
Mutation Analysis of SARS-CoV-2 Variants Isolated from Symptomatic Cases from Andhra Pradesh, India
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and S-Gene Amplification
2.2. Sequencing Procedure
2.3. Sequence Analysis
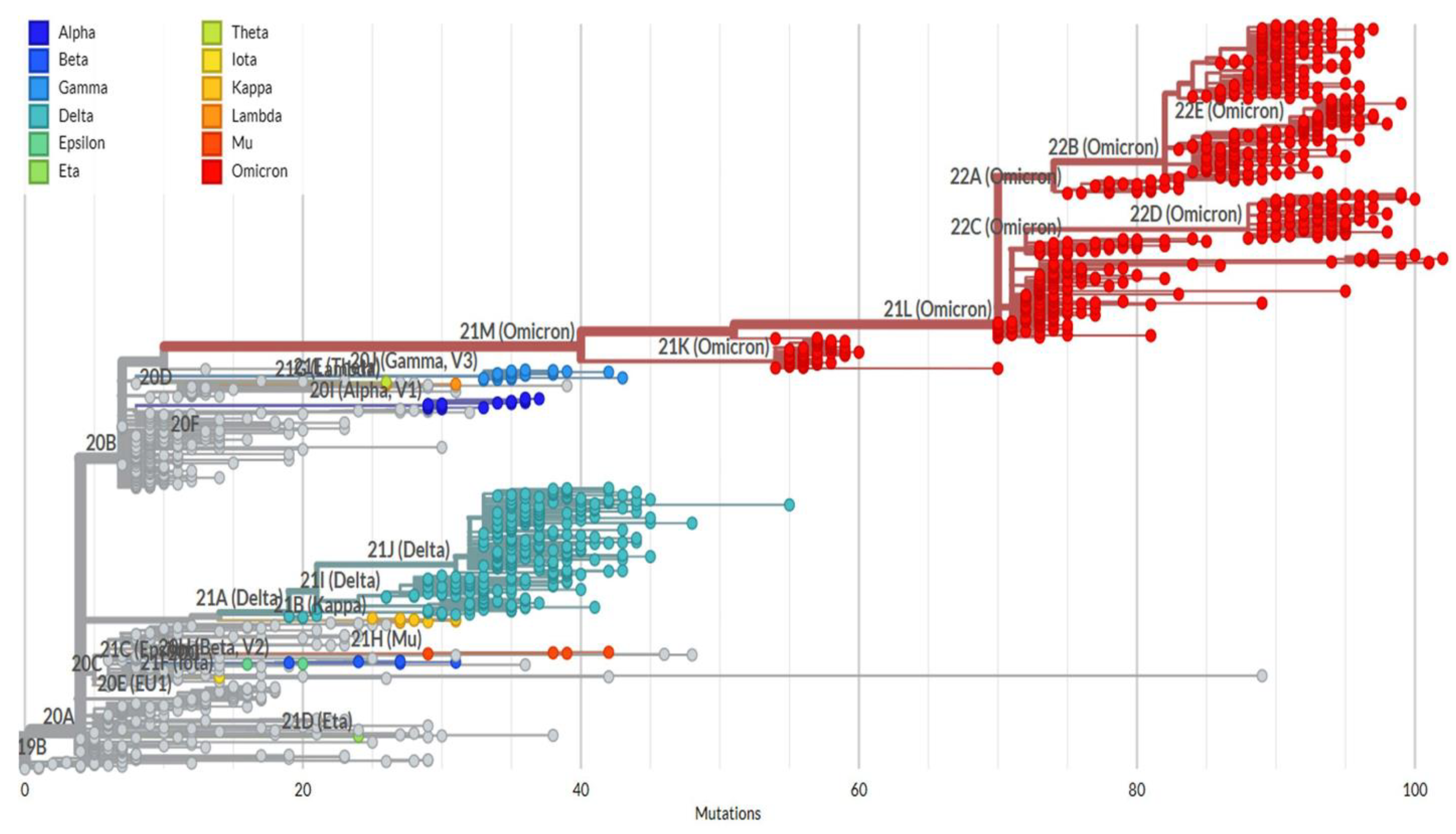
2.4. Phylogeny Tree Construction
2.5. Computational Biology Workup
2.6. Statistical Analysis
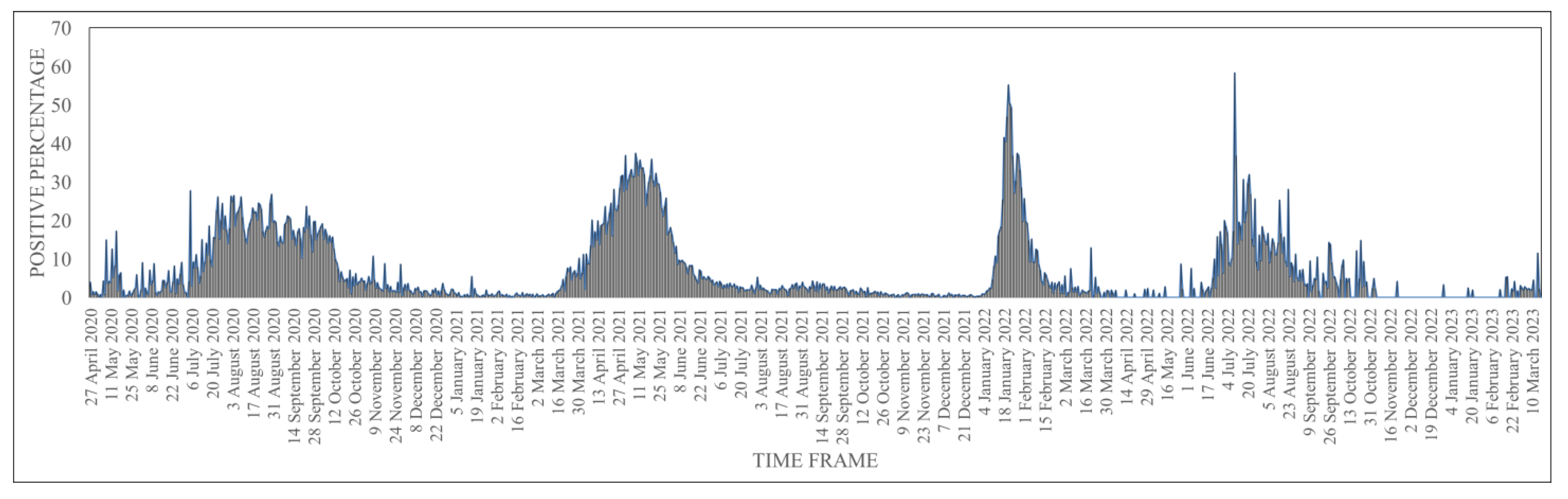
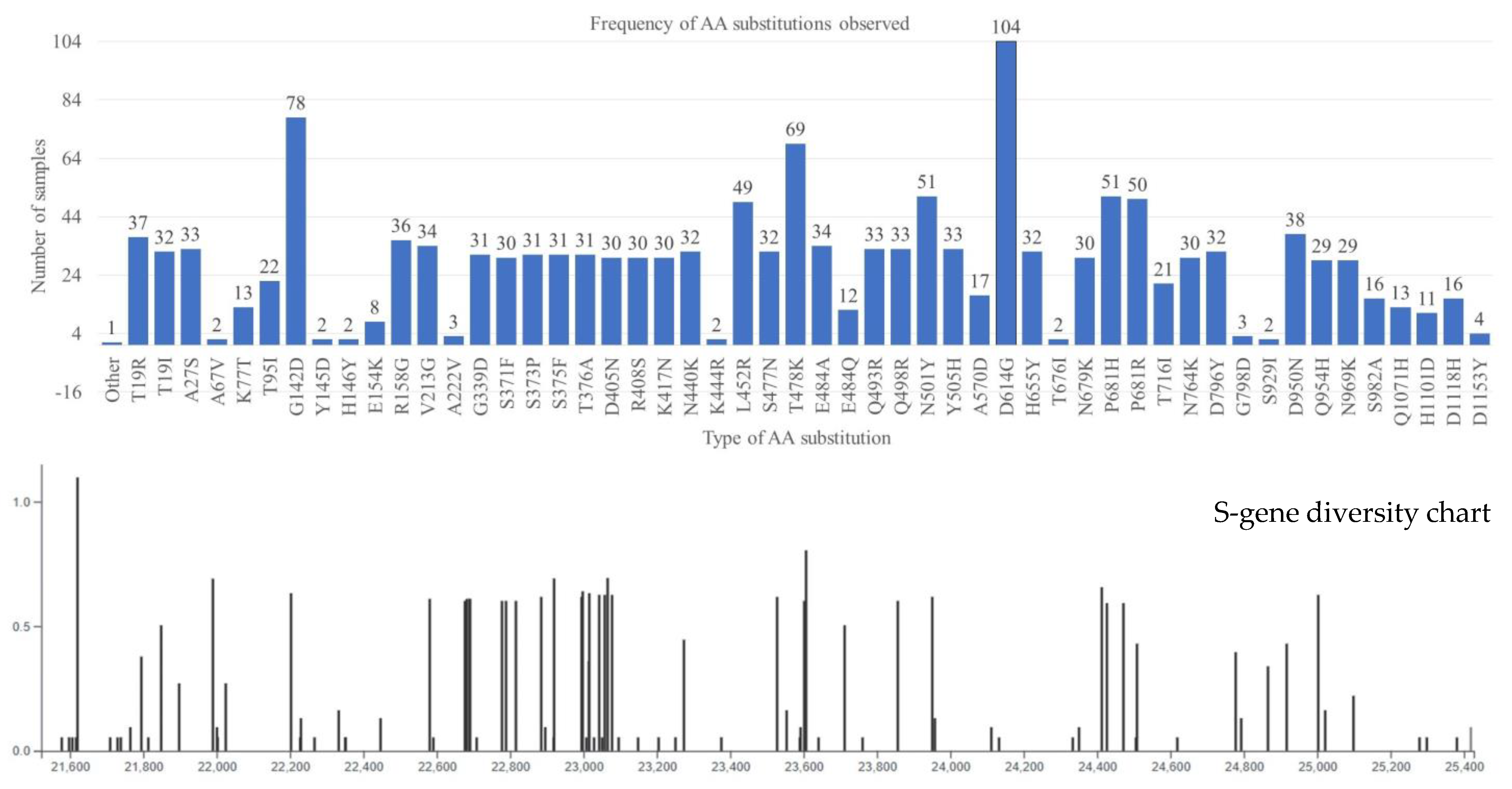
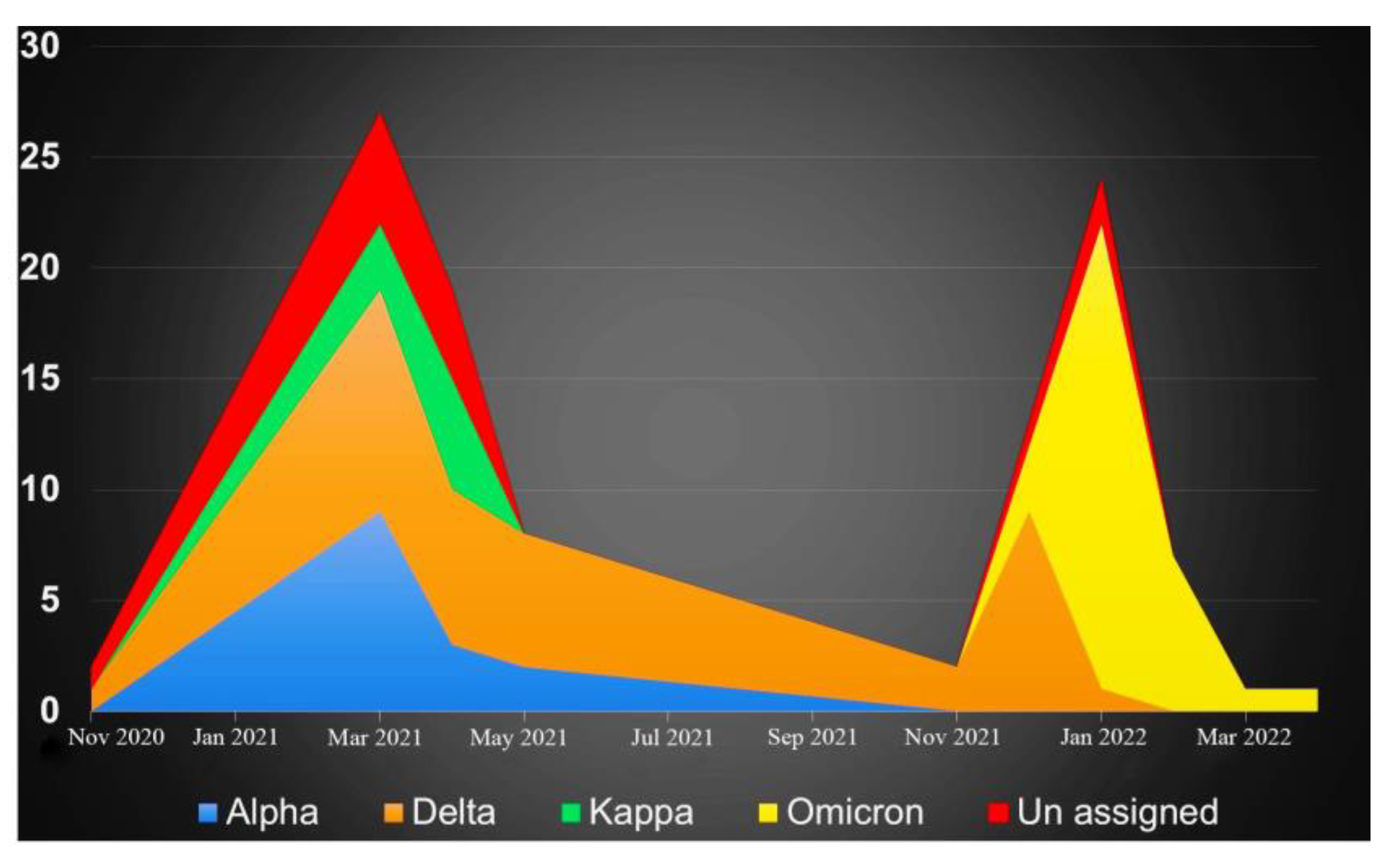
3. Results and Discussion
Effect of Vaccination on Mutations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [PubMed] [Green Version]
- Why Genomic Sequencing Is Crucial in COVID-19 Response. Available online: https://www.afro.who.int/news/why-genomic-sequencing-crucial-covid-19-response (accessed on 29 December 2022).
