
The Ubiquitin-Proteasome System Facilitates Membrane Fusion and Uncoating during Coronavirus Entry
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Chemicals and Antibodies
2.3. Cell Viability Assay
2.4. Virus Infection and Drug Treatment
2.5. Virus Internalization Assay
2.6. RNA Preparation and Real Time RT-qPCR
2.7. Western Blot Analysis
2.8. Virus Titration
2.9. R18 Labeling and R18/DIOC Labeling of Virus
2.10. Statistical Analysis
2.11. Densitometry
3. Results
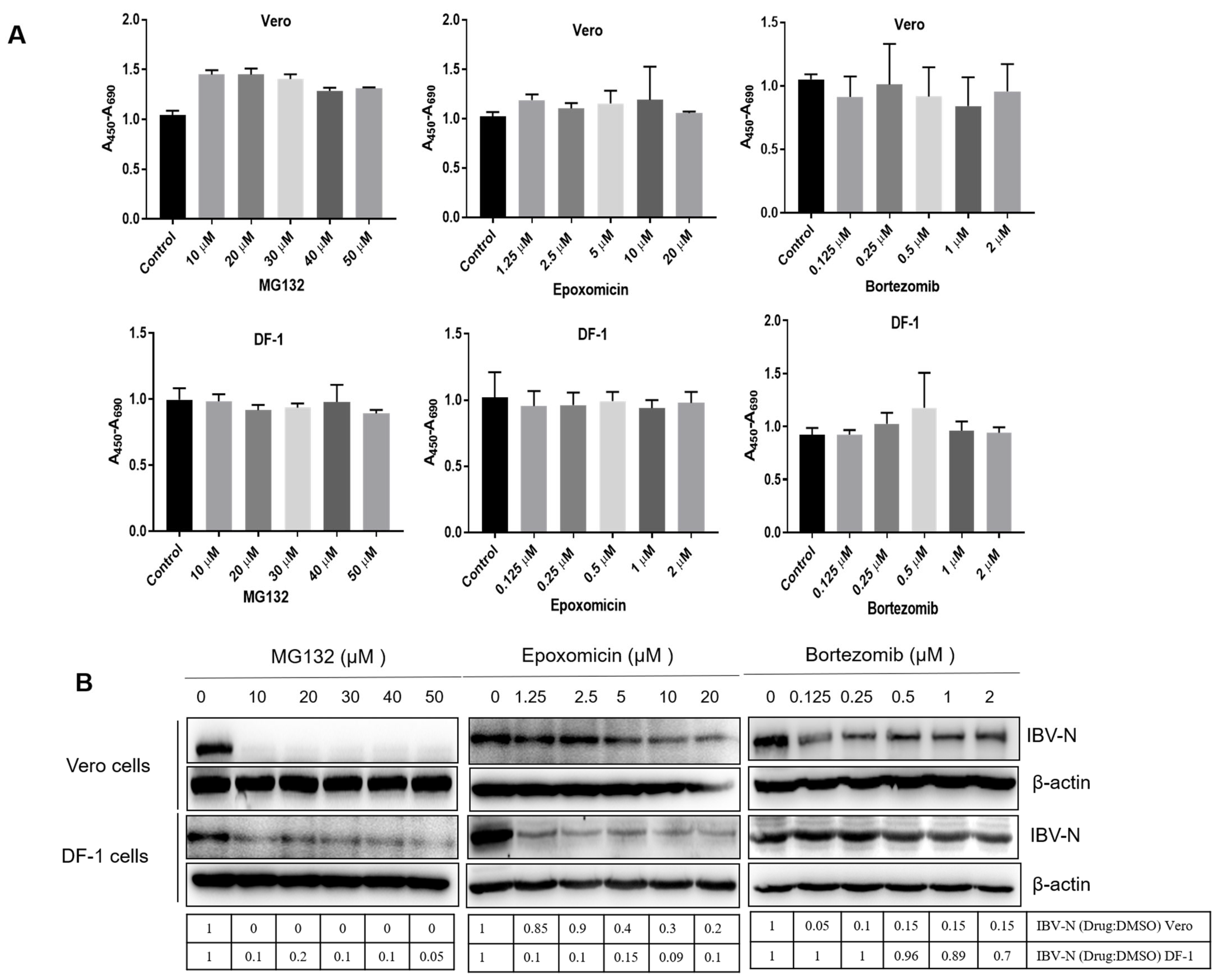
3.1. Proteasome Inhibitors Interfere with IBV Infection
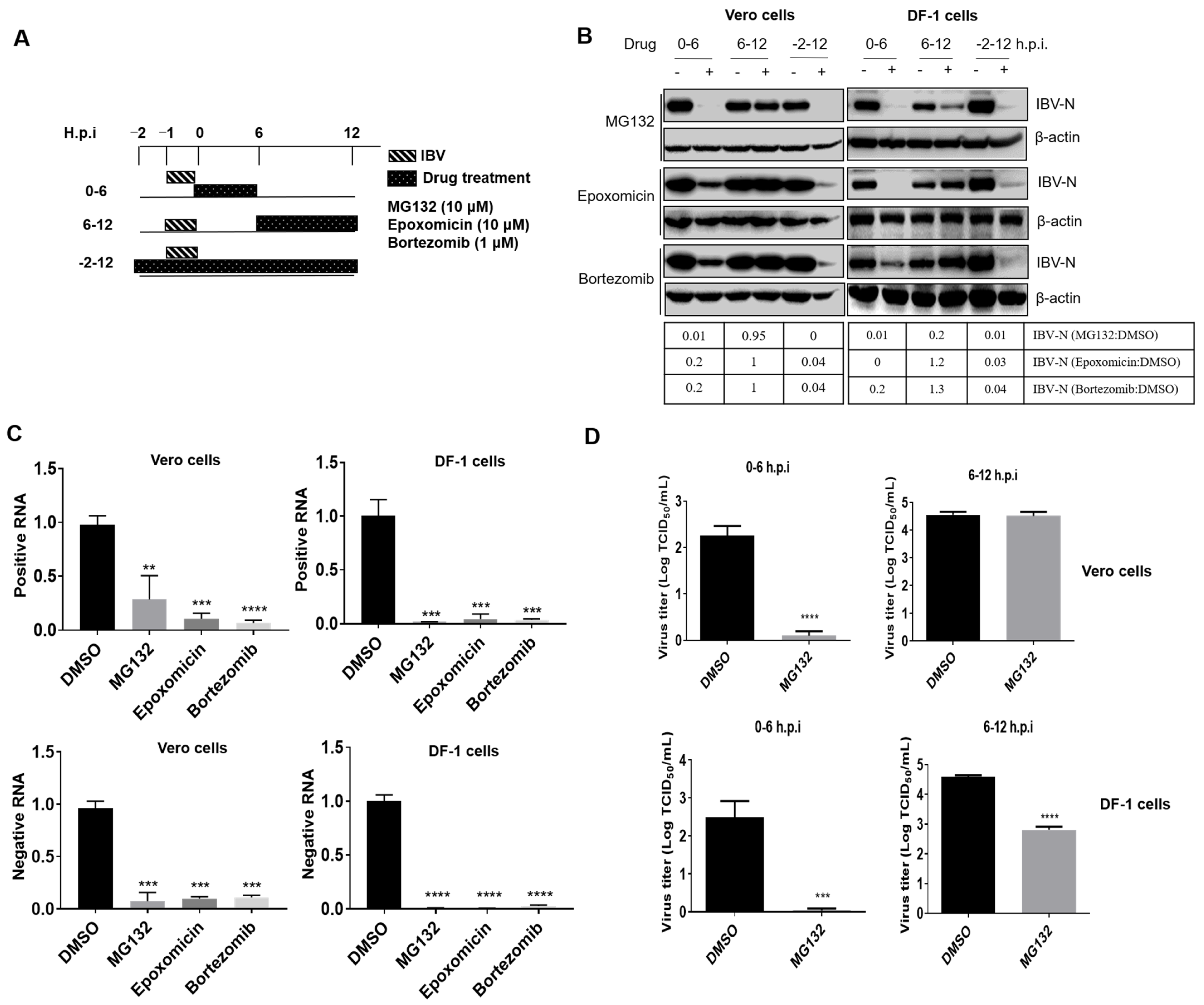
3.2. Proteasome Inhibitors Act at An Early Step of IBV Infection
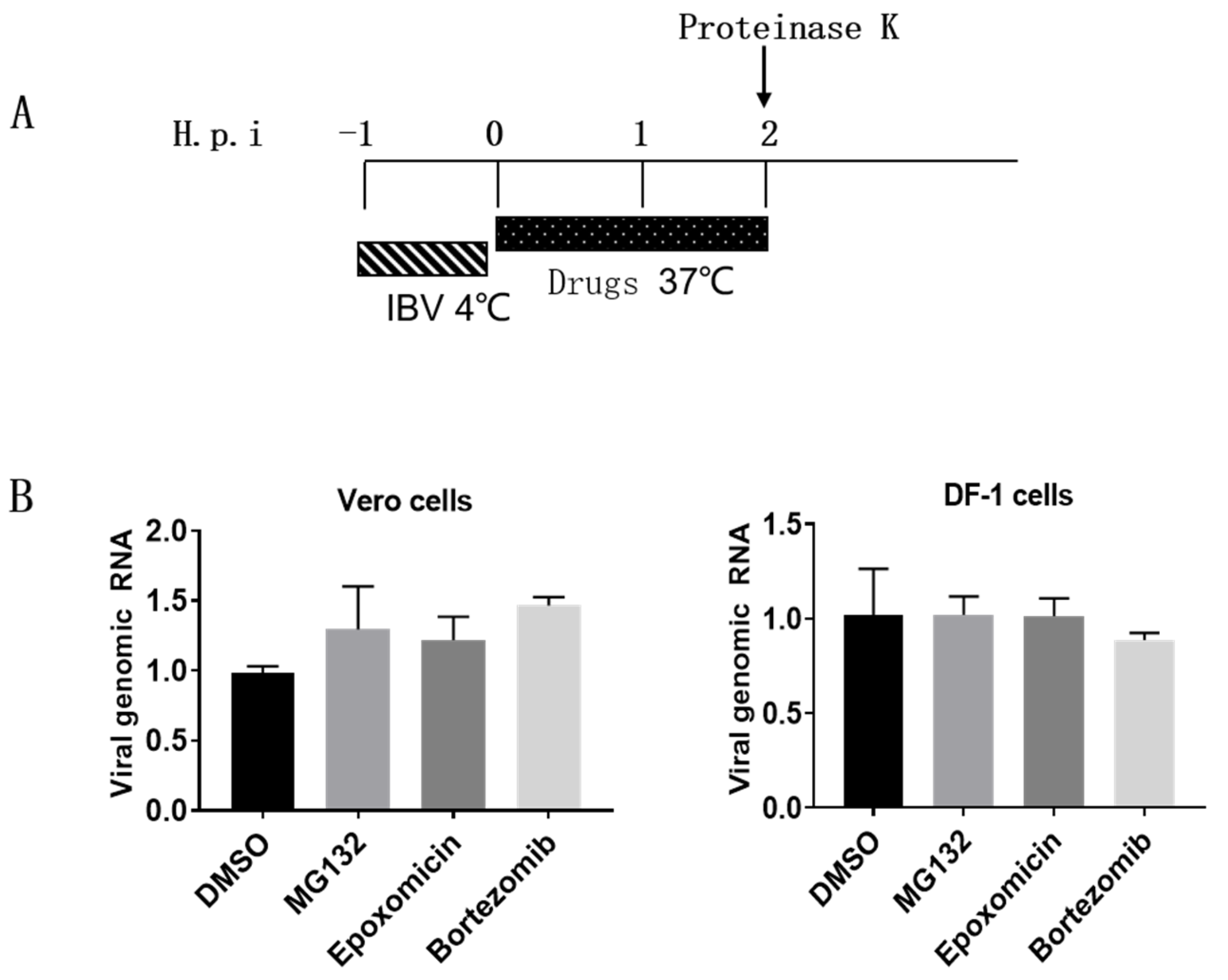
3.3. Proteasome Inhibitors Do Not Interfere with IBV Internalization
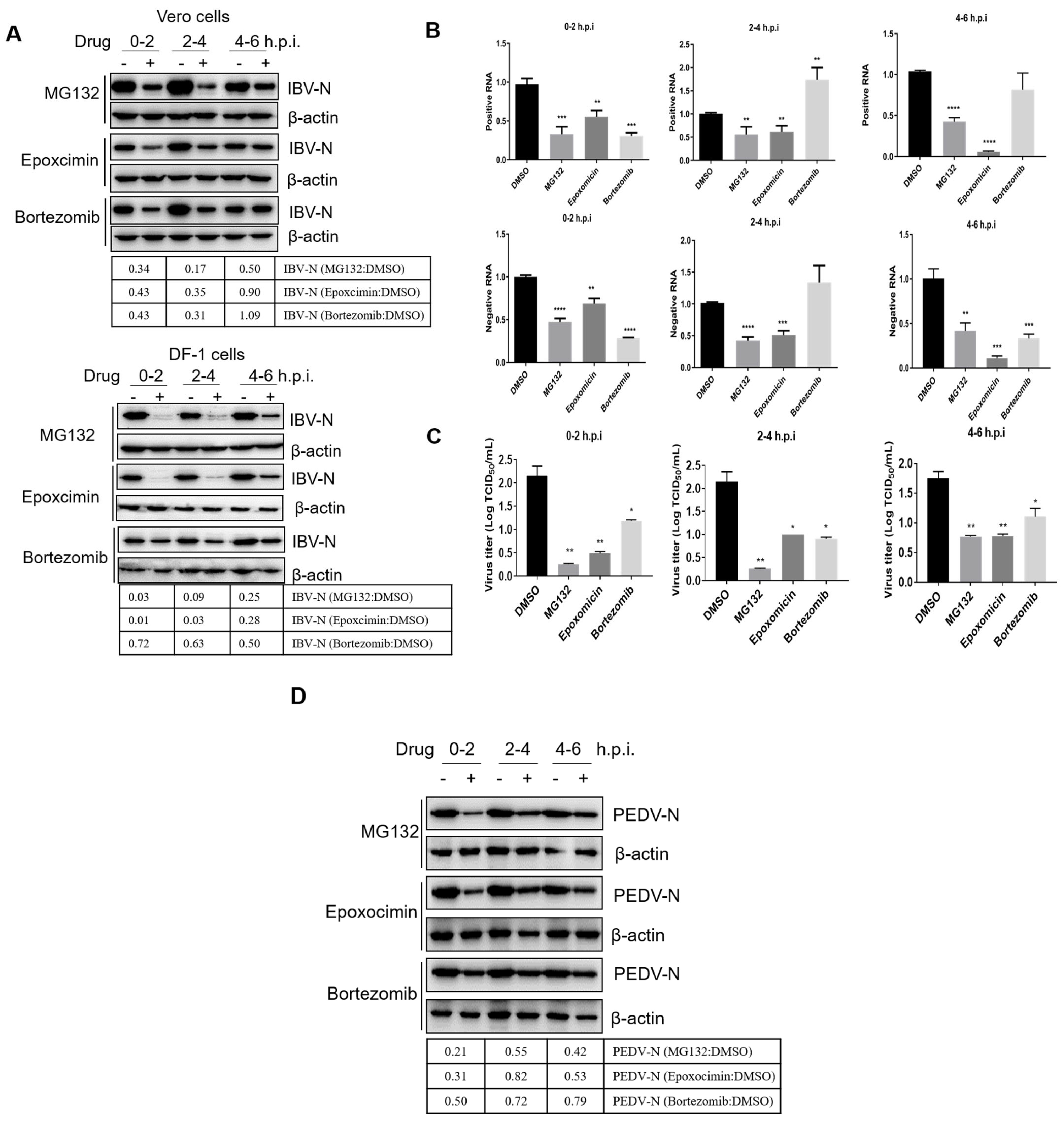
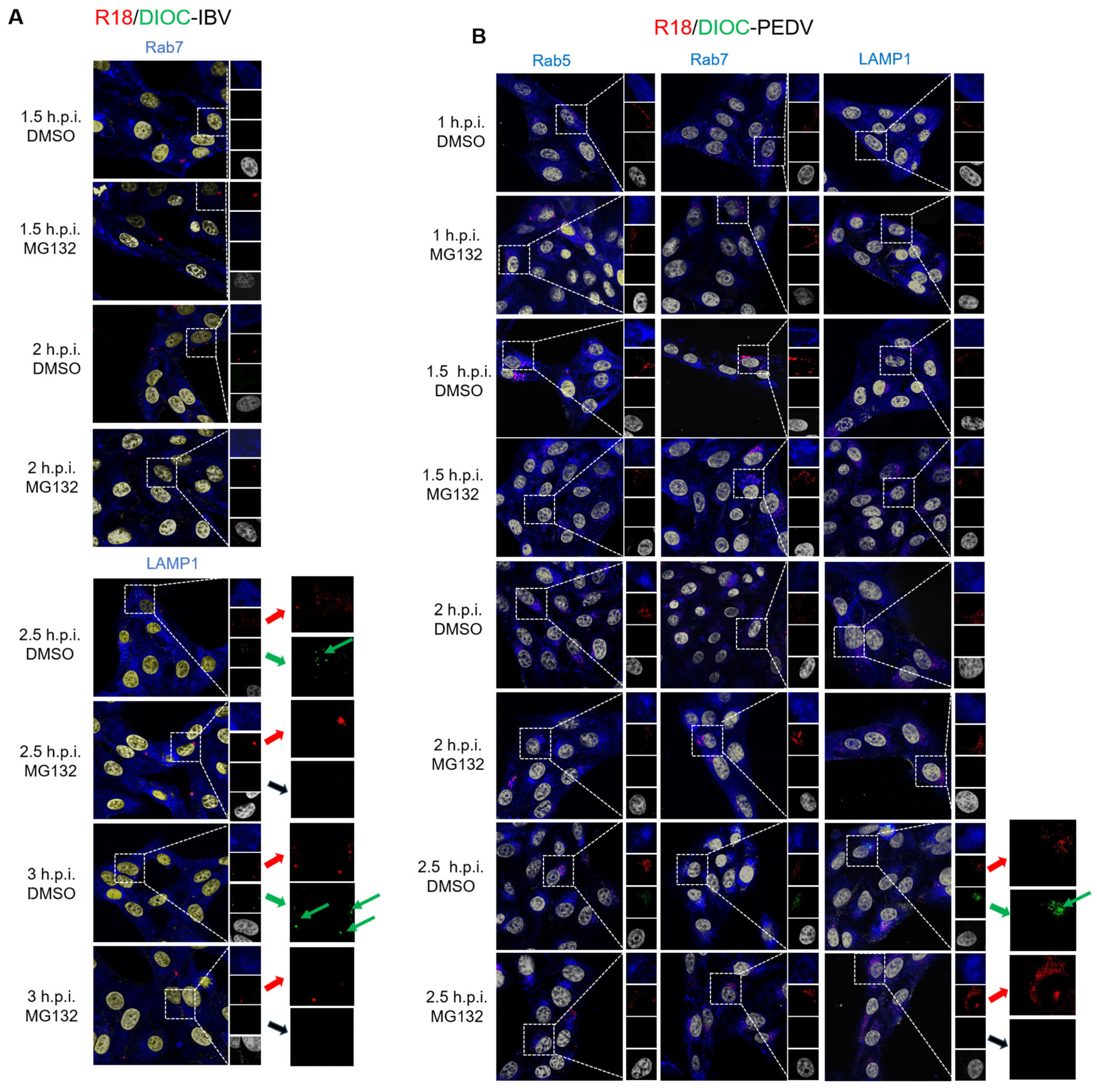
3.4. Proteasome Inhibitors Interfere with IBV and PEDV Membrane Fusion
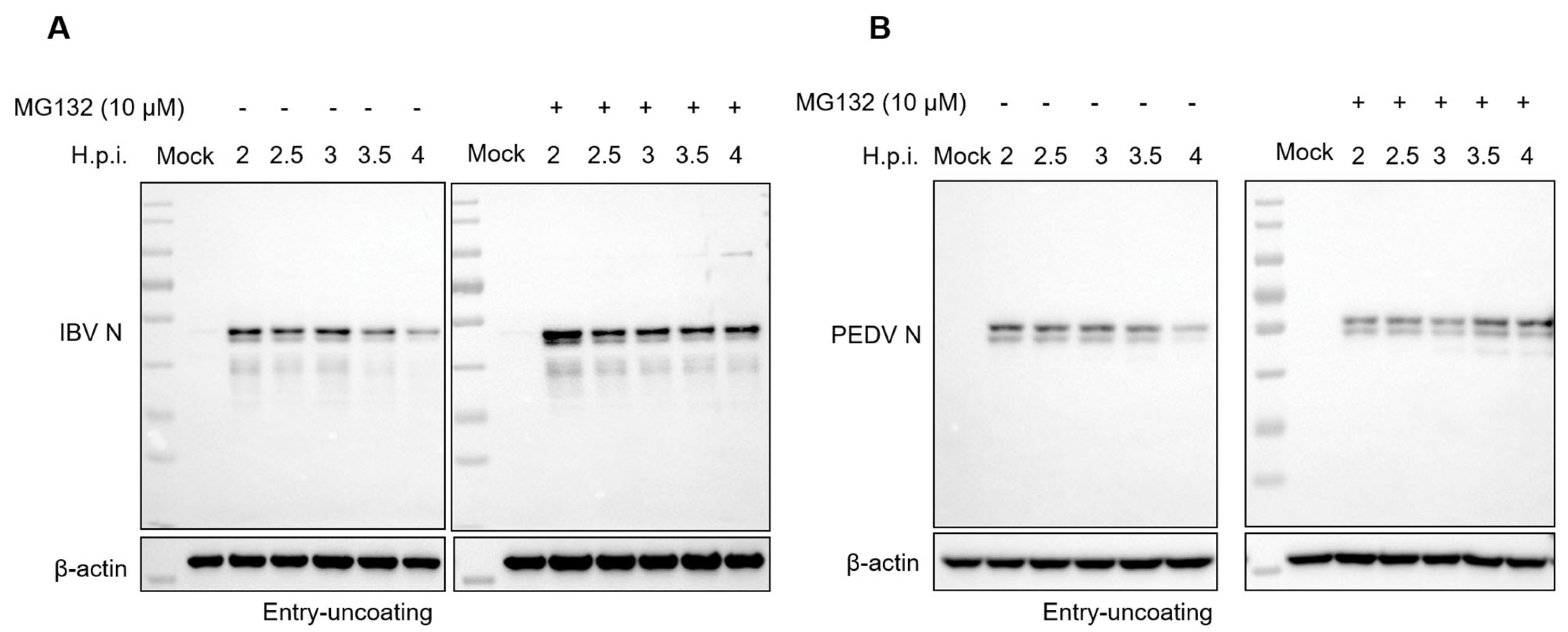
3.5. Proteasome Inhibitors Interfere with IBV and PEDV N Protein Degradation and Uncoating
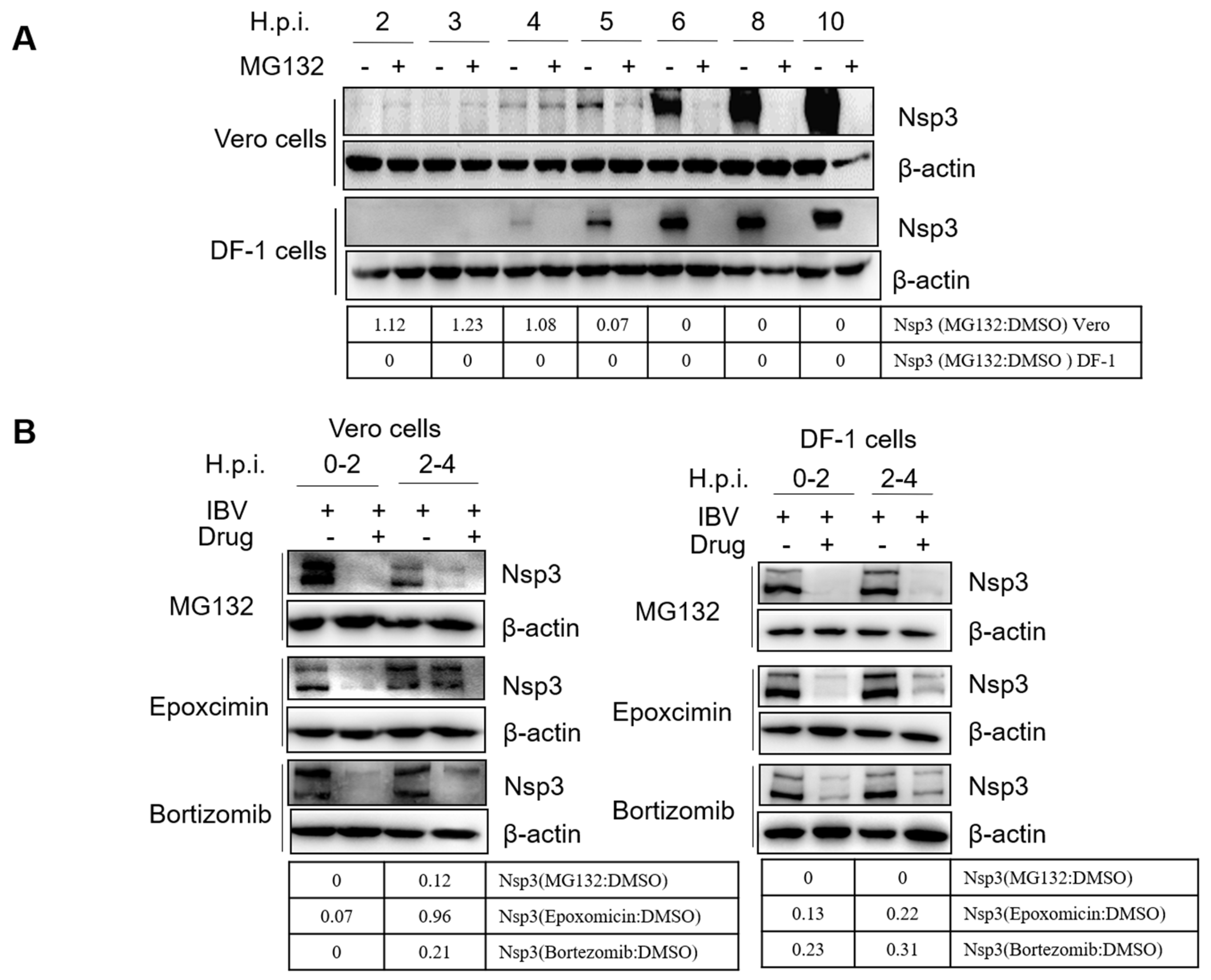
3.6. Proteasome Inhibitors Interfere with IBV Initial Translation
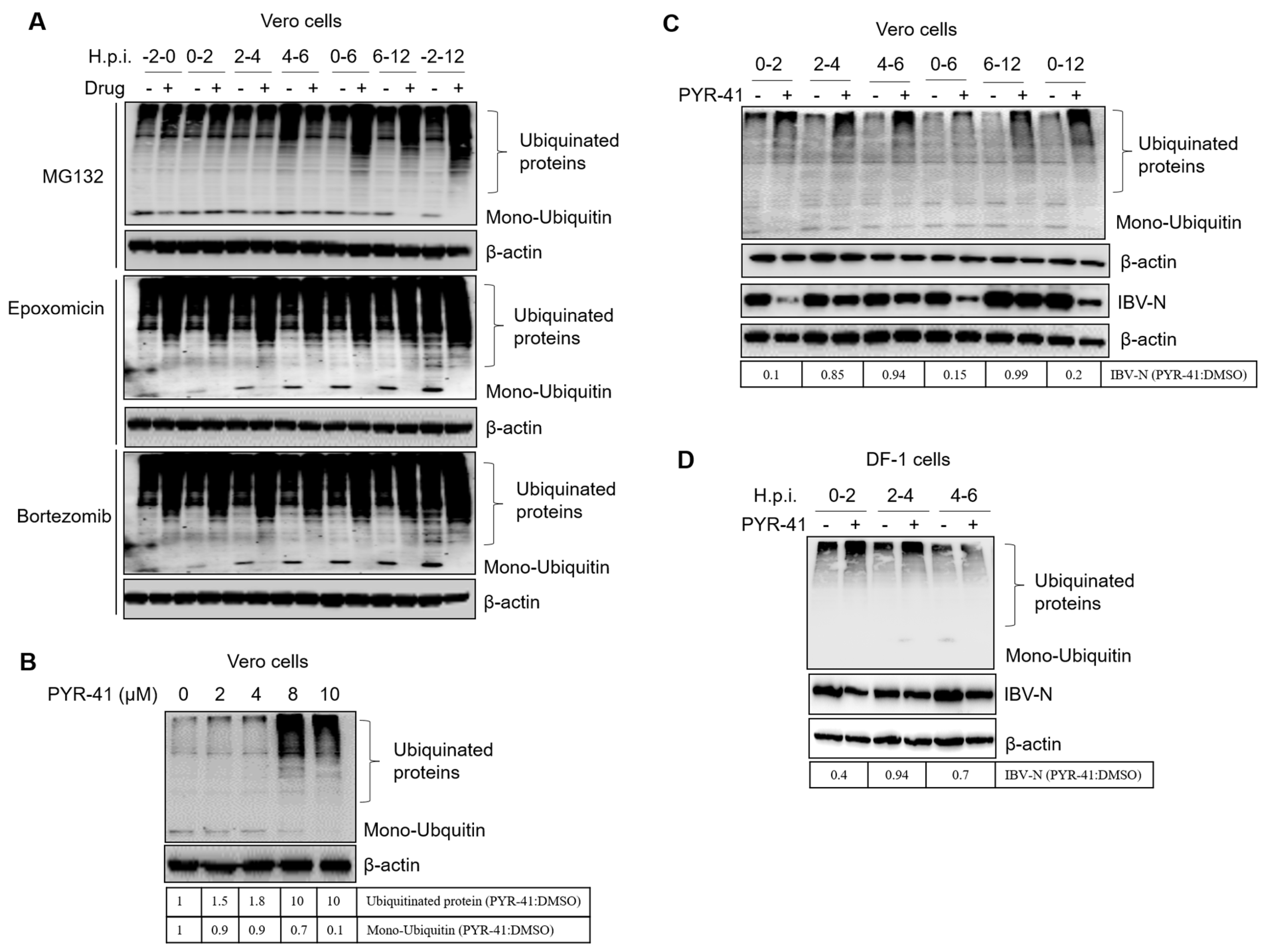
3.7. Ubiquitination of Cellular or Viral Proteins Is Necessary for IBV Entry
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The molecular virology of coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Jackwood, M.W. Review of infectious bronchitis virus around the world. Avian Dis. 2012, 56, 634–641. [Google Scholar] [CrossRef]
- Pensaert, M.B.; de Bouck, P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978, 58, 243–247. [Google Scholar] [CrossRef]
- Puranaveja, S.; Poolperm, P.; Lertwatcharasarakul, P.; Kesdaengsakonwut, S.; Boonsoongnern, A.; Urairong, K.; Kitikoon, P.; Choojai, P.; Kedkovid, R.; Teankum, K.; et al. Chinese-like strain of porcine epidemic diarrhea virus, Thailand. Emerg. Infect. Dis. 2009, 15, 1112–1115. [Google Scholar] [CrossRef]
- Takahashi, K.; Okada, K.; Ohshima, K. An outbreak of swine diarrhea of a new-type associated with coronavirus-like particles in Japan. Jpn. J. Vet. Sci. 1983, 45, 829–832. [Google Scholar] [CrossRef]
- Park, S.J.; Moon, H.J.; Yang, J.S.; Lee, C.S.; Song, D.S.; Kang, B.K.; Park, B.K. Sequence analysis of the partial spike glycoprotein gene of porcine epidemic diarrhea viruses isolated in Korea. Virus Genes 2007, 35, 321–332. [Google Scholar] [CrossRef]
