
Identification of Host Factors for Rift Valley Fever Phlebovirus
 , , , ,
, , , ,  , and
, and 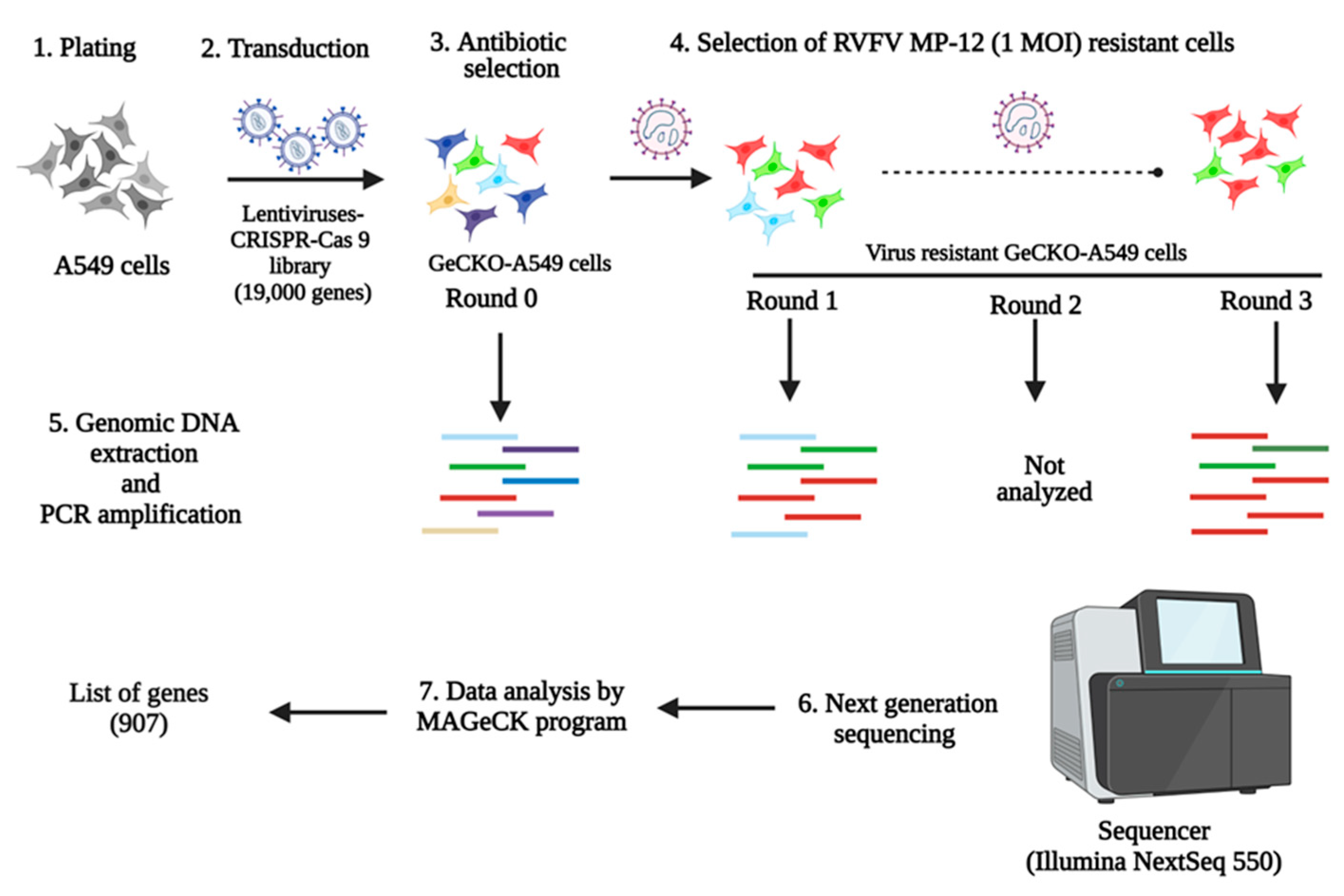{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Virus Strains
2.3. Generation of GeCKO-A549 Cell Line and RVFV Screening
2.4. siRNA Transfection
2.5. RT-qPCR for Host Gene Expression
2.6. Generation of WDR7 Knock-Out (KO) Cells
2.7. Western Blot Analysis
2.8. Testing of WDR7 KO Cells for Virus Replication
2.9. Intracellular Viral RNA Accumulation Assay
2.10. Plaque Assay
2.11. TCID50-CPE Assay
2.12. Statistical Analysis
3. Results
3.1. Identification of Host Factors Involved in RVFV Replication
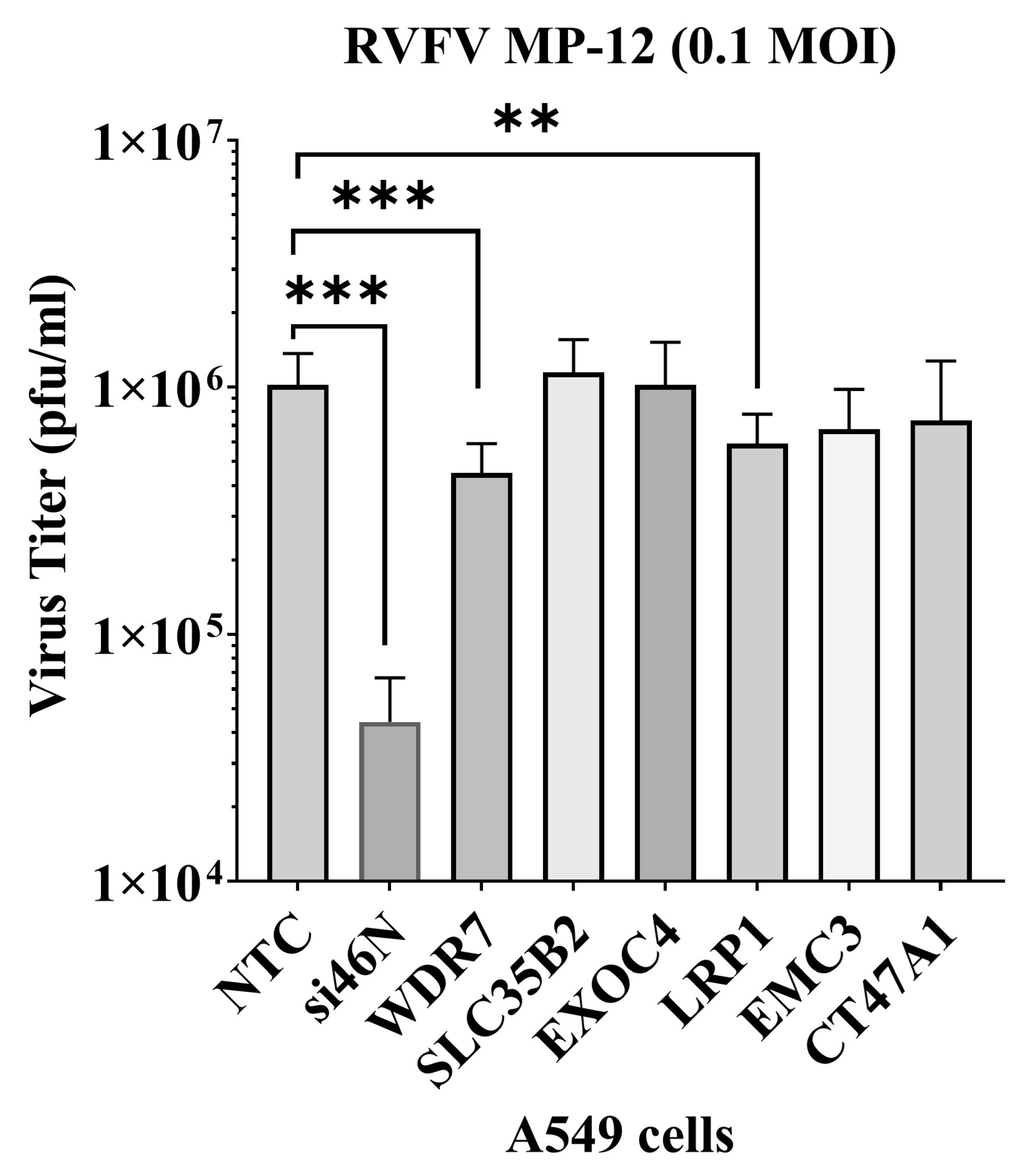
3.2. Validation of Genes from the Pooled GeCKO-A549 Cell Screening
3.3. Generation and Characterization of Knockout Cells
3.4. Effect of WDR7 Gene Knockout on RVFV and LACV Infection
3.5. WDR7 Gene Knock-Out Impairs RVFV and LACV Intracellular RNA Accumulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Daubney, R.; Hudson, J.R.; Garnham, P.C. Enzootic hepatitis or rift valley fever. An undescribed virus disease of sheep cattle and man from east africa. J. Pathol. Bacteriol. 1931, 34, 545–579. [Google Scholar] [CrossRef]
- Morvan, J.; Saluzzo, J.-F.; Fontenille, D.; Rollin, P.E.; Coulanges, P. Rift valley fever on the east coast of Madagascar. Res. Virol. 1991, 142, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, T.; Boulianne, C.; Vincent, M.J.; Pezzanite, L.; Al-Qahtani, M.M.; Al-Mazrou, Y.; Khan, A.S.; Rollin, P.E.; Swanepoel, R.; Ksiazek, T.G.; et al. Genetic analysis of viruses associated with emergence of Rift Valley fever in Saudi Arabia and Yemen, 2000–2001. Emerg. Infect. Dis. 2002, 8, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- Sissoko, D.; Giry, C.; Gabrie, P.; Tarantola, A.; Pettinelli, F.; Collet, L.; D’ortenzio, E.; Renault, P.; Pierre, V. Rift valley fever, mayotte, 2007–2008. Emerg. Infect. Dis. 2009, 15, 568–570. [Google Scholar] [CrossRef] [PubMed]
- Coetzer, J.A. Natural Cases in New-Born Lambs. Onderstepoort J.Vet.Res. 1977, 44, 205–211. [Google Scholar]
