
A Single Oral Immunization with Replication-Competent Adenovirus-Vectored Vaccine Induces a Neutralizing Antibody Response in Mice against Canine Distemper Virus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
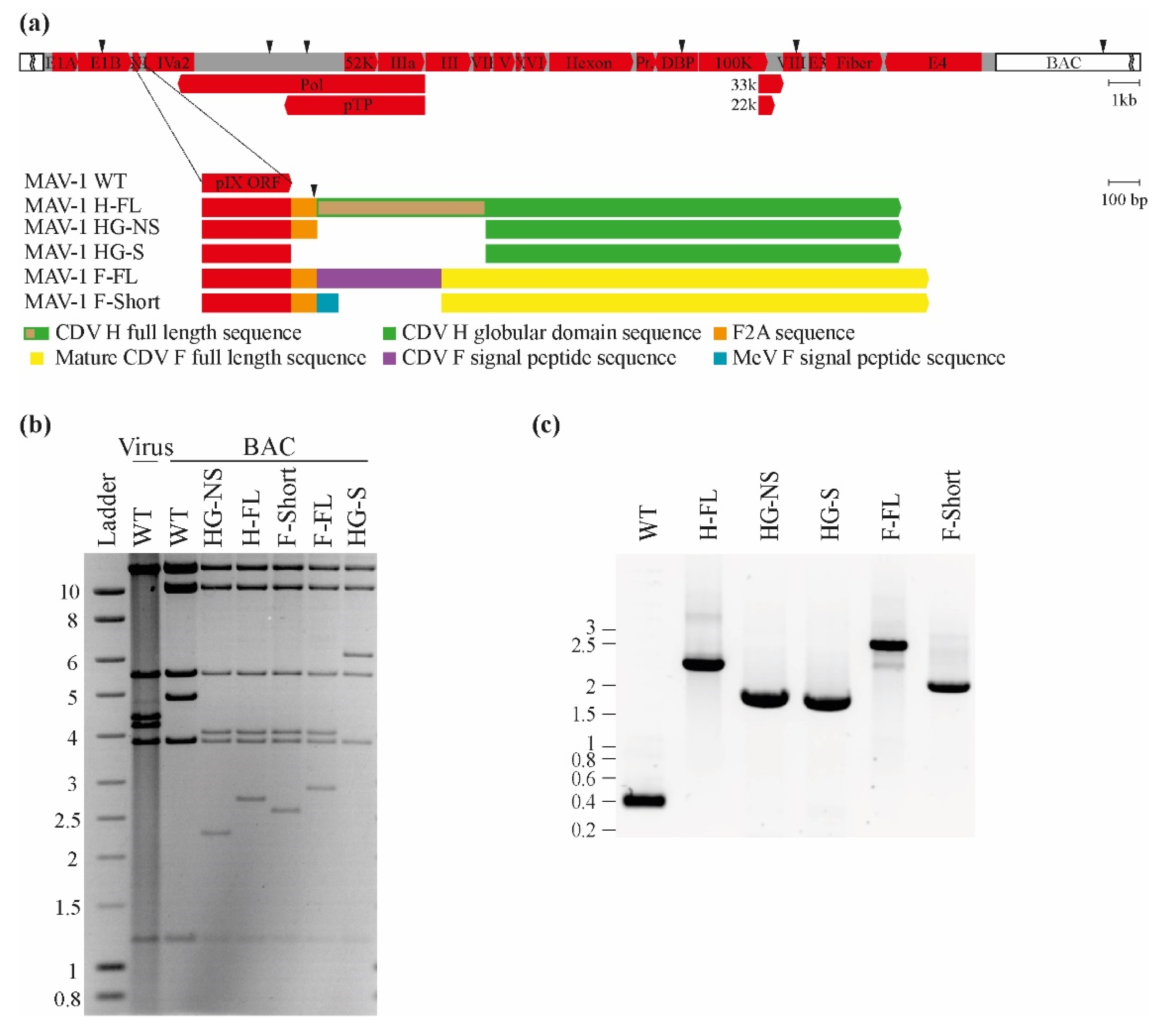
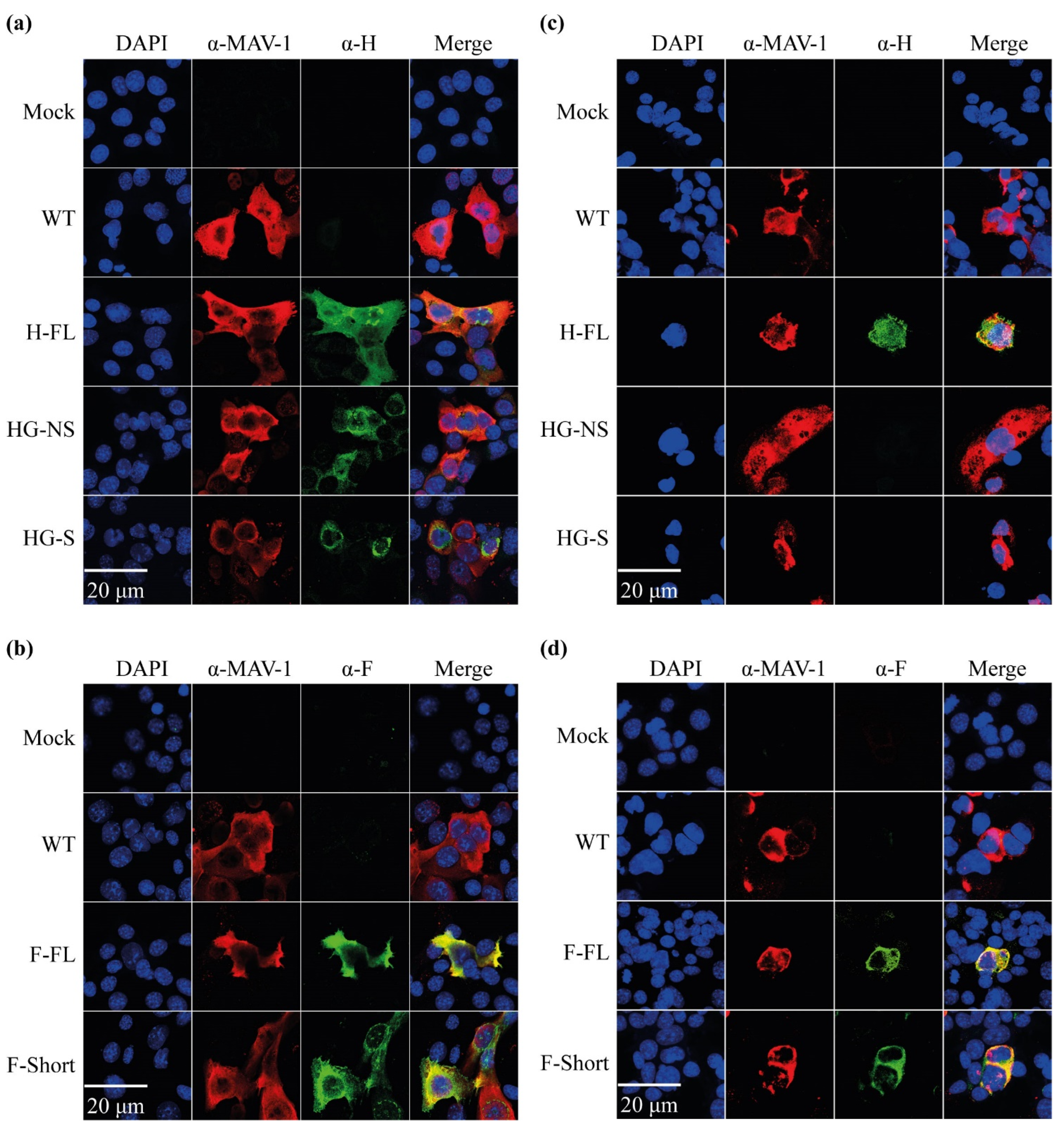
3.1. Design and Generation of MAV-1 Recombinants Expressing CDV Antigens
3.2. Oral Immunization with the MAV-1 H-FL Strain Induces Anti-CDV Immune Response
3.3. Impact of Pre-Existing Immunity against MAV-1 on the Capacity of MAV-1 H-FL Strain to Induce Anti-CDV Immune Response
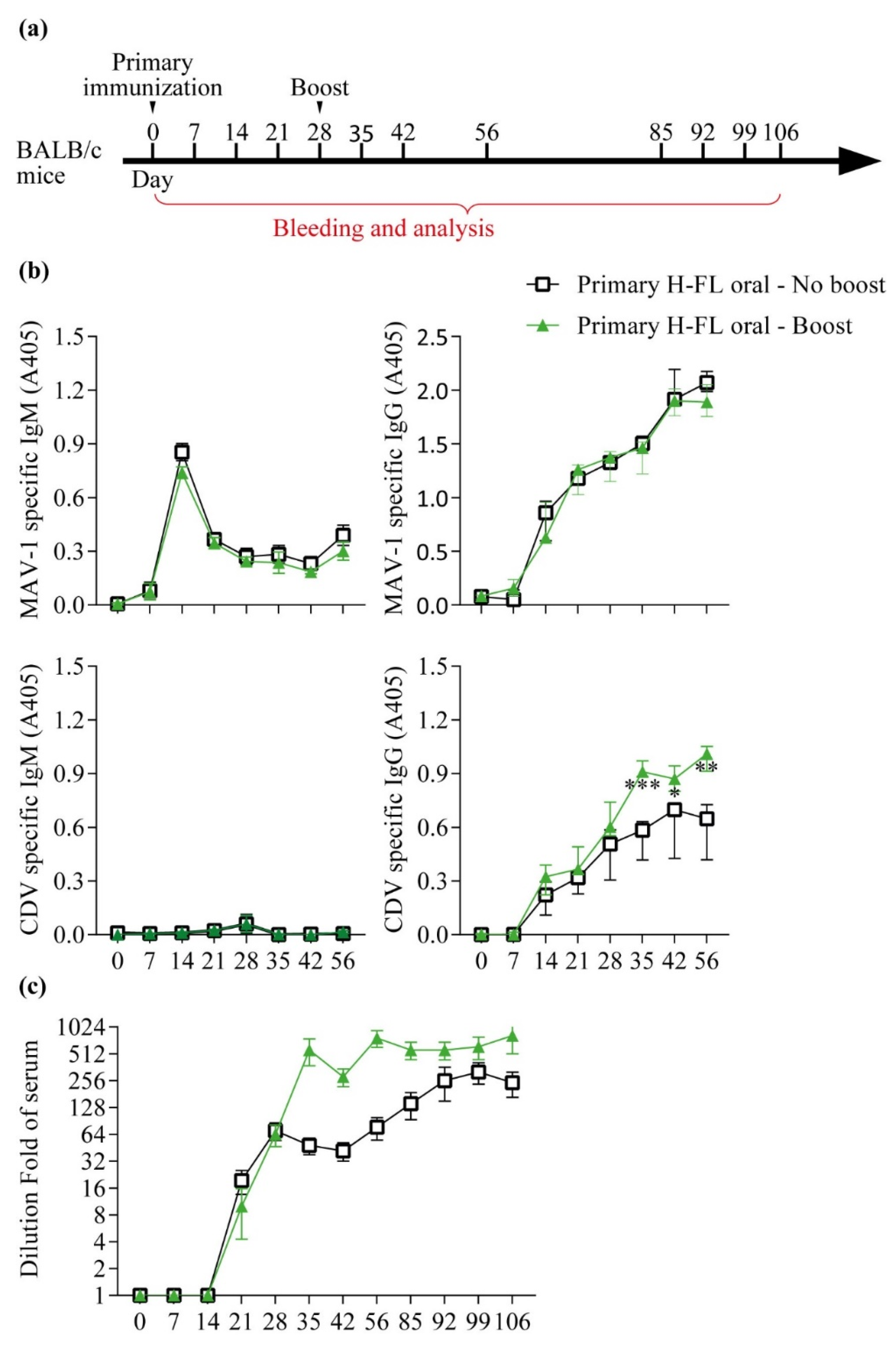
3.4. Pre-Existing Immunity Does Not Block the Ability of a Second Administration of the Oral Vaccine to Boost Immunity against the Transgene
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Loots, A.K.; Mitchell, E.; Dalton, D.L.; Kotze, A.; Venter, E.H. Advances in Canine Distemper Virus Pathogenesis Research: A Wildlife Perspective. J. Gen. Virol. 2017, 98, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Ke, G.M.; Ho, C.H.; Chiang, M.J.; Sanno-Duanda, B.; Chung, C.S.; Lin, M.Y.; Shi, Y.Y.; Yang, M.H.; Tyan, Y.C.; Liao, P.C.; et al. Phylodynamic Analysis of the Canine Distemper Virus Hemagglutinin Gene. BMC Vet. Res. 2015, 11, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krakowka, S.; Cockerell, G.; Koestner, A. Effects of Canine Distemper Virus Infection on Lymphoid Function in vitro and in vivo. Infect. Immun. 1975, 11, 1069–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lempp, C.; Spitzbarth, I.; Puff, C.; Cana, A.; Kegler, K.; Techangamsuwan, S.; Baumgärtner, W.; Seehusen, F. New Aspects of the Pathogenesis of Canine Distemper Leukoencephalitis. Viruses 2014, 6, 2571–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beineke, A.; Baumgärtner, W.; Wohlsein, P.; Baumgartner, W.; Wohlsein, P. Cross-Species Transmission of Canine Distemper Virus-an Update. One Health 2015, 1, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Seki, F.; Yamamoto, Y.; Nao, N.; Tokiwa, H. Animal Morbilliviruses and Their Cross-Species Transmission Potential. Curr. Opin. Virol. 