- Chen, J.F.; Sun, D.B.; Wang, C.B.; Shi, H.Y.; Cui, X.C.; Liu, S.W.; Qiu, H.J.; Feng, L. Molecular characterization and phylogenetic analysis of membrane protein genes of porcine epidemic diarrhea virus isolates in China. Virus Genes 2008, 36, 355–364. [Google Scholar] [CrossRef]
- Chen, Q.; Li, G.; Stasko, J.; Thomas, J.T.; Stensland, W.R.; Pillatzki, A.E.; Gauger, P.C.; Schwartz, K.J.; Madson, D.; Yoon, K.J.; et al. Isolation and characterization of porcine epidemic diarrhea viruses associated with the 2013 disease outbreak among swine in the United States. J. Clin. Microbiol. 2014, 52, 234–243. [Google Scholar] [CrossRef]
- Song, D.; Park, B. Porcine epidemic diarrhoea virus: A comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes 2012, 44, 167–175. [Google Scholar] [CrossRef]
- Lomniczi, B.; Kennedy, I. Genome of infectious bronchitis virus. J. Virol. 1977, 24, 99–107. [Google Scholar] [CrossRef]
- Quinteros, J.A.; Noormohammadi, A.H.; Lee, S.W.; Browning, G.F.; Diaz-Mendez, A. Genomics and pathogenesis of the avian coronavirus infectious bronchitis virus. Aust. Vet. J. 2022, 100, 496–512. [Google Scholar] [CrossRef]
- Compton, S.R.; Barthold, S.W.; Smith, A.L. The cellular and molecular pathogenesis of coronaviruses. Lab. Anim. Sci. 1993, 43, 15–28. [Google Scholar]
- Wang, H.; Yuan, X.; Sun, Y.; Mao, X.; Meng, C.; Tan, L.; Song, C.; Qiu, X.; Ding, C.; Liao, Y. Infectious bronchitis virus entry mainly depends on clathrin mediated endocytosis and requires classical endosomal/lysosomal system. Virology 2019, 528, 118–136. [Google Scholar] [CrossRef]
- Wei, X.; She, G.; Wu, T.; Xue, C.; Cao, Y. PEDV enters cells through clathrin-, caveolae-, and lipid raft-mediated endocytosis and traffics via the endo-/lysosome pathway. Vet. Res. 2020, 51, 10. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Hou, W.; Shan, Y.; Wang, S.; Liu, F. Dynamic Dissection of the Endocytosis of Porcine Epidemic Diarrhea Coronavirus Cooperatively Mediated by Clathrin and Caveolae as Visualized by Single-Virus Tracking. mBio 2021, 12, e00256-21. [Google Scholar] [CrossRef]
- Brierley, I.; Digard, P.; Inglis, S.C. Characterization of an efficient coronavirus ribosomal frameshifting signal: Requirement for an RNA pseudoknot. Cell 1989, 57, 537–547. [Google Scholar] [CrossRef]
- Liu, D.X.; Inglis, S.C. Association of the infectious bronchitis virus 3c protein with the virion envelope. Virology 1991, 185, 911–917. [Google Scholar] [CrossRef]
- Yang, D.; Leibowitz, J.L. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015, 206, 120–133. [Google Scholar] [CrossRef]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. Curr. Top. Microbiol. Immunol. 2005, 287, 1–30. [Google Scholar]