- CDC Coronavirus Disease 2019 (COVID-19). Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html (accessed on 29 December 2022).
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 30 December 2022).
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Fiser, A.; Šali, A. Modeller: Generation and refinement of homology-based protein structure models. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2003; Volume 374, pp. 461–491. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Bagcchi, S. The world’s largest COVID-19 vaccination campaign. Lancet Infect. Dis. 2021, 21, 323. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- WHO Coronavirus (COVID-19) Dashboard. WHO Coronavirus (COVID-19) Dashboard with Vaccination Data. Available online: https://covid19.who.int/data (accessed on 3 July 2023).
- Tada, T.; Zhou, H.; Dcosta, B.M.; Samanovic, M.I.; Mulligan, M.J.; Landau, N.R. The spike proteins of SARS-CoV-2 B. 1.617 and B. 1.618 variants identified in India provide partial resistance to vaccine-elicited and therapeutic monoclonal antibodies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Krueger, N.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Sidarovich, A.; Moldenhauer, A.-S.; Winkler, M.S.; Schulz, S. SARS-CoV-2 variant B. 1.617 is resistant to Bamlanivimab and evades antibodies induced by infection and vaccination. Cell Rep. 2021, 36, 109415. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar]
- Rani, P.R.; Imran, M.; Lakshmi, J.V.; Jolly, B.; Afsar, S.; Jain, A.; Divakar, M.K.; Suresh, P.; Sharma, D.; Rajesh, N. Insights from genomes and genetic epidemiology of SARS-CoV-2 isolates from the state of Andhra Pradesh. Epidemiol. Infect. 2021, 149, e181. [Google Scholar] [CrossRef]
- Sarkar, P.; Banerjee, S.; Saha, S.A.; Mitra, P.; Sarkar, S. Genome surveillance of SARS-CoV-2 variants and their role in pathogenesis focusing on second wave of COVID-19 in India. Sci. Rep. 2023, 13, 4692. [Google Scholar] [CrossRef]
- Singh, U.B.; Deb, S.; Rani, L.; Bhardwaj, D.; Gupta, R.; Kabra, M.; Bala, K.; Kumari, L.; Perumalla, S.; Shukla, J. Genomic surveillance of SARS-CoV-2 upsurge in India due to Omicron sub-lineages BA. 2.74, BA. 2.75 and BA. 2.76. Lancet Reg. Health-Southeast Asia 2023, 11, 100148. [Google Scholar] [CrossRef]
- COVID-19 Portfolio. Search. Available online: https://icite.od.nih.gov/covid19/search/#search:searchId=64abd7893089f55f5254fa25 (accessed on 11 July 2023).