2020, 41, 38–45. [Google Scholar] [CrossRef]
- Roelke-Parker, M.E.; Munson, L.; Packer, C.; Kock, R.; Cleaveland, S.; Carpenter, M.; O’Brien, S.J.; Pospischil, A.; Hofmann-Lehmann, R.; Lutz, H.; et al. A Canine Distemper Virus Epidemic in Serengeti Lions (Panthera leo). Nature 1996, 379, 441–445. [Google Scholar] [CrossRef]
- Seimon, T.A.; Miquelle, D.G.; Chang, T.Y.; Newton, A.L.; Korotkova, I.; Ivanchuk, G.; Lyubchenko, E.; Tupikov, A.; Slabe, E.; McAloose, D. Canine Distemper Virus: An Emerging Disease in Wild Endangered Amur Tigers (Panthera tigris altaica). MBio 2013, 4, e00410-13. [Google Scholar] [CrossRef] [Green Version]
- Feng, N.; Yu, Y.; Wang, T.; Wilker, P.; Wang, J.; Li, Y.; Sun, Z.; Gao, Y.; Xia, X. Fatal Canine Distemper Virus Infection of Giant Pandas in China. Sci. Rep. 2016, 6, 27518. [Google Scholar] [CrossRef]
- Kennedy, S.; Kuiken, T.; Jepson, P.D.; Deaville, R.; Forsyth, M.; Barrett, T.; Van De Bildt, M.W.G.; Osterhaus, A.D.M.E.; Eybatov, T.; Duck, C.; et al. Mass Die-Off of Caspian Seals Caused by Canine Distemper Virus. Emerg. Infect. Dis. 2000, 6, 637–639. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Zheng, Y.; Zhang, S.; Fan, Q.; Liu, H.; Zhang, F.; Wang, W.; Liao, G.; Hu, R. Canine Distemper Outbreak in Rhesus Monkeys, China. Emerg. Infect. Dis. 2011, 17, 1541–1543. [Google Scholar] [CrossRef]
- Sun, Z.; Li, A.; Ye, H.; Shi, Y.; Hu, Z.; Zeng, L. Natural Infection with Canine Distemper Virus in Hand-Feeding Rhesus Monkeys in China. Vet. Microbiol. 2010, 141, 374–378. [Google Scholar] [CrossRef]