- Bickerton, E.; Keep, S.M.; Britton, P. Reverse Genetics System for the Avian Coronavirus Infectious Bronchitis Virus. Methods Mol. Biol. 2017, 1602, 83–102. [Google Scholar]
- Graham, R.L.; Sparks, J.S.; Eckerle, L.D.; Sims, A.C.; Denison, M.R. SARS coronavirus replicase proteins in pathogenesis. Virus Res. 2008, 133, 88–100. [Google Scholar] [CrossRef]
- Pasternak, A.O.; Spaan, W.J.; Snijder, E.J. Nidovirus transcription: How to make sense…? J. Gen. Virol. 2006, 87 Pt 6, 1403–1421. [Google Scholar] [CrossRef]
- Ruch, T.R.; Machamer, C.E. The Coronavirus E Protein: Assembly and Beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef]
- Wang, J.; Fang, S.; Xiao, H.; Chen, B.; Tam, J.P.; Liu, D.X. Interaction of the coronavirus infectious bronchitis virus membrane protein with beta-actin and its implication in virion assembly and budding. PLoS ONE 2009, 4, e4908. [Google Scholar]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 1994, 79, 13–21. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Seissler, T.; Marquet, R.; Paillart, J.C. Hijacking of the Ubiquitin/Proteasome Pathway by the HIV Auxiliary Proteins. Viruses 2017, 9, 322. [Google Scholar] [CrossRef]
- Hicke, L. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2001, 2, 195–201. [Google Scholar] [CrossRef]
- Verchot, J. Plant Virus Infection and the Ubiquitin Proteasome Machinery: Arms Race along the Endoplasmic Reticulum. Viruses 2016, 8, 314. [Google Scholar] [CrossRef]
- Banks, L.; Pim, D.; Thomas, M. Viruses and the 26S proteasome: Hacking into destruction. Trends Biochem. Sci. 2003, 28, 452–459. [Google Scholar] [CrossRef]
- Bailey-Elkin, B.A.; Knaap, R.C.M.; Kikkert, M.; Mark, B.L. Structure and Function of Viral Deubiquitinating Enzymes. J. Mol. Biol. 2017, 429, 3441–3470. [Google Scholar] [CrossRef]
- Clague, M.J.; Urbe, S. Ubiquitin: Same molecule, different degradation pathways. Cell 2010, 143, 682–685. [Google Scholar] [CrossRef]
- Rock, K.L.; Gramm, C.; Rothstein, L.; Clark, K.; Stein, R.; Dick, L.; Hwang, D.; Goldberg, A.L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78, 761–771. [Google Scholar] [CrossRef]
- Eytan, E.; Ganoth, D.; Armon, T.; Hershko, A. ATP-dependent incorporation of 20S protease into the 26S complex that degrades proteins conjugated to ubiquitin. Proc. Natl. Acad. Sci. USA 1989, 86, 7751–7755. [Google Scholar] [CrossRef]
- Kleiger, G.; Mayor, T. Perilous journey: A tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014, 24, 352–359. [Google Scholar] [CrossRef]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef]