- Coetzer, J.A. The pathology of Rift Valley fever. II. Lesions occurring in field cases in adult cattle, calves and aborted foetuses. Onderstepoort J. Vet. Res. 1982, 49, 11–17. [Google Scholar]
- Pepin, M.; Bouloy, M.; Bird, B.H.; Kemp, A.; Paweska, J. Rift Valley fever virus (Bunyaviridae: Phlebovirus): An update on pathogenesis, molecular epidemiology, vectors, diagnostics and prevention. Vet. Res. 2010, 41, 61. [Google Scholar] [CrossRef]
- Wilson, W.C.; Kim, I.J.; Trujillo, J.D.; Sunwoo, S.Y.; Noronha, L.E.; Urbaniak, K.; McVey, D.S.; Drolet, B.S.; Morozov, I.; Faburay, B.; et al. Susceptibility of white-tailed deer to Rift valley fever virus. Emerg. Infect. Dis. 2018, 24, 1717–1719. [Google Scholar] [CrossRef]
- Boushab, B.M.; Fall-Malick, F.Z.; Baba, S.E.W.O.; Salem, M.L.O.; Belizaire, M.R.D.; Ledib, H.; Ahmed, M.M.O.B.; Basco, L.K.; Ba, H. Severe human illness caused by rift valley fever virus in Mauritania, 2015. Open Forum Infect. Dis. 2016, 3, 2–5. [Google Scholar] [CrossRef]
- Ikegami, T.; Makino, S. The pathogenesis of rift valley fever. Viruses 2011, 3, 493–519. [Google Scholar] [CrossRef]
- Linthicum, K.J.; Davies, F.G.; Kairo, A.; Bailey, C.L. Rift Valley fever virus (family Bunyaviridae, genus Phlebovirus). Isolations from Diptera collected during an inter-epizootic period in Kenya. J. Hyg. 1985, 95, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Gommet, C.; Billecocq, A.; Jouvion, G.; Hasan, M.; do Valle, T.Z.; Guillemot, L.; Blanchet, C.; van Rooijen, N.; Montagutelli, X.; Bouloy, M.; et al. Tissue tropism and target cells of NSs-deleted rift valley fever virus in live immunodeficient mice. PLoS Negl. Trop. Dis. 2011, 5, e1421. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Powell, D.S.; Bethel, L.M.; Caroline, A.L.; Schmid, R.J.; Oury, T.; Reed, D.S. Aerosolized Rift Valley Fever Virus Causes Fatal Encephalitis in African Green Monkeys and Common Marmosets. J. Virol. 2014, 88, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Scharton, D.; Van Wettere, A.J.; Bailey, K.W.; Vest, Z.; Westover, J.B.; Siddharthan, V.; Gowen, B.B. Rift valley fever virus infection in golden Syrian hamsters. PLoS ONE 2015, 10, e0116722. [Google Scholar] [CrossRef]