- Sakai, K.; Yoshikawa, T.; Seki, F.; Fukushi, S.; Tahara, M.; Nagata, N.; Ami, Y.; Mizutani, T.; Kurane, I.; Yamaguchi, R.; et al. Canine Distemper Virus Associated with a Lethal Outbreak in Monkeys Can Readily Adapt to Use Human Receptors. J. Virol. 2013, 87, 7170–7175. [Google Scholar] [CrossRef] [Green Version]
- Bieringer, M.; Han, J.W.; Kendl, S.; Khosravi, M.; Plattet, P.; Schneider-Schaulies, J. Experimental Adaptation of Wild-Type Canine Distemper Virus (CDV) to the Human Entry Receptor CD150. PLoS ONE 2013, 8, e57488. [Google Scholar] [CrossRef] [Green Version]
- Di Sabatino, D.; Savini, G.; Lorusso, A. Canine Distemper and Endangered Wildlife: Is It Time for Mandatory Vaccination of Dogs? Vaccine 2015, 33, 6519. [Google Scholar] [CrossRef]
- Day, M.J.; Horzinek, M.C.; Schultz, R.D.; Squires, R.A.; Vaccination Guidelines Group of the World Small Animal Veterinary Association. WSAVA Guidelines for the Vaccination of Dogs and Cats. J. Small Anim. Pract. 2016, 57, 4–8. [Google Scholar] [CrossRef]
- Freuling, C.M.; Hampson, K.; Selhorst, T.; Schroder, R.; Meslin, F.X.; Mettenleiter, T.C.; Muller, T. The Elimination of Fox Rabies from Europe: Determinants of Success and Lessons for the Future. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120142. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.F.; Schröder, R.; Wysocki, P.; Mettenleiter, T.C.; Freuling, C.M.; Muller, T.F.; Schroder, R.; Wysocki, P.; Mettenleiter, T.C.; Freuling, C.M. Spatio-Temporal Use of Oral Rabies Vaccines in Fox Rabies Elimination Programmes in Europe. PLoS Negl. Trop. Dis. 2015, 9, e0003953. [Google Scholar] [CrossRef] [Green Version]
- Wimsatt, J.; Biggins, D.; Innes, K.; Taylor, B.; Garell, D. Evaluation of Oral and Subcutaneous Delivery of an Experimental Canarypox Recombinant Canine Distemper Vaccine in the Siberian Polecat (Mustela eversmanni). J. Zoo Wildl. Med. 2003, 34, 25–35. [Google Scholar] [CrossRef]
