
Genomic and Temporal Analysis of Deletions Correlated to qRT-PCR Dropout in N Gene in Alpha, Delta and Omicron Variants
 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Collection
2.2. RT-PCR Amplification
2.3. Viral RNA Extraction and Next-Generation Sequencing
2.4. Data Analysis
2.5. Reference Consensus Sequences Generation
2.6. N Protein Structure Prediction
3. Results
3.1. Ct Values
3.2. Sequence Analysis
3.3. LCS Comparison
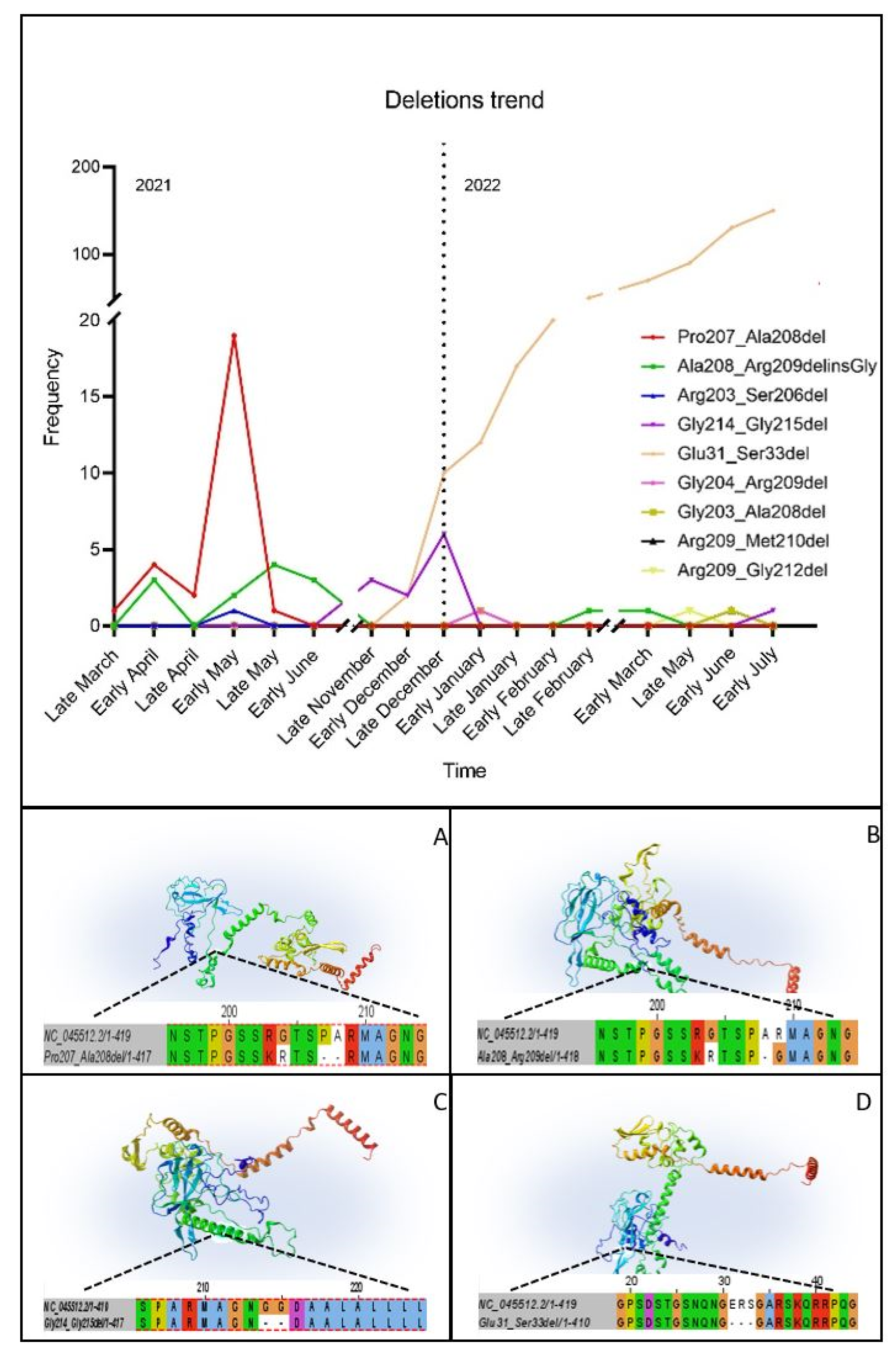
3.4. Temporal Analysis
3.5. D Structure Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Niemi, M.E.K.; Daly, M.J.; Ganna, A. The human genetic epidemiology of COVID-19. Nat. Rev. Genet. 2022, 23, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, O.A.; Uppal, S.; Huang, W.; Kane, M.A.; Villasmil, R.; Rogozin, I.B.; Poliakov, E.; Redmond, T.M. The Functional Consequences of the Novel Ribosomal Pausing Site in SARS-CoV-2 Spike Glycoprotein RNA. Int. J. Mol. Sci. 2021, 22, 6490. [Google Scholar] [CrossRef] [PubMed]
- DeGrace, M.M.; Ghedin, E.; Frieman, M.B.; Krammer, F.; Grifoni, A.; Alisoltani, A.; Alter, G.; Amara, R.R.; Baric, R.S.; Barouch, D.H.; et al. Defining the risk of SARS-CoV-2 variants on immune protection. Nature 2022, 605, 640–652. [Google Scholar] [CrossRef]
- Flores-Vega, V.R.; Monroy-Molina, J.V.; Jiménez-Hernández, L.E.; Torres, A.G.; Santos-Preciado, J.I.; Rosales-Reyes, R. SARS-CoV-2: Evolution and Emergence of New Viral Variants. Viruses 2022, 14, 653. [Google Scholar] [CrossRef]
- World Health Organization. Diagnostic Testing for SARS-CoV-2: Interim Guidance. World Health Organization. License: CC BY-NC-SA 3.0 IGO. 11 September 2020. Available online: https://apps.who.int/iris/handle/10665/334254 (accessed on 11 January 2023).
- Hui, K.P.Y.; Ho, J.C.W.; Cheung, M.C.; Ng, K.C.; Ching, R.H.H.; Lai, K.L.; Kam, T.T.; Gu, H.; Sit, K.Y.; Hsin, M.K.Y.; et al. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 2022, 603, 715–720. [Google Scholar] [CrossRef]