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon III, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F. Evaluating the effects of SARS-CoV-2 spike mutation D614G on transmissibility and pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Triche, T.J.; Bard, J.D.; Biegel, J.A.; Judkins, A.R.; Gai, X. Spike Protein NTD mutation G142D in SARS-CoV-2 Delta VOC lineages is associated with frequent back mutations, increased viral loads, and immune evasion. medRxiv 2021. [Google Scholar]
- Wang, Q.; Ye, S.-B.; Zhou, Z.-J.; Song, A.-L.; Zhu, X.; Peng, J.-M.; Liang, R.-M.; Yang, C.-H.; Yu, X.-W.; Huang, X. Key mutations in the spike protein of SARS-CoV-2 affecting neutralization resistance and viral internalization. J. Med. Virol. 2023, 95, e28407. [Google Scholar] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D.; Alameh, M.-G.; de Silva, T.; Collini, P.; Hornsby, H.; Brown, R.; LaBranche, C.C.; Edwards, R.J.; Sutherland, L.; Santra, S. D614G spike mutation increases SARS CoV-2 susceptibility to neutralization. Cell Host Microbe 2021, 29, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D. Antibody resistance of SARS-CoV-2 variants B. 1.351 and B. 1.1. 7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef]
- Asif, A.; Ilyas, I.; Abdullah, M.; Sarfraz, S.; Mustafa, M.; Mahmood, A. The Comparison of Mutational Progression in SARS-CoV-2: A Short Updated Overview. J. Mol. Pathol. 2022, 3, 201–218. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by the novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020, 94, 10–1128. [Google Scholar]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Socher, E.; Heger, L.; Paulsen, F.; Zunke, F.; Arnold, P. Molecular dynamics simulations of the delta and omicron SARS-CoV-2 spike–ACE2 complexes reveal distinct changes between both variants. Comput. Struct. Biotechnol. J. 2022, 20, 1168–1176. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Zhou, D.; Ginn, H.M.; Duyvesteyn, H.M.; Supasa, P.; Case, J.B.; Zhao, Y.; Walter, T.S.; Mentzer, A.J.; Liu, C. The antigenic anatomy of SARS-CoV-2 receptor binding domain. Cell 2021, 184, 2183–2200. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, J.; Gonçalves, P.F.B.; Netz, P.A. Why does the novel coronavirus spike protein interact so strongly with the human ACE2? A thermodynamic answer. ChemBioChem 2021, 22, 865–875. [Google Scholar]
- Watson, O.J.; Barnsley, G.; Toor, J.; Hogan, A.B.; Winskill, P.; Ghani, A.C. Global impact of the first year of COVID-19 vaccination: A mathematical modelling study. Lancet Infect. Dis. 2022, 22, 1293–1302. [Google Scholar] [PubMed]
- Baig, R.; Mateen, M.A.; Aborode, A.T.; Novman, S.; Matheen, I.A.; Siddiqui, O.S.; Ahmed, F.A. Third wave in India and an update on vaccination: A short communication. Ann. Med. Surg. 2022, 75, 103414. [Google Scholar]
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [PubMed]
- De Wilde, A.H.; Snijder, E.J.; Kikkert, M.; van Hemert, M.J. Host factors in coronavirus replication. Roles Host Gene Non-Coding RNA Expr. Virus Infect. 2018, 419, 1–42. [Google Scholar]