- Gaudreault, N.N.; Indran, S.V.; Bryant, P.K.; Richt, J.A.; Wilson, W.C. Comparison of Rift Valley fever virus replication in North American livestock and wildlife cell lines. Front. Microbiol. 2015, 6, 664. [Google Scholar] [CrossRef]
- Odendaal, L.; Davis, A.S.; Fosgate, G.T.; Clift, S.J. Lesions and Cellular Tropism of Natural Rift Valley Fever Virus Infection in Young Lambs. Vet. Pathol. 2020, 57, 66–81. [Google Scholar] [CrossRef]
- Rissmann, M.; Lenk, M.; Stoek, F.; Szentiks, C.A.; Eiden, M.; Groschup, M.H. Replication of rift valley fever virus in amphibian and reptile-derived cell lines. Pathogens 2021, 10, 681. [Google Scholar] [CrossRef]
- Lumley, S.; Horton, D.L.; Hernandez-Triana, L.L.M.; Johnson, N.; Fooks, A.R.; Hewson, R. Rift valley fever virus: Strategies for maintenance, survival and vertical transmission in mosquitoes. J. Gen. Virol. 2017, 98, 875–887. [Google Scholar] [CrossRef]
- Wichgers Schreur, P.J.; Vloet, R.P.M.; Kant, J.; van Keulen, L.; Gonzales, J.L.; Visser, T.M.; Koenraadt, C.J.M.; Vogels, C.B.F.; Kortekaas, J. Reproducing the Rift Valley fever virus mosquito-lamb-mosquito transmission cycle. Sci. Rep. 2021, 11, 1477. [Google Scholar] [CrossRef]
- Caroline, A.L.; Powell, D.S.; Bethel, L.M.; Oury, T.D.; Reed, D.S.; Hartman, A.L. Broad Spectrum Antiviral Activity of Favipiravir (T-705): Protection from Highly Lethal Inhalational Rift Valley Fever. PLoS Negl. Trop. Dis. 2014, 8, 2–9. [Google Scholar] [CrossRef]
- Kwaśnik, M.; Rożek, W.; Rola, J. Rift Valley fever—A growing threat to humans and animals. J. Vet. Res. 2021, 65, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Turell, M.J.; Wilson, W.C.; Bennett, K.E. Potential for North American mosquitoes (Diptera: Culicidae) to transmit rift Valley fever virus. J. Med. Entomol. 2010, 47, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Brustolin, M.; Talavera, S.; NuÑez, A.; SantamarÍa, C.; Rivas, R.; Pujol, N.; Valle, M.; Verdún, M.; Brun, A.; Pagès, N.; et al. Rift Valley fever virus and European mosquitoes: Vector competence of Culex pipiens and Stegomyia albopicta (=Aedes albopictus). Med. Vet. Entomol. 2017, 31, 365–372. [Google Scholar] [CrossRef]