- Colosimo, M.; Minchella, P.; Tallerico, R.; Talotta, I.; Peronace, C.; Gallelli, L.; Di Mizio, G.; Cione, E. Comparison of AllplexTM 2019-nCoV and TaqPathTM COVID-19 Assays. Reports 2022, 5, 14. [Google Scholar] [CrossRef]
- Hasan, M.R.; Sundararaju, S.; Manickam, C.; Mirza, F.; Al-Hail, H.; Lorenz, S.; Tang, P. A Novel Point Mutation in the N Gene of SARS-CoV-2 May Affect the Detection of the Virus by Reverse Transcription-Quantitative PCR. J. Clin. Microbiol. 2021, 59, e03278-20. [Google Scholar] [CrossRef] [PubMed]
- Ko, K.K.K.; Abdul Rahman, N.B.; Tan, S.Y.L.; Chan, K.X.L.; Goh, S.S.; Sim, J.H.C.; Lim, K.L.; Tan, W.L.; Chan, K.S.; Oon, L.L.E.; et al. SARS-CoV-2 N Gene G29195T Point Mutation May Affect Diagnostic Reverse Transcription-PCR Detection. Microbiol. Spectr. 2022, 10, e0222321. [Google Scholar] [CrossRef]
- Jonguitud-Borrego, N.; Malcı, K.; Anand, M.; Baluku, E.; Webb, C.; Liang, L.; Barba-Ostria, C.; Guaman, L.P.; Hui, L.; Rios-Solis, L. High-throughput and automated screening for COVID-19. Front. Med. Technol. 2022, 4, 969203. [Google Scholar] [CrossRef]
- Tombuloglu, H.; Sabit, H.; Al-Khallaf, H.; Kabanja, J.H.; Alsaeed, M.; Al-Saleh, N.; Al-Suhaimi, E. Multiplex real-time RT-PCR method for the diagnosis of SARS-CoV-2 by targeting viral N, RdRP and human RP genes. Sci. Rep. 2022, 12, 2853. [Google Scholar] [CrossRef]
- Hossain, M.W.; Hossain, M.; Arafath, K.; Ety, S.S.; Shetu MM, H.; Kabir, M.; Noor, F.A.; Mannoor, K. Real-Time fast PCR amplification using designated and conventional real time thermal cycler systems: COVID-19 perspective. PLoS ONE 2022, 17, e0276464. [Google Scholar] [CrossRef]
- Seegene Inc. Allplex SARS-CoV-2 Assay. Available online: https://www.seegene.com/assays/allplex_2019_ncov_assay (accessed on 12 January 2023).
- Cepheid Xpert Xpress SARS-CoV-2. Available online: https://www.cepheid.com/it/tests/Critical-Infectious-Diseases/Xpert-Xpress-SARS-CoV-2 (accessed on 10 January 2023).