- Delboy, M.G.; Roller, D.G.; Nicola, A.V. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J. Virol. 2008, 82, 3381–3390. [Google Scholar] [CrossRef]
- Burch, A.D.; Weller, S.K. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J. Virol. 2004, 78, 7175–7185. [Google Scholar] [CrossRef]
- Satheshkumar, P.S.; Anton, L.C.; Sanz, P.; Moss, B. Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J. Virol. 2009, 83, 2469–2479. [Google Scholar] [CrossRef]
- Isaacson, M.K.; Ploegh, H.L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 2009, 5, 559–570. [Google Scholar] [CrossRef]
- Gao, G.; Luo, H. The ubiquitin-proteasome pathway in viral infections. Can. J. Physiol. Pharmacol. 2006, 84, 5–14. [Google Scholar] [CrossRef]
- Yuan, Z.; Hu, B.; Xiao, H.; Tan, X.; Li, Y.; Tang, K.; Zhang, Y.; Cai, K.; Ding, B. The E3 Ubiquitin Ligase RNF5 Facilitates SARS-CoV-2 Membrane Protein-Mediated Virion Release. mBio 2021, 13, e0316821. [Google Scholar] [CrossRef]
- Giraldo, M.I.; Xia, H.; Aguilera-Aguirre, L.; Hage, A.; van Tol, S.; Shan, C.; Xie, X.; Sturdevant, G.L.; Robertson, S.J.; McNally, K.L.; et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 2020, 585, 414–419. [Google Scholar] [CrossRef]
- Schneider, S.M.; Lee, B.H.; Nicola, A.V. Viral entry and the ubiquitin-proteasome system. Cell. Microbiol. 2021, 23, e13276. [Google Scholar] [CrossRef]
- Wang, S.; Liu, H.; Zu, X.; Liu, Y.; Chen, L.; Zhu, X.; Zhang, L.; Zhou, Z.; Xiao, G.; Wang, W. The ubiquitin-proteasome system is essential for the productive entry of Japanese encephalitis virus. Virology 2016, 498, 116–127. [Google Scholar] [CrossRef]
- Byk, L.A.; Iglesias, N.G.; De Maio, F.A.; Gebhard, L.G.; Rossi, M.; Gamarnik, A.V. Dengue Virus Genome Uncoating Requires Ubiquitination. mBio 2016, 7, e00804-16. [Google Scholar] [CrossRef]
- Raaben, M.; Posthuma, C.C.; Verheije, M.H.; te Lintelo, E.G.; Kikkert, M.; Drijfhout, J.W.; Snijder, E.J.; Rottier, P.J.; de Haan, C.A. The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J. Virol. 2010, 84, 7869–7879. [Google Scholar] [CrossRef]
- Yu, G.Y.; Lai, M.M. The ubiquitin-proteasome system facilitates the transfer of murine coronavirus from endosome to cytoplasm during virus entry. J. Virol. 2005, 79, 644–648. [Google Scholar] [CrossRef]
- Choi, A.G.; Wong, J.; Marchant, D.; Luo, H. The ubiquitin-proteasome system in positive-strand RNA virus infection. Rev. Med. Virol. 2013, 23, 85–96. [Google Scholar] [CrossRef]