- Hartman, D.A.; Rice, L.M.; DeMaria, J.; Borland, E.M.; Bergren, N.A.; Fagre, A.C.; Robb, L.L.; Webb, C.T.; Kading, R.C. Entomological risk factors for potential transmission of Rift Valley fever virus around concentrations of livestock in Colorado. Transbound. Emerg. Dis. 2019, 66, 1709–1717. [Google Scholar] [CrossRef] [PubMed]
- Cakir, M.; Obernier, K.; Forget, A.; Krogan, N.J. Target Discovery for Host-Directed Antiviral Therapies: Application of Proteomics Approaches. mSystems 2021, 6, e0038821. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.W.; Slone, T.W.; Peters, C.J. Pathogenesis of Rift Valley fever virus (RVFV) in inbred rats. Microb. Pathog. 1987, 2, 283–293. [Google Scholar] [CrossRef]
- Kainulainen, M.; Lau, S.; Samuel, C.E.; Hornung, V.; Weber, F. NSs Virulence Factor of Rift Valley Fever Virus Engages the F-Box Proteins FBXW11 and β-TRCP1 To Degrade the Antiviral Protein Kinase PKR. J. Virol. 2016, 90, 6140–6147. [Google Scholar] [CrossRef]
- Hartman, A. Rift Valley Fever virus. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef]
- Ganaie, S.S.; Schwarz, M.M.; McMillen, C.M.; Price, D.A.; Feng, A.X.; Albe, J.R.; Wang, W.; Miersch, S.; Orvedahl, A.; Cole, A.R.; et al. Lrp1 is a host entry factor for Rift Valley fever virus. Cell 2021, 184, 5163–5178.e24. [Google Scholar] [CrossRef]
- Léger, P.; Tetard, M.; Youness, B.; Cordes, N.; Rouxel, R.N.; Flamand, M.; Lozach, P. Differential Use of the C-Type Lectins L-SIGN and DC-SIGN for Phlebovirus Endocytosis. Traffic 2016, 17, 639–656. [Google Scholar] [CrossRef]
