activated Macrophages and the Occurrence of Tumor-Associated Macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Choksi, S.; Qu, J.; Jang, J.; Choe, M.; Banfi, B.; Engelhardt, J.F.; Liu, Z. NADPH Oxidases Are Essential for Macrophage Differentiation *. J. Biol. Chem. 2016, 291, 20030–20041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Prete, A.; Zaccagnino, P.; Di Paola, M.; Saltarella, M.; Oliveros Celis, C.; Nico, B.; Santoro, G.; Lorusso, M. Role of Mitochondria and Reactive Oxygen Species in Dendritic Cell Differentiation and Functions. Free Radic. Biol. Med. 2008, 44, 1443–1451. [Google Scholar] [CrossRef] [Green Version]
- Robins, M.; Wyatt, H.A. Hyperbaric Treatment of Ischemia Reperfusion Injury. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Granger, D.N.; Kvietys, P.R. Reperfusion Injury and Reactive Oxygen Species: The Evolution of a Concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Speir, E.; Shibutani, T.; Yu, Z.X.; Ferrans, V.; Epstein, S.E. Role of Reactive Oxygen Intermediates in Cytomegalovirus Gene Expression and in the Response of Human Smooth Muscle Cells to Viral Infection. Circ. Res. 1996, 79, 1143–1152. [Google Scholar] [CrossRef]
- Combs, J.A.; Norton, E.B.; Saifudeen, Z.R.; Bentrup, K.H.Z.; Katakam, P.V.; Morris, C.A.; Myers, L.; Kaur, A.; Sullivan, D.E.; Zwezdaryk, K.J. Human Cytomegalovirus Alters Host Cell Mitochondrial Function during Acute Infection. J. Virol. 2020, 94, e01183-19. [Google Scholar] [CrossRef] [Green Version]
- Karniely, S.; Weekes, M.P.; Antrobus, R.; Rorbach, J.; van Haute, L.; Umrania, Y.; Smith, D.L.; Stanton, R.J.; Minczuk, M.; Lehner, P.J.; et al. Human Cytomegalovirus Infection Upregulates the Mitochondrial Transcription and Translation Machineries. mBio 2016, 7, e00029-16. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-Interacting Protein Links Oxidative Stress to Inflammasome Activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Dosunmu-Ogunbi, A.M.; Wood, K.C.; Novelli, E.M.; Straub, A.C. Decoding the Role of SOD2 in Sickle Cell Disease. Blood Adv. 2019, 3, 2679–2687. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Avdic, S.; Hodkinson, J.; Jackson, S.; Wills, M.; Slobedman, B.; Sinclair, J. Latency-Associated Viral Interleukin-10 (IL-10) Encoded by Human Cytomegalovirus Modulates Cellular IL-10 and CCL8 Secretion during Latent Infection through Changes in the Cellular MicroRNA Hsa-MiR-92a. J. Virol. 2014, 88, 13947–13955. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.E.; Chen, K.C.; Groves, I.J.; Sedikides, G.X.; Gandhi, A.; Houldcroft, C.J.; Poole, E.L.; Montanuy, I.; Mason, G.M.; Okecha, G.; et al. Latent Cytomegalovirus-Driven Recruitment of Activated CD4+ T Cells Promotes Virus Reactivation. Front. Immunol. 2021, 12, 657945. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene Regulation by Long Non-Coding RNAs and Its Biological Functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Ponath, V.; Kaina, B. Death of Monocytes through Oxidative Burst of Macrophages and Neutrophils: Killing in Trans. PLoS ONE 2017, 12, e0170347. [Google Scholar] [CrossRef]
- Dahlgren, C.; Karlsson, A.; Bylund, J. Intracellular Neutrophil Oxidants: From Laboratory Curiosity to Clinical Reality. J. Immunol. 2019, 202, 3127–3134. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| galK insertion F | CATCCCAAGCACTCCACACGCTATCACAGACCACGGACACGGCAAAAAATCCTGTTGACAATTAATCATCGGCA |
| galK insertion R | ACGTCTTTCCGCTTACTCAACGCGTCAGCCCGCGCTCGGCAGAGCTACCATCAGCACTGTCCTGCTCCTT |
| Reversion primer F | CAAGCACTCCACACGCTATCACAGACCACGGACACGGCAAAAAATTGGTAGCTCTGCCGAGCGCGGGCTGACGCGTTGAGTAAGCGGAAA |
| Reversion primer R | TTTCCGCTTACTCAACGCGTCAGCCCGCGCTCGGCAGAGCTACCAATTTTTTGCCGTGTCCGTGGTCTGTGATAGCGTGTGGAGTGCTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, M.R.; Roche, K.L.; Murphy, E.A.; Sinclair, J.H. A Viral Long Non-Coding RNA Protects against Cell Death during Human Cytomegalovirus Infection of CD14+ Monocytes. Viruses 2022, 14, 246. https://0-doi-org.brum.beds.ac.uk/10.3390/v14020246
Perera MR, Roche KL, Murphy EA, Sinclair JH. A Viral Long Non-Coding RNA Protects against Cell Death during Human Cytomegalovirus Infection of CD14+ Monocytes. Viruses. 2022; 14(2):246. https://0-doi-org.brum.beds.ac.uk/10.3390/v14020246
Chicago/Turabian StylePerera, Marianne R., Kathryn L. Roche, Eain A. Murphy, and John H. Sinclair. 2022. "A Viral Long Non-Coding RNA Protects against Cell Death during Human Cytomegalovirus Infection of CD14+ Monocytes" Viruses 14, no. 2: 246. https://0-doi-org.brum.beds.ac.uk/10.3390/v14020246