
Low Intrahost and Interhost Genetic Diversity of Carnivore Protoparvovirus 1 in Domestic Cats during a Feline Panleukopenia Outbreak

 ,
,  , , , and
, , , and 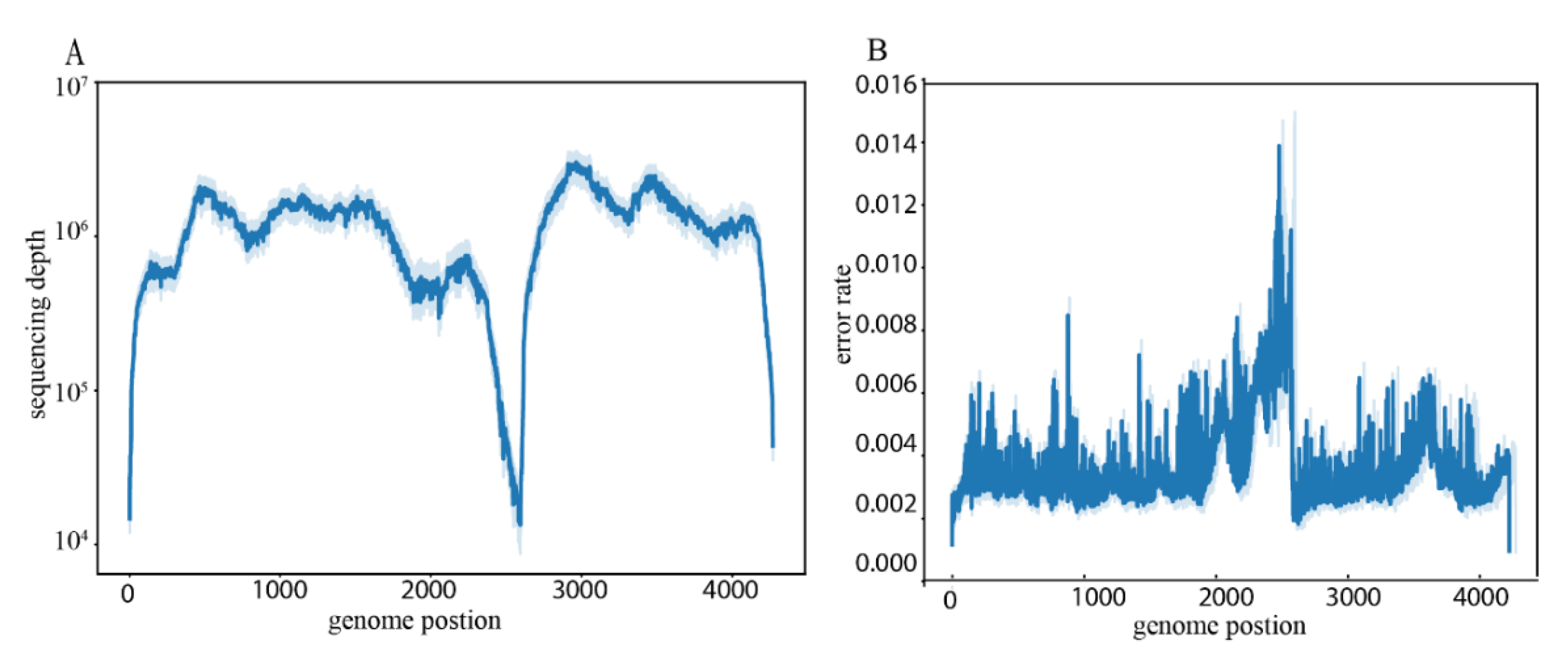{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, Baiting and Sequencing
2.3. Detection and Characterisation of FPV and CPV
2.4. Evolutionary Analysis
2.5. Nucleotide Diversity Analysis
3. Results
3.1. Overview
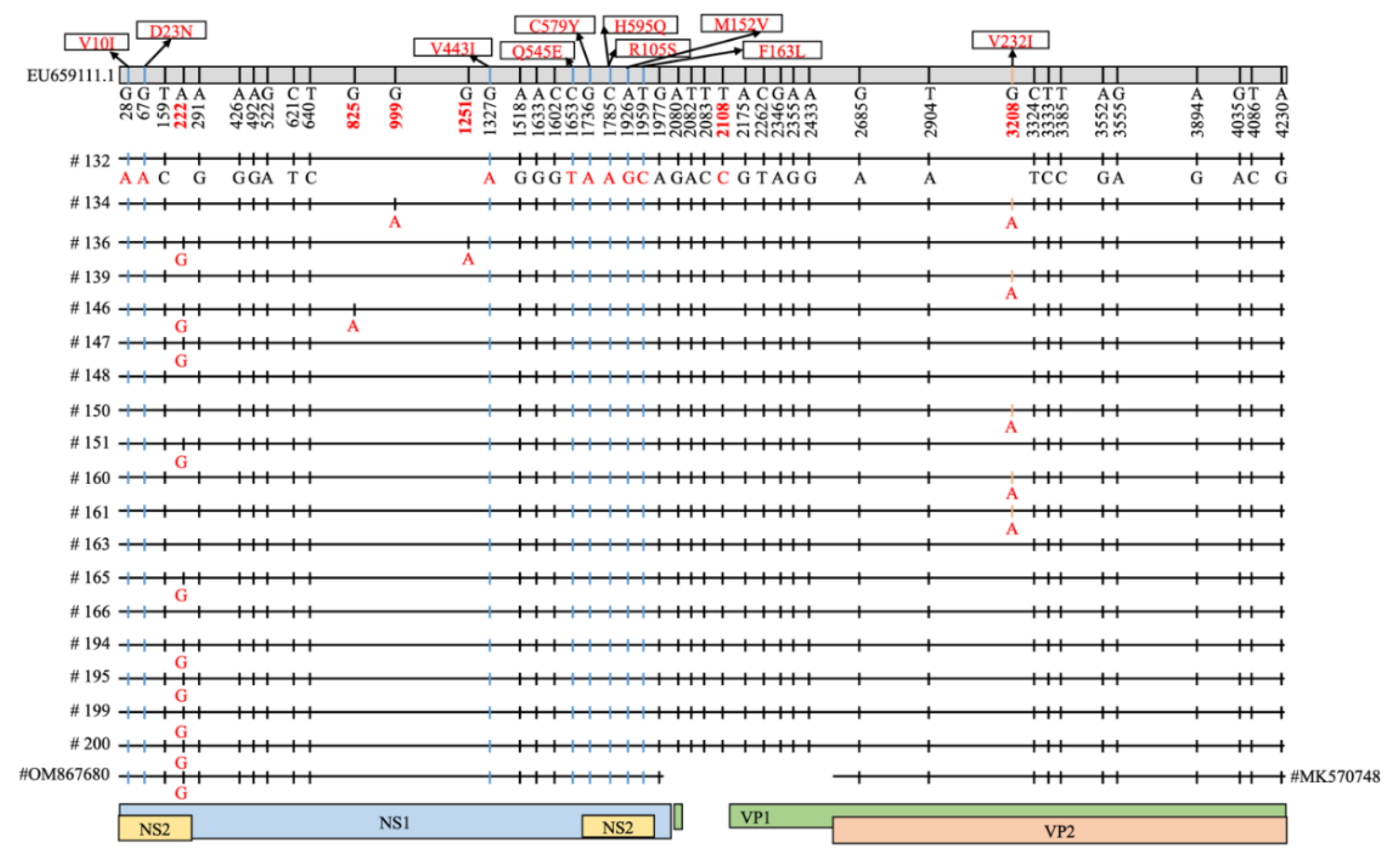
3.2. Characterization of Carnivore Protoparvovirus 1 and Mutation Analysis
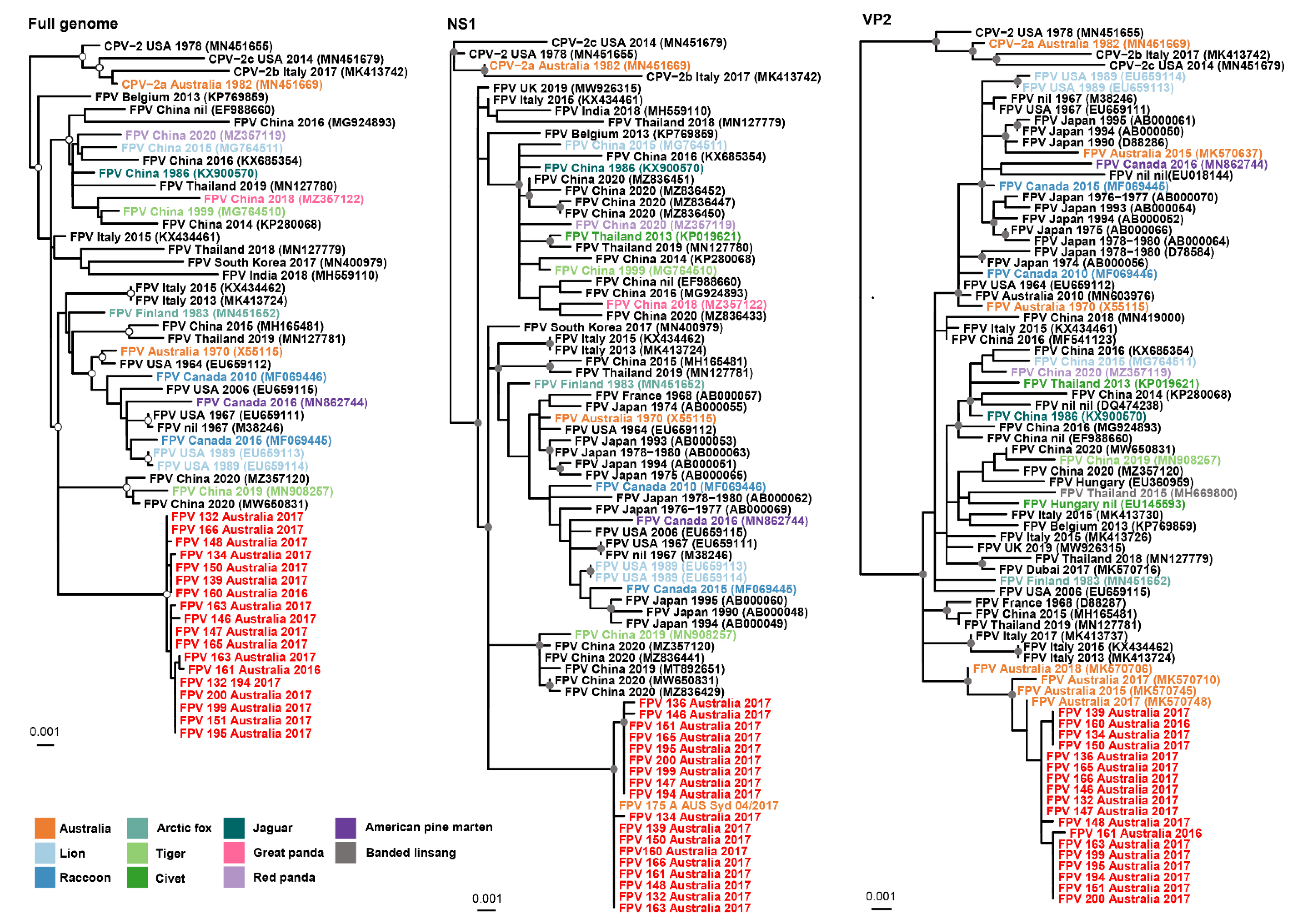
3.3. Interhost FPV Diversity
3.4. Evolutionary Analysis
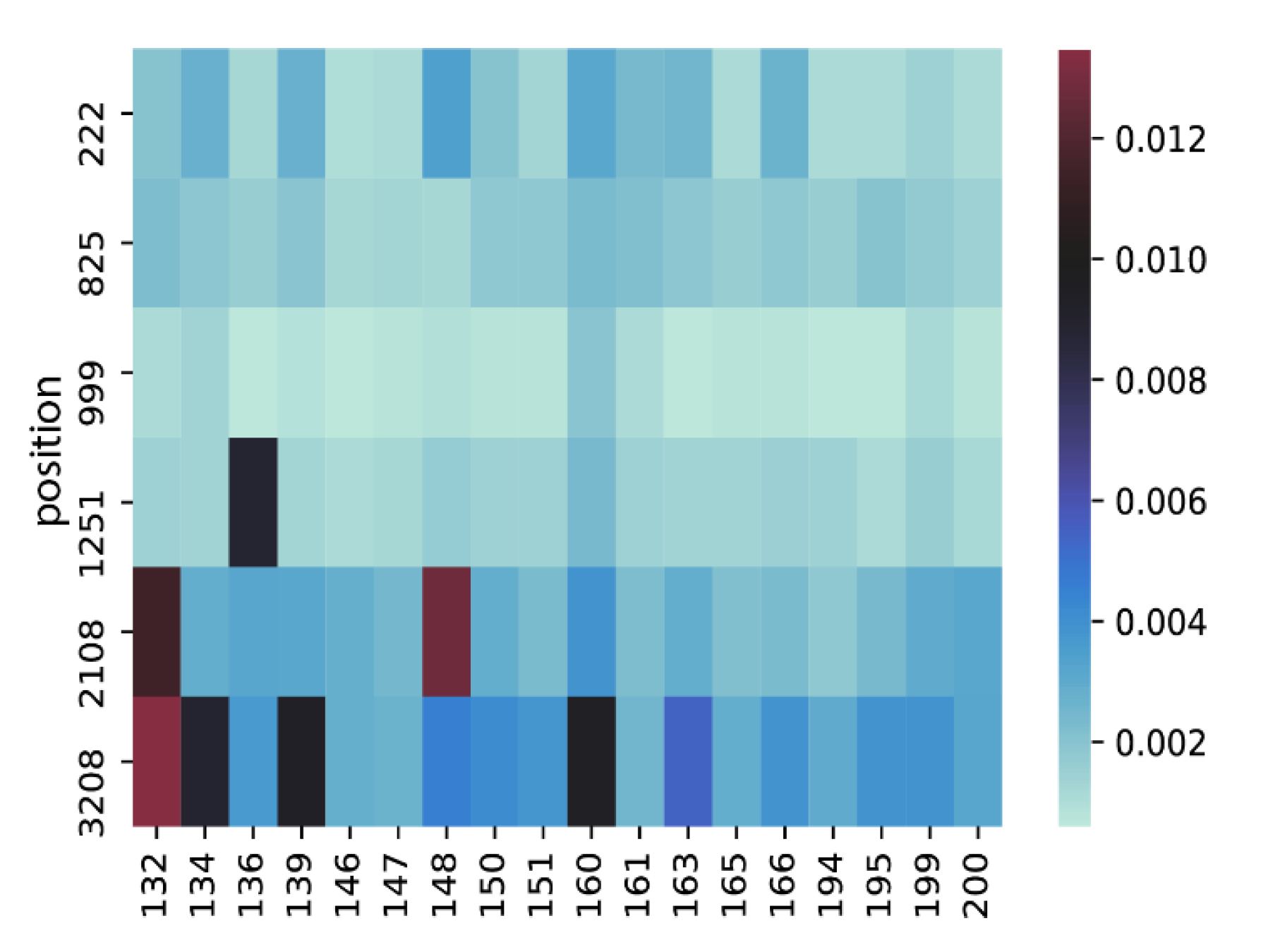
3.5. Intrahost Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J. ICTV virus taxonomy profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367. [Google Scholar] [CrossRef] [PubMed]
- Barrs, V.R. Feline Panleukopenia: A Re-emergent Disease. Vet. Clin. N. Am. Small Anim. Pract. 2019, 49, 651–670. [Google Scholar] [CrossRef] [PubMed]
- Van Brussel, K.; Carrai, M.; Lin, C.; Kelman, M.; Setyo, L.; Aberdein, D.; Brailey, J.; Lawler, M.; Maher, S.; Plaganyi, I. Distinct lineages of feline parvovirus associated with epizootic outbreaks in Australia, New Zealand and the United Arab Emirates. Viruses 2019, 11, 1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, E.; Davis, C.; Carrai, M.; Ward, M.P.; O’Keeffe, S.; van Boeijen, M.; Beveridge, L.; Desario, C.; Buonavoglia, C.; Beatty, J.A. Feline parvovirus seroprevalence is high