- Genomics, P. CleanPlex®SARS-CoV-2 FLEX Research and Surveillance Panels. Available online: www.paragongenomics.com (accessed on 12 January 2023).
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Pangolin—CoV-Lineages. Available online: https://cov-lineages.org/resources/pangolin.html (accessed on 22 January 2023).
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Nextclade. Available online: https://clades.nextstrain.org (accessed on 27 January 2023).
- Integrative Genomics Viewer. Available online: https://software.broadinstitute.org/software/igv/home (accessed on 5 July 2023).
- Clustal Omega < Multiple Sequence Alignment < E.M.B.L.-E.B.I. Available online: https://www.ebi.ac.uk/Tools/msa/clustalo/ (accessed on 14 January 2023).
- EMBOSSPrograms E.M.B.L.-E.B.I. Available online: https://www.ebi.ac.uk/Tools/st/emboss_transeq/ (accessed on 12 February 2023).
- Robetta—Baker Lab. Available online: https://robetta.bakerlab.org/ (accessed on 23 February 2023).
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; O’Sullivan, D.M.; Shah, D.; Atkinson, L.; Pereira RP, A.; Whale, A.S.; Busby, E.J.; Huggett, J.F.; Harris, K. Comparison of SARS-CoV-2 N gene real-time RT-PCR targets and commercially available mastermixes. J. Virol. Methods 2021, 295, 114215. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Ceci, A.; Roby, C.; Briggs, R.; Ziolo, D.; Korba, R.; Mejia, R.; Kelly, S.T.; Toney, D.; Friedlander, M.J.; et al. A comparative analysis exposes an amplification delay distinctive to SARS-CoV-2 Omicron variants of clinical and public health relevance. Emerg. Microbes Infect. 2023, 12, 2154617. [Google Scholar] [CrossRef] [PubMed]
- Isabel, S.; Abdulnoor, M.; Boissinot, K.; Isabel, M.R.; de Borja, R.; Zuzarte, P.C.; Sjaarda, C.P.; RBarker, K.; Sheth, P.M.; Matukas, L.M.; et al. Emergence of a mutation in the nucleocapsid gene of SARS-CoV-2 interferes with PCR detection in Canada. Sci. Rep. 2022, 12, 10867. [Google Scholar] [CrossRef] [PubMed]
- Han, A.X.; Toporowski, A.; Sacks, J.A.; Perkins, M.D.; Briand, S.; van Kerkhove, M.; Hannay, E.; Carmona, S.; Rodriguez, B.; Parker, E.; et al. SARS-CoV-2 diagnostic testing rates determine the sensitivity of genomic surveillance programs. Nat. Genet. 2023, 55, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Ippoliti, C.; De Maio, F.; Santarelli, G.; Marchetti, S.; Vella, A.; Santangelo, R.; Sanguinetti, M.; Posteraro, B. Rapid Detection of the Omicron (B.1.1.529) SARS-CoV-2 Variant Using a COVID-19 Diagnostic PCR Assay. Microbiol. Spectr. 2022, 10, e0099022. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, M.; Bellocchi, M.C.; Marchegiani, G.; Grelli, S.; Micheli, V.; Stella, D.; Zerillo, B.; Carioti, L.; Svicher, V.; Rogliani, P.; et al. First Case of a COVID-19 Patient Infected by Delta AY.4 with a Rare Deletion Leading to a N Gene Target Failure by a Specific Real Time PCR Assay: Novel Omicron VOC Might Be Doing Similar Scenario? Microorganisms 2022, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Laine, P.; Nihtilä, H.; Mustanoja, E.; Lyyski, A.; Ylinen, A.; Hurme, J.; Paulin, L.; Jokiranta, S.; Auvinen, P.; Meri, T. SARS-CoV-2 variant with mutations in N gene affecting detection by widely used PCR primers. J. Med. Virol. 2022, 94, 1227–1231. [Google Scholar] [CrossRef]