- Alvarez, E.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Marcos-Villar, L.; Enjuanes, L. The envelope protein of severe acute respiratory syndrome coronavirus interacts with the non-structural protein 3 and is ubiquitinated. Virology 2010, 402, 281–291. [Google Scholar] [CrossRef]
- Li, Z.; Hao, P.; Zhao, Z.; Gao, W.; Huan, C.; Li, L.; Chen, X.; Wang, H.; Jin, N.; Luo, Z.Q.; et al. Correction To: The E3 ligase RNF5 restricts SARS-CoV-2 replication by targeting its envelope protein for degradation. Signal Transduct. Target. Ther. 2023, 8, 85. [Google Scholar] [CrossRef]
- Ko, A.; Lee, E.W.; Yeh, J.Y.; Yang, M.R.; Oh, W.; Moon, J.S.; Song, J. MKRN1 induces degradation of West Nile virus capsid protein by functioning as an E3 ligase. J. Virol. 2010, 84, 426–436. [Google Scholar] [CrossRef]
- Gilfoy, F.; Fayzulin, R.; Mason, P.W. West Nile virus genome amplification requires the functional activities of the proteasome. Virology 2009, 385, 74–84. [Google Scholar] [CrossRef]
- Luo, H. Interplay between the virus and the ubiquitin-proteasome system: Molecular mechanism of viral pathogenesis. Curr. Opin. Virol. 2016, 17, 1–10. [Google Scholar] [CrossRef]
- Calistri, A.; Munegato, D.; Carli, I.; Parolin, C.; Palu, G. The ubiquitin-conjugating system: Multiple roles in viral replication and infection. Cells 2014, 3, 386–417. [Google Scholar] [CrossRef]
- Schubert, U.; Ott, D.E.; Chertova, E.N.; Welker, R.; Tessmer, U.; Princiotta, M.F.; Bennink, J.R.; Krausslich, H.G.; Yewdell, J.W. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 2000, 97, 13057–13062. [Google Scholar] [CrossRef]
- Seyoum Tola, F. The Role of Ubiquitin-Proteasome System in the Pathogenesis of Severe Acute Respiratory Syndrome Coronavirus-2 Disease. Int. J. Inflamm. 2023, 2023, 6698069. [Google Scholar] [CrossRef]
- Schneider, M.; Ackermann, K.; Stuart, M.; Wex, C.; Protzer, U.; Schatzl, H.M.; Gilch, S. Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J. Virol. 2012, 86, 10112–10122. [Google Scholar] [CrossRef]
- Bolland, S.; Pierce, S.K. Ups and downs in the search for a Herpes simplex virus vaccine. elife 2015, 4, e06883. [Google Scholar] [CrossRef]
- Cheng, S.; Yan, W.; Gu, W.; He, Q. The ubiquitin-proteasome system is required for the early stages of porcine circovirus type 2 replication. Virology 2014, 456–457, 198–204. [Google Scholar] [CrossRef]
- Widjaja, I.; de Vries, E.; Tscherne, D.M.; Garcia-Sastre, A.; Rottier, P.J.; de Haan, C.A. Inhibition of the ubiquitin-proteasome system affects influenza A virus infection at a postfusion step. J. Virol. 2010, 84, 9625–9631. [Google Scholar] [CrossRef]