in domestic cats from disease outbreak and non-outbreak regions in Australia. Viruses 2020, 12, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochizuki, M.; Horiuchi, M.; Hiragi, H.; San Gabriel, M.C.; Yasuda, N.; Uno, T. Isolation of canine parvovirus from a cat manifesting clinical signs of feline panleukopenia. J. Clin. Microbiol. 1996, 34, 2101–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, Y.; Nakamura, K.; Miyazawa, T.; Tohya, Y.; Takahashi, E.; Mochizuki, M. Feline host range of canine parvovirus: Recent emergence of new antigenic types in cats. Emerg. Infect. Dis. 2002, 8, 341. [Google Scholar] [CrossRef]
- Gamoh, K.; Shimazaki, Y.; Makie, H.; Senda, M.; Itoh, O.; Inoue, Y. The pathogenicity of canine parvovirus type-2b, FP84 strain isolated from a domestic cat, in domestic cats. J. Vet. Med. Sci. 2003, 65, 1027–1029. [Google Scholar] [CrossRef] [Green Version]
- Decaro, N.; Desario, C.; Amorisco, F.; Losurdo, M.; Colaianni, M.L.; Greco, M.F.; Buonavoglia, C. Canine parvovirus type 2c infection in a kitten associated with intracranial abscess and convulsions. J. Feline Med. Surg. 2011, 13, 231–236. [Google Scholar] [CrossRef]
- Hueffer, K.; Govindasamy, L.; Agbandje-McKenna, M.; Parrish, C.R. Combinations of two capsid regions controlling canine host range determine canine transferrin receptor binding by canine and feline parvoviruses. J. Virol. 2003, 77, 10099–10105. [Google Scholar] [CrossRef] [Green Version]