- Lesbon, J.C.C.; Poleti, M.D.; de Mattos Oliveira, E.C.; Patané, J.S.L.; Clemente, L.G.; Viala, V.L.; Ribeiro, G.; Giovanetti, M.; de Alcantara, L.C.J.; Teixeira, O.; et al. Nucleocapsid (N) Gene Mutations of SARS-CoV-2 Can Affect Real-Time RT-PCR Diagnostic and Impact False-Negative Results. Viruses 2021, 13, 2474. [Google Scholar] [CrossRef]
- Peronace, C.; Tallerico, R.; Colosimo, M.; Fazio, M.; Pasceri, F.; Talotta, I.; Panduri, G.; Pintomalli, L.; Oteri, R.; Calantoni, V.; et al. The First Identification in Italy of SARS-CoV-2 Omicron BA.4 Harboring KSF141_del: A Genomic Comparison with Omicron Sub-Variants. Biomedicines 2022, 10, 1839. [Google Scholar] [CrossRef]
- Lee, S.; Won, D.; Kim, C.K.; Ahn, J.; Lee, Y.; Na, H.; Kim, Y.T.; Lee, M.K.; Choi, J.R.; Lim, H.S.; et al. Novel indel mutation in the N gene of SARS-CoV-2 clinical samples that were diagnosed positive in a commercial RT-PCR assay. Virus Res. 2021, 297, 198398. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Sample Number | Variant | Allplex SARS-CoV-2 Extraction-Free Assay | Xpert Xpress SARS-CoV-2 Assay | |||||
|---|---|---|---|---|---|---|---|---|
| E Gene | RdRP/S Gene | N Gene | IC * | E Gene | N2 Gene | IC * | ||
| 1 | B.1.1.7 | 25.98 | 29.02 | N/A | 22.05 | 20.9 | 24.1 | 27.6 |
| 2 | B.1.1.7 | 21.23 | 22.34 | N/A | 22.45 | 17.1 | 17.9 | 27.5 |
| 3 | B.1.1.7 | 19.18 | 21.25 | N/A | 22.12 | 14.9 | 16.3 | 27.5 |
| 4 | B.1.1.7 | 19.12 | 22.01 | N/A | 21.98 | 15.8 | 17.1 | 27.1 |
| 5 | B.1.1.7 | 24.45 | 26.68 | N/A | 22.01 | 20 | 21.7 | 27.2 |
| 6 | B.1.1.7 | 19.04 | 21.32 | N/A | 21.77 | 15 | 16.6 | 28.3 |
| 7 | B.1.1.7 | 23.12 | 24.23 | N/A | 22.13 | 18.6 | 19.3 | 27.4 |
| 8 | B.1.1.7 | 25.98 | 28.1 | N/A | 22.31 | 21.8 | 23.4 | 27.7 |
| 9 | B.1.1.7 | 27.98 | 30.01 | N/A | 21.89 | 22.9 | 25.4 | 28.1 |
| 10 | B.1.1.7 | 26.78 | 29.07 | N/A | 22.52 | 21.8 | 24.7 | 28.5 |
| 11 | B.1.1.7 | 24.98 | 28.13 | N/A | 22.33 | 20.7 | 23.3 | 28.2 |
| 12 | B.1.1.7 | 23.37 | 23.98 | N/A | 21.88 | 19.7 | 19.8 | 27.3 |
| 13 | B.1.1.7 | 23.01 | 26.25 | N/A | 21.91 | 18.5 | 21.4 | 27.1 |
| 14 | B.1.1.7 | 24.55 | 27.32 | N/A | 22.12 | 20.2 | 23.1 | 28.3 |
| 15 | B.1.1.7 | 23.72 | 26.03 | N/A | 22.37 | 18.7 | 21.3 | 27.5 |
| 16 | B.1.1.7 | 20.34 | 22.22 | N/A | 22.41 | 16.12 | 17.6 | 28.0 |
| 17 | B.1.1.7 | 21.23 | 24.37 | N/A | 22.4 | 17.06 | 19.5 | 28.1 |
| 18 | B.1.1.7 | 18.11 | 21.01 | N/A | 21.83 | 14.94 | 16.3 | 27.9 |
| 19 | B.1.1.7 | 19.23 | 19.99 | N/A | 21.85 | 14.78 | 15.3 | 27.4 |
| 20 | B.1.1.7 | 22.12 | 23.92 | N/A | 22.01 | 17.63 | 19.5 | 28.0 |