- Connolly, M.; Thomas, P.; Woodroffe, R.; Raphael, B.L. Comparison of Oral and Intramuscular Recombinant Canine Distemper Vaccination in African Wild Dogs (Lycaon pictus). J. Zoo Wildl. Med. 2013, 44, 882–888. [Google Scholar] [CrossRef]
- Goffin, E.; Javaux, J.; Destexhe, E.; Pretto, C.D.; Spindler, K.R.; Machiels, B.; Gillet, L. Oral Vaccination with Replication-Competent Adenovirus in Mice Reveals Dissemination of the Viral Vaccine beyond the Gastrointestinal Tract. J. Virol. 2019, 93, e00237-19. [Google Scholar] [CrossRef] [Green Version]
- Beard, C.W.; Spindler, K.R. Analysis of Early Region 3 Mutants of Mouse Adenovirus Type 1. J. Virol. 1996, 70, 5867–5874. [Google Scholar] [CrossRef] [Green Version]
- Cauthen, A.N.; Brown, C.C.; Spindler, K.R. In vitro and in vivo Characterization of a Mouse Adenovirus Type 1 Early Region 3 Null Mutant. J. Virol. 1999, 73, 8640–8646. [Google Scholar] [CrossRef] [Green Version]
- Tirumuru, N.; Pretto, C.D.; Castro Jorge, L.A.; Spindler, K.R. Mouse Adenovirus Type 1 Early Region 1A Effects on the Blood-Brain Barrier. mSphere 2016, 1, e00079-16. [Google Scholar] [CrossRef] [Green Version]
- Orlando, E.A.; Imbschweiler, I.; Gerhauser, I.; Baumgärtner, W.; Wewetzer, K. In vitro Characterization and Preferential Infection by Canine Distemper Virus of Glial Precursors with Schwann Cell Characteristics from Adult Canine Brain. Neuropathol. Appl. Neurobiol. 2008, 34, 621–637. [Google Scholar] [CrossRef]
- Cherpillod, P.; Beck, K.; Zurbriggen, A.; Wittek, R. Sequence Analysis and Expression of the Attachment and Fusion Proteins of Canine Distemper Virus Wild-Type Strain A75/17. J. Virol. 1999, 73, 2263–2269. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.; Li, B.; Ge, Y.; Ko, D.; Yendluri, S.; Harding, T.; VanRoey, M.; Spindler, K.R.; Jooss, K. Novel Immunocompetent Murine Tumor Model for Evaluation of Conditionally Replication-Competent (Oncolytic) Murine Adenoviral Vectors. J. Virol. 