- Parrish, C.R.; Have, P.; Foreyt, W.J.; Evermann, J.F.; Senda, M.; Carmichael, L.E. The global spread and replacement of canine parvovirus strains. J. Gen. Virol. 1988, 69, 1111–1116. [Google Scholar] [CrossRef]
- Parrish, C.R.; O’Connell, P.H.; Evermann, J.F.; Carmichael, L.E. Natural variation of canine parvovirus. Science 1985, 230, 1046–1048. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R.; Aquadro, C.F.; Strassheim, M.; Evermann, J.; Sgro, J.; Mohammed, H. Rapid antigenic-type replacement and DNA sequence evolution of canine parvovirus. J. Virol. 1991, 65, 6544–6552. [Google Scholar] [CrossRef] [Green Version]
- Buonavoglia, C.; Martella, V.; Pratelli, A.; Tempesta, M.; Cavalli, A.; Buonavoglia, D.; Bozzo, G.; Elia, G.; Decaro, N.; Carmichael, L. Evidence for evolution of canine parvovirus type 2 in Italy. J. Gen. Virol. 2001, 82, 3021–3025. [Google Scholar] [CrossRef] [PubMed]
- Balboni, A.; Bassi, F.; De Arcangeli, S.; Zobba, R.; Dedola, C.; Alberti, A.; Battilani, M. Molecular analysis of Carnivore Protoparvovirus detected in white blood cells of naturally infected cats. BMC Vet. Res. 2018, 14, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battilani, M.; Balboni, A.; Ustulin, M.; Giunti, M.; Scagliarini, A.; Prosperi, S. Genetic complexity and multiple infections with more Parvovirus species in naturally infected cats. Vet. Res. 2011, 42, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battilani, M.; Gallina, L.; Vaccari, F.; Morganti, L. Co-infection with multiple variants of canine parvovirus type 2 (CPV-2). Vet. Res. Commun. 2007, 31, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Battilani, M.; Balboni, A.; Giunti, M.; Prosperi, S. Co-infection with feline and canine parvovirus in a cat. Vet. Ital. 2013, 49, 127–129. [Google Scholar]