- Salehi, N.; Amiri-Yekta, A.; Totonchi, M. Profiling of Initial available SARS-CoV-2 sequences from Iranian related COVID-19 patients. Cell J. Yakhteh 2020, 22, 148. [Google Scholar]
- Gómez, C.E.; Perdiguero, B.; Esteban, M. Emerging SARS-CoV-2 variants and impact in global vaccination programs against SARS-CoV-2/COVID-19. Vaccines 2021, 9, 243. [Google Scholar] [CrossRef]
- Kumar, S.; Karuppanan, K.; Subramaniam, G. Omicron (BA. 1) and Sub-Variants (BA. 1.1, BA. 2 and BA. 3) of SARS-CoV-2 Spike Infectivity and Pathogenicity: A Comparative Sequence and Structural-based Computational Assessment. J. Med. Virol. 2022, 94, 4780–4791. [Google Scholar]
- Mejdani, M.; Haddadi, K.; Pham, C.; Mahadevan, R. SARS-CoV-2 receptor-binding mutations and antibody contact sites. Antib. Ther. 2021, 4, 149–158. [Google Scholar]
- Yahi, N.; Chahinian, H.; Fantini, J. Infection-enhancing anti-SARS-CoV-2 antibodies recognize both the original Wuhan/D614G strain and Delta variants. A potential risk for mass vaccination? J. Infect. 2021, 83, 607–635. [Google Scholar] [CrossRef] [PubMed]
- Kubik, S.; Arrigo, N.; Bonet, J.; Xu, Z. Mutational hotspot in the SARS-CoV-2 Spike protein N-terminal domain conferring immune escape potential. Viruses 2021, 13, 2114. [Google Scholar] [PubMed]







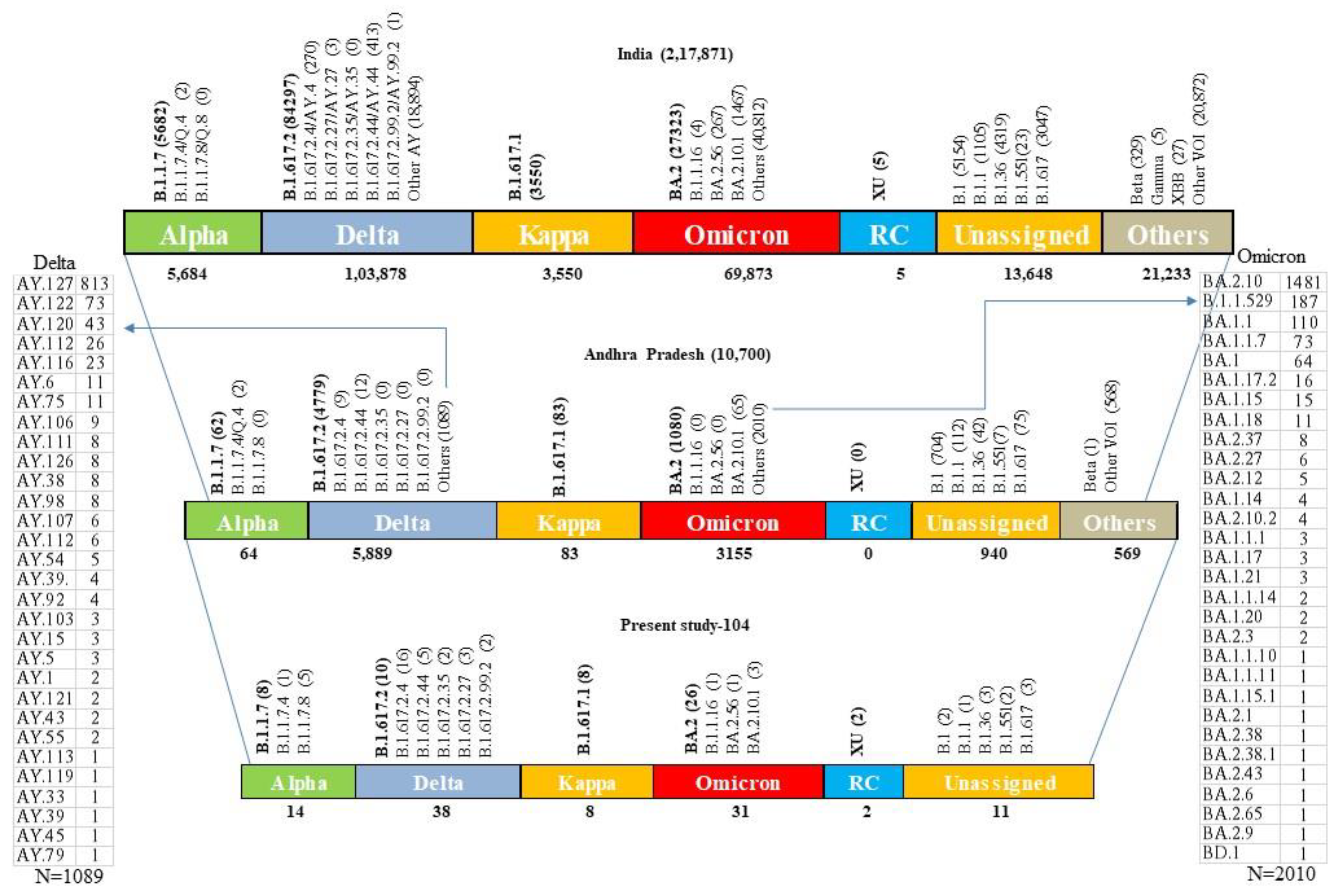
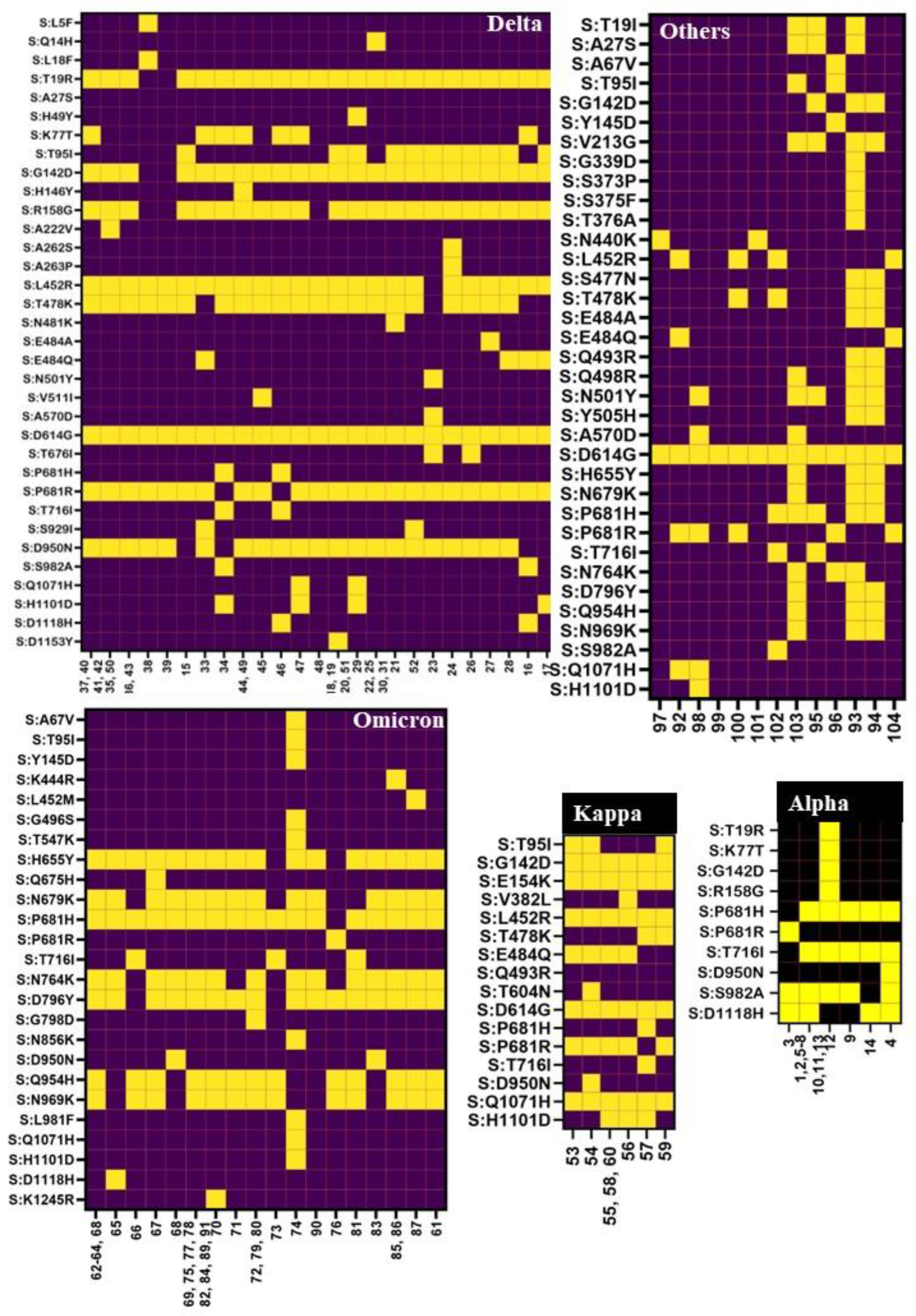
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primers | Primer Binding | Target Size | Overlap | ||
|---|---|---|---|---|---|---|
| Start | End | |||||
| T1 | FP * | ACAAATCCAATTCAGTTGTCTTCCTATTC | 21,357 | 22,326 | 969 | 364 |
| RP | CACCAGCTGTCCAACCTGAAGA | |||||