- Casorla-Perez, L.A.; Lopez, T.; Lopez, S.; Arias, C.F. The Ubiquitin-Proteasome System Is Necessary for Efficient Replication of Human Astrovirus. J. Virol. 2018, 92, e01809-17. [Google Scholar] [CrossRef]
- Wang, Z.; Ni, J.; Li, J.; Shi, B.; Xu, Y.; Yuan, Z. Inhibition of hepatitis B virus replication by cIAP2 involves accelerating the ubiquitin-proteasome-mediated destruction of polymerase. J. Virol. 2011, 85, 11457–11467. [Google Scholar] [CrossRef]
- Neznanov, N.; Dragunsky, E.M.; Chumakov, K.M.; Neznanova, L.; Wek, R.C.; Gudkov, A.V.; Banerjee, A.K. Different effect of proteasome inhibition on vesicular stomatitis virus and poliovirus replication. PLoS ONE 2008, 3, e1887. [Google Scholar] [CrossRef]
- Fung, T.S.; Liao, Y.; Liu, D.X. The endoplasmic reticulum stress sensor IRE1alpha protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J. Virol. 2014, 88, 12752–12764. [Google Scholar] [CrossRef]
- McHenry, E.W.; Reedman, E.J.; Sheppard, M. The physiological properties of ascorbic acid: An effect upon the weights of guinea-pigs. Biochem. J. 1938, 32, 1302–1304. [Google Scholar] [CrossRef]
- Chu, V.C.; McElroy, L.J.; Chu, V.; Bauman, B.E.; Whittaker, G.R. The avian coronavirus infectious bronchitis virus undergoes direct low-pH-dependent fusion activation during entry into host cells. J. Virol. 2006, 80, 3180–3188. [Google Scholar] [CrossRef]
- Krzyzaniak, M.A.; Zumstein, M.T.; Gerez, J.A.; Picotti, P.; Helenius, A. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog. 2013, 9, e1003309. [Google Scholar] [CrossRef]
- Tsubuki, S.; Saito, Y.; Tomioka, M.; Ito, H.; Kawashima, S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J. Biochem. 1996, 119, 572–576. [Google Scholar] [CrossRef]
- Meng, L.; Mohan, R.; Kwok, B.H.; Elofsson, M.; Sin, N.; Crews, C.M. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA 1999, 96, 10403–10408. [Google Scholar] [CrossRef]
- Adams, J.; Palombella, V.J.; Sausville, E.A.; Johnson, J.; Destree, A.; Lazarus, D.D.; Maas, J.; Pien, C.S.; Prakash, S.; Elliott, P.J. Proteasome inhibitors: A novel class of potent and effective antitumor agents. Cancer Res. 1999, 59, 2615–2622. [Google Scholar]
- Sawicki, S.G.; Sawicki, D.L.; Siddell, S.G. A Contemporary View of Coronavirus Transcription. J. Virol. 2006, 81, 20–29. [Google Scholar] [CrossRef]
- Bentley, K.; Keep, S.M.; Armesto, M.; Britton, P. Identification of a noncanonically transcribed subgenomic mRNA of infectious bronchitis virus and other gammacoronaviruses. J. Virol. 2013, 87, 2128–2136. [Google Scholar] [CrossRef]