- Tang, P.; Chiu, C. Metagenomics for the discovery of novel human viruses. Future Microbiol. 2010, 5, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.Y. Viral pathogen discovery. Curr. Opin. Microbiol. 2013, 16, 468–478. [Google Scholar] [CrossRef] [Green Version]
- Pallen, M. Diagnostic metagenomics: Potential applications to bacterial, viral and parasitic infections. Parasitology 2014, 141, 1856–1862. [Google Scholar] [CrossRef] [Green Version]
- Conceição-Neto, N.; Zeller, M.; Lefrère, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Van Ranst, M. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, R.; Shi, M.; Grueber, C.E.; Holmes, E.C.; Hogg, C.J.; Belov, K.; Barrs, V.R. Fecal viral diversity of captive and wild Tasmanian devils characterized using virion-enriched metagenomics and metatranscriptomics. J. Virol. 2019, 93, e00205–e00219. [Google Scholar] [CrossRef] [Green Version]
- Van Brussel, K.; Wang, X.; Shi, M.; Carrai, M.; Feng, S.; Li, J.; Holmes, E.; Beatty, J.; Barrs, V.R. The enteric virome of cats with feline panleukopenia differs in abundance and diversity from healthy cats. Transbound. Emerg. Dis. 2022. [CrossRef]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5448. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Rettedal, E.A.; Van Der Helm, E.; Ellabaan, M.; Panagiotou, G.; Sommer, M.O. Antibiotic treatment drives the diversification of the human gut resistome. Genom. Proteom. Bioinform. 2019, 17, 39–51. [Google Scholar] [CrossRef]