2009, 83, 3450–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion Protein Linkers: Property, Design and Functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiels, B.; Lete, C.; Guillaume, A.; Mast, J.; Stevenson, P.G.; Vanderplasschen, A.; Gillet, L. Antibody Evasion by a Gammaherpesvirus O-Glycan Shield. PLoS Pathog. 2011, 7, e1002387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latif, M.B.; Machiels, B.; Xiao, X.; Mast, J.; Vanderplasschen, A.; Gillet, L. Deletion of Murid Herpesvirus 4 ORF63 Affects the Trafficking of Incoming Capsids toward the Nucleus. J. Virol. 2015, 90, 2455–2472. [Google Scholar] [CrossRef] [Green Version]
- Bett, A.J.; Prevec, L.; Graham, F.L. Packaging Capacity and Stability of Human Adenovirus Type 5 Vectors. J. Virol. 1993, 67, 5911–5921. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Alía, M.A.; Russell, S.J. Probing Morbillivirus Antisera Neutralization Using Functional Chimerism between Measles Virus and Canine Distemper Virus Envelope Glycoproteins. Viruses 2019, 11, 688. [Google Scholar] [CrossRef] [Green Version]
- Plattet, P.; Alves, L.; Herren, M.; Aguilar, H.C. Measles Virus Fusion Protein: Structure, Function and Inhibition. Viruses 2016, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, S.A. Correlates of Protection Induced by Vaccination. Am. Soc. Microbiol. 2010, 17, 1055–1065. [Google Scholar] [CrossRef] [Green Version]
- Jensen, W.A.; Totten, J.S.; Lappin, M.R.; Schultz, R.D. Use of Serologic Tests to Predict Resistance to Canine Distemper Virus-Induced Disease in Vaccinated Dogs. J. Vet. Diagn. Investig. 2015, 27, 576–580. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, M.; Freisl, M.; Zablotski, Y.; Khan, M.A.A.; Speck, S.; Truyen, U.; Hartmann, K. Prevalence of Neutralizing Antibodies to Canine Distemper Virus and Response to Vaccination in Client-Owned Adult Healthy Dogs. Viruses 2021, 13, 945. [Google Scholar] [CrossRef]