- Sakai, T.; Ohuchi, M.; Imai, M.; Mizuno, T.; Kawasaki, K.; Kuroda, K.; Yamashina, S. Dual wavelength imaging allows analysis of membrane fusion of influenza virus inside cells. J. Virol. 2006, 80, 2013–2018. [Google Scholar] [CrossRef]
- Mao, S.; Cai, X.; Niu, S.; Wei, J.; Jiang, N.; Deng, H.; Wang, W.; Zhang, J.; Shen, S.; Ma, Y.; et al. TRIM21 promotes ubiquitination of SARS-CoV-2 nucleocapsid protein to regulate innate immunity. J. Med. Virol. 2023, 95, e28719. [Google Scholar] [CrossRef]
- Wang, H.; Chen, X.; Kong, N.; Jiao, Y.; Sun, D.; Dong, S.; Qin, W.; Zhai, H.; Yu, L.; Zheng, H.; et al. TRIM21 inhibits porcine epidemic diarrhea virus proliferation by proteasomal degradation of the nucleocapsid protein. Arch. Virol. 2021, 166, 1903–1911. [Google Scholar] [CrossRef]
- Nakagawa, K.; Lokugamage, K.G.; Makino, S. Viral and Cellular mRNA Translation in Coronavirus-Infected Cells. Adv. Virus Res. 2016, 96, 165–192. [Google Scholar]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef]
- Yang, Y.; Kitagaki, J.; Dai, R.M.; Tsai, Y.C.; Lorick, K.L.; Ludwig, R.L.; Pierre, S.A.; Jensen, J.P.; Davydov, I.V.; Oberoi, P.; et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007, 67, 9472–9481. [Google Scholar] [CrossRef]
- Yang, B.; Jia, Y.; Meng, Y.; Xue, Y.; Liu, K.; Li, Y.; Liu, S.; Li, X.; Cui, K.; Shang, L.; et al. SNX27 suppresses SARS-CoV-2 infection by inhibiting viral lysosome/late endosome entry. Proc. Natl. Acad. Sci. USA 2022, 119, e2117576119. [Google Scholar] [CrossRef]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef]
- Milewska, A.; Nowak, P.; Owczarek, K.; Szczepanski, A.; Zarebski, M.; Hoang, A.; Berniak, K.; Wojarski, J.; Zeglen, S.; Baster, Z.; et al. Entry of Human Coronavirus NL63 into the Cell. J. Virol. 2018, 92, e01933-17. [Google Scholar] [CrossRef]
- Kawase, M.; Shirato, K.; Matsuyama, S.; Taguchi, F. Protease-mediated entry via the endosome of human coronavirus 229E. J. Virol. 2009, 83, 712–721. [Google Scholar] [CrossRef]
- Zhou, N.; Pan, T.; Zhang, J.; Li, Q.; Zhang, X.; Bai, C.; Huang, F.; Peng, T.; Zhang, J.; Liu, C.; et al. Glycopeptide Antibiotics Potently Inhibit Cathepsin L in the Late Endosome/Lysosome and Block the Entry of Ebola Virus, Middle East Respiratory Syndrome Coronavirus (MERS-CoV), and Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV). J. Biol. Chem. 2016, 291, 9218–9232. [Google Scholar] [CrossRef]