| T2 | FP * | CAATTTTGTAATGATCCATTTTTGGGTGT | 21,962 | 22,903 | 941 | 386 |
| RP | ACCACCAACCTTAGAATCAAGATTGT | |||||
| T3 | FP * | AGAGTCCAACCAACAGAATCTATTGT | 22,517 | 23,522 | 1005 | 399 |
| RP | CAGCCCCTATTAAACAGCCTGC | |||||
| T4 | FP * | CCAGCAACTGTTTGTGGACCTA | 23,123 | 24,126 | 1003 | 337 |
| RP | CATTTCATCTGTGAGCAAAGGTGG | |||||
| T5 | FP * | GTGGTGATTCAACTGAATGCAGC | 23,789 | 24,789 | 1000 | 398 |
| RP | GTGAAGTTCTTTTCTTGTGCAGGG | |||||
| T6 | FP * | GCACTTGGAAAACTTCAAGATGTGG | 24,391 | 25,673 | 1282 | 390 |
| RP | AGGTGTGAGTAAACTGTTACAAACAAC | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagaraja, M.; Sireesha, K.; Srikar, A.; Sudheer Kumar, K.; Mohan, A.; Vengamma, B.; Tirumala, C.; Verma, A.; Kalawat, U. Mutation Analysis of SARS-CoV-2 Variants Isolated from Symptomatic Cases from Andhra Pradesh, India. Viruses 2023, 15, 1656. https://0-doi-org.brum.beds.ac.uk/10.3390/v15081656
Nagaraja M, Sireesha K, Srikar A, Sudheer Kumar K, Mohan A, Vengamma B, Tirumala C, Verma A, Kalawat U. Mutation Analysis of SARS-CoV-2 Variants Isolated from Symptomatic Cases from Andhra Pradesh, India. Viruses. 2023; 15(8):1656. https://0-doi-org.brum.beds.ac.uk/10.3390/v15081656
Chicago/Turabian StyleNagaraja, Mudhigeti, Kodavala Sireesha, Anagoni Srikar, Katari Sudheer Kumar, Alladi Mohan, Bhuma Vengamma, Chejarla Tirumala, Anju Verma, and Usha Kalawat. 2023. "Mutation Analysis of SARS-CoV-2 Variants Isolated from Symptomatic Cases from Andhra Pradesh, India" Viruses 15, no. 8: 1656. https://0-doi-org.brum.beds.ac.uk/10.3390/v15081656
{kind=link}