- Schultz, R.D.; Thiel, B.; Mukhtar, E.; Sharp, P.; Larson, L.J. Age and Long-Term Protective Immunity in Dogs and Cats. J. Comp. Pathol. 2010, 142 (Suppl. 1), S102–S108. [Google Scholar] [CrossRef]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-Existing Immunity against Ad Vectors: Humoral, Cellular, and Innate Response, What’s Important? Hum. Vaccines Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.Q.; Gao, G.P.; Reyes-Sandoval, A.; Li, Y.; Wilson, J.M.; Ertl, H.C. Oral Vaccination of Mice with Adenoviral Vectors Is Not Impaired by Preexisting Immunity to the Vaccine Carrier. J. Virol. 2003, 77, 10780–10789. [Google Scholar] [CrossRef] [Green Version]
- Gurwith, M.; Lock, M.; Taylor, E.M.; Ishioka, G.; Alexander, J.; Mayall, T.; Ervin, J.E.; Greenberg, R.N.; Strout, C.; Treanor, J.J.; et al. Safety and Immunogenicity of an Oral, Replicating Adenovirus Serotype 4 Vector Vaccine for H5N1 Influenza: A Randomised, Double-Blind, Placebo-Controlled, Phase 1 Study. Lancet Infect. Dis. 2013, 13, 238–250. [Google Scholar] [CrossRef] [Green Version]
- De Vleeschauwer, A.R.; Zhou, X.; Lefebvre, D.J.; Garnier, A.; Watier, F.; Pignon, C.; Lacour, S.A.; Zientara, S.; Bakkali-Kassimi, L.; De Clercq, K.; et al. A Canine Adenovirus Type 2 Vaccine Vector Confers Protection against Foot-and-Mouth Disease in Guinea Pigs. Vaccine 2018, 36, 2193–2198. [Google Scholar] [CrossRef]
- Henderson, H.; Jackson, F.; Bean, K.; Panasuk, B.; Niezgoda, M.; Slate, D.; Li, J.; Dietzschold, B.; Mattis, J.; Rupprecht, C.E. Oral Immunization of Raccoons and Skunks with a Canine Adenovirus Recombinant Rabies Vaccine. Vaccine 2009, 27, 7194–7197. [Google Scholar] [CrossRef]