- Mitra, P. Inhibiting fusion with cellular membrane system: Therapeutic options to prevent severe acute respiratory syndrome coronavirus-2 infection. Am. J. Physiol. Cell Physiol. 2020, 319, C500–C509. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Dong, S.; Kong, N.; Zhang, Y.; Li, Y.; Sun, D.; Qin, W.; Zhai, H.; Zhai, X.; Yang, X.; Ye, C.; et al. TARDBP Inhibits Porcine Epidemic Diarrhea Virus Replication through Degrading Viral Nucleocapsid Protein and Activating Type I Interferon Signaling. J. Virol. 2022, 96, e0007022. [Google Scholar] [CrossRef]
- Zhang, H.; Tu, J.; Cao, C.; Yang, T.; Gao, L. Proteasome activator PA28gamma-dependent degradation of coronavirus disease (COVID-19) nucleocapsid protein. Biochem. Biophys. Res. Commun. 2020, 529, 251–256. [Google Scholar] [CrossRef]
- Jiao, Y.; Kong, N.; Wang, H.; Sun, D.; Dong, S.; Chen, X.; Zheng, H.; Tong, W.; Yu, H.; Yu, L.; et al. PABPC4 Broadly Inhibits Coronavirus Replication by Degrading Nucleocapsid Protein through Selective Autophagy. Microbiol. Spectr. 2021, 9, e0090821. [Google Scholar] [CrossRef]
- Schmidt, F.I.; Bleck, C.K.; Reh, L.; Novy, K.; Wollscheid, B.; Helenius, A.; Stahlberg, H.; Mercer, J. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep. 2013, 4, 464–476. [Google Scholar] [CrossRef]
- Mercer, J.; Snijder, B.; Sacher, R.; Burkard, C.; Bleck, C.K.; Stahlberg, H.; Pelkmans, L.; Helenius, A. RNAi screening reveals proteasome- and Cullin3-dependent stages in vaccinia virus infection. Cell Rep. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Barrado-Gil, L.; Galindo, I.; Martinez-Alonso, D.; Viedma, S.; Alonso, C. The ubiquitin-proteasome system is required for African swine fever replication. PLoS ONE 2017, 12, e0189741. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.; Zhang, X.; Wang, H.; Mao, X.; Sun, Y.; Tan, L.; Song, C.; Qiu, X.; Ding, C.; Liao, Y. The Ubiquitin-Proteasome System Facilitates Membrane Fusion and Uncoating during Coronavirus Entry. Viruses 2023, 15, 2001. https://0-doi-org.brum.beds.ac.uk/10.3390/v15102001
Yuan X, Zhang X, Wang H, Mao X, Sun Y, Tan L, Song C, Qiu X, Ding C, Liao Y. The Ubiquitin-Proteasome System Facilitates Membrane Fusion and Uncoating during Coronavirus Entry. Viruses. 2023; 15(10):2001. https://0-doi-org.brum.beds.ac.uk/10.3390/v15102001
Chicago/Turabian StyleYuan, Xiao, Xiaoman Zhang, Huan Wang, Xiang Mao, Yingjie Sun, Lei Tan, Cuiping Song, Xusheng Qiu, Chan Ding, and Ying Liao. 2023. "The Ubiquitin-Proteasome System Facilitates Membrane Fusion and Uncoating during Coronavirus Entry" Viruses 15, no. 10: 2001. https://0-doi-org.brum.beds.ac.uk/10.3390/v15102001