| 21 | B.1.1.7 | 19.15 | 22.32 | N/A | 21.99 | 15.06 | 17.8 | 28.4 |
| 22 | B.1.1.7 | 23.04 | 24.59 | N/A | 22.38 | 19.06 | 20.9 | 27.8 |
| 23 | B.1.1.7 | 24.46 | 27.12 | N/A | 22.52 | 20.1 | 23.0 | 27.1 |
| 24 | B.1.1.7 | 20.67 | 21.89 | N/A | 22.04 | 16.4 | 17.7 | 27.3 |
| 25 | B.1.1.7 | 28.47 | 30.15 | N/A | 22.56 | 24.1 | 25.6 | 28.2 |
| 26 | B.1.1.7 | 25.34 | 29.07 | N/A | 22.63 | 21.1 | 24.3 | 28.5 |
| 27 | B.1.1.7 | 23.34 | 25.87 | N/A | 22.43 | 19.5 | 22.1 | 27.3 |
| 28 | B.1.1.7 | 26.76 | 29.1 | N/A | 22.76 | 22.3 | 24.8 | 27.8 |
| 29 | B.1.1.7 | 23.53 | 26.67 | N/A | 22.51 | 19.7 | 22.4 | 27.3 |
| 30 | B.1.1.7 | 24.79 | 27.2 | N/A | 22.49 | 21.1 | 23.0 | 28.1 |
| 31 | B.1.1.7 | 19.55 | 21.68 | N/A | 21.81 | 15.2 | 17.5 | 27.7 |
| 32 | AY.102 | 22.13 | 23.54 | N/A | 22.06 | 18.1 | 19.1 | 27.4 |
| 33 | B.1.617.2 | 17.77 | 21.98 | N/A | 21.86 | 14.8 | 18.2 | 28.5 |
| 34 | AY.43 | 23.51 | 25.12 | N/A | 22.32 | 19.6 | 21.0 | 28.5 |
| 35 | AY.43 | 21.88 | 24.56 | N/A | 22.44 | 18.2 | 20.2 | 27.0 |
| 36 | AY.43 | 25.34 | 27.89 | N/A | 22.61 | 21.8 | 24.1 | 27.4 |
| 37 | AY.43 | 18.87 | 21.03 | N/A | 21.93 | 15 | 16.6 | 28.6 |
| 38 | AY.23 | 18.56 | 20.45 | N/A | 22.14 | 15.2 | 17.0 | 27.9 |
| 39 | AY.23 | 23.34 | 25.12 | N/A | 22.25 | 19.3 | 20.6 | 28.6 |
| 40 | AY.23 | 20.06 | 22.76 | N/A | 22.17 | 16 | 18.2 | 27.2 |
| 41 | AY.23 | 26.34 | 28.98 | N/A | 22.78 | 22.1 | 25.0 | 27.6 |
| 42 | BA.1.1 | 17.88 | 19.34 | N/A | 22.21 | 14.4 | 15.4 | 28.1 |
| 43 | BA.1 | 20.91 | 22.65 | N/A | 22.01 | 16.5 | 18.4 | 27.3 |
| 44 | BA.1.21 | 20.67 | 21.76 | N/A | 22.32 | 17.1 | 18.2 | 27.5 |
| 45 | BA.2 | 17.76 | 18.63 | N/A | 21.97 | 14.2 | 14.0 | 27.2 |
| 46 | BA.2.36 | 20.09 | 22.87 | N/A | 22.31 | 16.1 | 18.0 | 28.0 |
| 47 | BA.5 | 29.03 | 29.77 | N/A | 22.4 | 24.7 | 25.7 | 28.3 |
| 48 | BA.5.1 | 18.36 | 20.87 | N/A | 22.35 | 15.1 | 16.0 | 27.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gatti, G.; Brandolini, M.; Mancini, A.; Taddei, F.; Zannoli, S.; Dirani, G.; Manera, M.; Arfilli, V.; Denicolò, A.; Marzucco, A.; et al. Genomic and Temporal Analysis of Deletions Correlated to qRT-PCR Dropout in N Gene in Alpha, Delta and Omicron Variants. Viruses 2023, 15, 1630. https://0-doi-org.brum.beds.ac.uk/10.3390/v15081630
Gatti G, Brandolini M, Mancini A, Taddei F, Zannoli S, Dirani G, Manera M, Arfilli V, Denicolò A, Marzucco A, et al. Genomic and Temporal Analysis of Deletions Correlated to qRT-PCR Dropout in N Gene in Alpha, Delta and Omicron Variants. Viruses. 2023; 15(8):1630. https://0-doi-org.brum.beds.ac.uk/10.3390/v15081630
Chicago/Turabian StyleGatti, Giulia, Martina Brandolini, Andrea Mancini, Francesca Taddei, Silvia Zannoli, Giorgio Dirani, Martina Manera, Valentina Arfilli, Agnese Denicolò, Anna Marzucco, and et al. 2023. "Genomic and Temporal Analysis of Deletions Correlated to qRT-PCR Dropout in N Gene in Alpha, Delta and Omicron Variants" Viruses 15, no. 8: 1630. https://0-doi-org.brum.beds.ac.uk/10.3390/v15081630