- Le, L.P.; Everts, M.; Dmitriew, I.P.; Davydova, J.G.; Yamamoto, M.; Curiel, D.T. Fluorescently Labeled Adenovirus with PIX-EGFP for Vector Detection. Mol. Imaging 2004, 3, 105–116. [Google Scholar] [CrossRef]
- Wang, F.X.; Zhang, S.Q.; Zhu, H.W.; Yang, Y.; Sun, N.; Tan, B.; Li, Z.G.; Cheng, S.P.; Fu, Z.F.; Wen, Y.J. Recombinant Rabies Virus Expressing the H Protein of Canine Distemper Virus Protects Dogs from the Lethal Distemper Challenge. Vet. Microbiol. 2014, 174, 362–371. [Google Scholar] [CrossRef]
- Mendonça, S.A.; Lorincz, R.; Boucher, P.; Curiel, D.T. Adenoviral Vector Vaccine Platforms in the SARS-CoV-2 Pandemic. npj Vaccines 2021, 6, 97. [Google Scholar] [CrossRef]
- Le Gars, M.; Sadoff, J.; Struyf, F.; Heerwegh, D.; Truyers, C.; Hendriks, J.; Gray, G.; Grinsztejn, B.; Goepfert, P.A.; Schuitemaker, H.; et al. Impact of Preexisting Anti–Adenovirus 26 Humoral Immunity on Immunogenicity of the Ad26.COV2.S Coronavirus Disease 2019 Vaccine. J. Infect. Dis. 2022, jiac142. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, X.; Goffin, E.; Gillard, L.; Machiels, B.; Gillet, L. A Single Oral Immunization with Replication-Competent Adenovirus-Vectored Vaccine Induces a Neutralizing Antibody Response in Mice against Canine Distemper Virus. Viruses 2022, 14, 1847. https://0-doi-org.brum.beds.ac.uk/10.3390/v14091847
Du X, Goffin E, Gillard L, Machiels B, Gillet L. A Single Oral Immunization with Replication-Competent Adenovirus-Vectored Vaccine Induces a Neutralizing Antibody Response in Mice against Canine Distemper Virus. Viruses. 2022; 14(9):1847. https://0-doi-org.brum.beds.ac.uk/10.3390/v14091847
Chicago/Turabian StyleDu, Xiang, Emeline Goffin, Lucie Gillard, Bénédicte Machiels, and Laurent Gillet. 2022. "A Single Oral Immunization with Replication-Competent Adenovirus-Vectored Vaccine Induces a Neutralizing Antibody Response in Mice against Canine Distemper Virus" Viruses 14, no. 9: 1847. https://0-doi-org.brum.beds.ac.uk/10.3390/v14091847