- Zheng, T.; Li, J.; Ni, Y.; Kang, K.; Misiakou, M.-A.; Imamovic, L.; Chow, B.K.; Rode, A.A.; Bytzer, P.; Sommer, M. Mining, analyzing, and integrating viral signals from metagenomic data. Microbiome 2019, 7, 42. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Kwan, E.; Carrai, M.; Lanave, G.; Hill, J.; Parry, K.; Kelman, M.; Meers, J.; Decaro, N.; Beatty, J.A.; Martella, V. Analysis of canine parvoviruses circulating in Australia reveals predominance of variant 2b and identifies feline parvovirus-like mutations in the capsid proteins. Transbound. Emerg. Dis. 2021, 68, 656–666. [Google Scholar] [CrossRef]
- Ogbu, K.I.; Mira, F.; Purpari, G.; Nwosuh, C.; Loria, G.R.; Schirò, G.; Chiaramonte, G.; Tion, M.T.; Di Bella, S.; Ventriglia, G. Nearly full-length genome characterization of canine parvovirus strains circulating in Nigeria. Transbound. Emerg. Dis. 2020, 67, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Voorhees, I.E.; Lee, H.; Allison, A.B.; Lopez-Astacio, R.; Goodman, L.B.; Oyesola, O.O.; Omobowale, O.; Fagbohun, O.; Dubovi, E.J.; Hafenstein, S.L. Limited intrahost diversity and background evolution accompany 40 years of canine parvovirus host adaptation and spread. J. Virol. 2019, 94, e01162-19. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Rodriguez-Mueller, B.; Garud, N.; Pollard, K.S. An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res. 2016, 26, 1612–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Hoelzer, K.; Shackelton, L.A.; Holmes, E.C.; Parrish, C.R. Within-host genetic diversity of endemic and emerging parvoviruses of dogs and cats. J. Virol. 2008, 82, 11096–11105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, M.; Wu, C.-N.; Lin, C.-F.; Nguyen, H.T.T.; Chiou, M.-T.; Lin, C.-N. Genetic characterization of feline panleukopenia virus from dogs in Vietnam reveals a unique Thr101 mutation in VP2. PeerJ 2020, 8, e9752. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, Q.M.K.; Alam, S.; Chowdhury, M.S.R.; Hasan, M.; Uddin, M.B.; Hossain, M.M.; Islam, M.R.; Rahman, M.M.; Rahman, M.M. First molecular characterization and phylogenetic analysis of the VP2 gene of feline panleukopenia virus in Bangladesh. Arch. Virol. 2021, 166, 2273–2278. [Google Scholar] [CrossRef] [PubMed]
- Mira, F.; Canuti, M.; Purpari, G.; Cannella, V.; Di Bella, S.; Occhiogrosso, L.; Schirò, G.; Chiaramonte, G.; Barreca, S.; Pisano, P. Molecular characterization and evolutionary analyses of Carnivore protoparvovirus 1 NS1 gene. Viruses 2019, 11, 308. [Google Scholar] [CrossRef] [Green Version]
- Allison, A.B.; Kohler, D.J.; Ortega, A.; Hoover, E.A.; Grove, D.M.; Holmes, E.C.; Parrish, C.R. Host-specific parvovirus evolution in nature is recapitulated by in vitro adaptation to different carnivore species. PLoS Pathog. 2014, 10, e1004475. [Google Scholar] [CrossRef]
- Chang, A.-M.; Chen, C.-C. Molecular characteristics of Carnivore protoparvovirus 1 with high sequence similarity between wild and domestic carnivores in Taiwan. Pathogens 2021, 10, 671. [Google Scholar] [CrossRef]
- Carrai, M.; Decaro, N.; Van Brussel, K.; Dall’Ara, P.; Desario, C.; Fracasso, M.; Šlapeta, J.; Colombo, E.; Bo, S.; Beatty, J.A. Canine parvovirus is shed infrequently by cats without diarrhoea in multi-cat environments. Vet. Microbiol. 2021, 261, 109204. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Carrai, M.; Van Brussel, K.; Feng, S.; Beatty, J.A.; Shi, M.; Holmes, E.C.; Li, J.; Barrs, V.R. Low Intrahost and Interhost Genetic Diversity of Carnivore Protoparvovirus 1 in Domestic Cats during a Feline Panleukopenia Outbreak. Viruses 2022, 14, 1412. https://0-doi-org.brum.beds.ac.uk/10.3390/v14071412
Wang X, Carrai M, Van Brussel K, Feng S, Beatty JA, Shi M, Holmes EC, Li J, Barrs VR. Low Intrahost and Interhost Genetic Diversity of Carnivore Protoparvovirus 1 in Domestic Cats during a Feline Panleukopenia Outbreak. Viruses. 2022; 14(7):1412. https://0-doi-org.brum.beds.ac.uk/10.3390/v14071412
Chicago/Turabian StyleWang, Xiuwan, Maura Carrai, Kate Van Brussel, Shuo Feng, Julia A. Beatty, Mang Shi, Edward C. Holmes, Jun Li, and Vanessa R. Barrs. 2022. "Low Intrahost and Interhost Genetic Diversity of Carnivore Protoparvovirus 1 in Domestic Cats during a Feline Panleukopenia Outbreak" Viruses 14, no. 7: 1412. https://0-doi-org.brum.beds.ac.uk/10.3390/v14071412