- Lozach, P.Y.; Kühbacher, A.; Meier, R.; Mancini, R.; Bitto, D.; Bouloy, M.; Helenius, A. DC-SIGN as a receptor for phleboviruses. Cell Host Microbe 2011, 10, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, I.; Lokugamage, N.; Nishiyama, S.; Ikegami, T. Mutational analysis of the rift valley fever virus glycoprotein precursor proteins for Gn protein expression. Viruses 2016, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Riblett, A.M.; Blomen, V.A.; Jae, L.T.; Altamura, L.A.; Doms, R.W.; Brummelkamp, T.R.; Wojcechowskyj, J.A. A Haploid Genetic Screen Identifies Heparan Sulfate Proteoglycans Supporting Rift Valley Fever Virus Infection. J. Virol. 2016, 90, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- De Boer, S.M.; Kortekaas, J.; de Haan, C.A.M.; Rottier, P.J.M.; Moormann, R.J.M.; Bosch, B.J. Heparan Sulfate Facilitates Rift Valley Fever Virus Entry into the Cell. J. Virol. 2012, 86, 13767–13771. [Google Scholar] [CrossRef]
- Hofmann, H.; Li, X.; Zhang, X.; Liu, W.; Kühl, A.; Kaup, F.; Soldan, S.S.; González-Scarano, F.; Weber, F.; He, Y.; et al. Severe Fever with Thrombocytopenia Virus Glycoproteins Are Targeted by Neutralizing Antibodies and Can Use DC-SIGN as a Receptor for pH-Dependent Entry into Human and Animal Cell Lines. J. Virol. 2013, 87, 4384–4394. [Google Scholar] [CrossRef]
- Bracci, N.; de la Fuente, C.; Saleem, S.; Pinkham, C.; Narayanan, A.; García-Sastre, A.; Balaraman, V.; Richt, J.A.; Wilson, W.; Kehn-Hall, K. Rift Valley fever virus Gn V5-epitope tagged virus enables identification of UBR4 as a Gn interacting protein that facilitates Rift Valley fever virus production. Virology 2022, 567, 65–76. [Google Scholar] [CrossRef]
- Devignot, S.; Sha, T.W.; Burkard, T.; Schmerer, P.; Hagelkruys, A.; Mirazimi, A.; Elling, U.; Penninger, J.M.; Weber, F. Low Density Lipoprotein Receptor-Related Protein 1 (LRP1) as an auxiliary host factor for RNA viruses. Life Sci. Alliance 2023, 6, e202302005. [Google Scholar] [CrossRef]
- Caplen, H.; Peters, C.J.; Bishop, D.H.L. Mutagen-directed attenuation of Rift Valley fever virus as a method for vaccine development. J. Gen. Virol. 1985, 66, 2271–2277. [Google Scholar] [CrossRef]
- Shivanna, V.; McDowell, C.; Wilson, W.C.; Richt, J.A. Complete Genome Sequence of Two Rift Valley Fever Virus Strains Isolated from Outbreaks in Saudi Arabia (2000) and Kenya (2006 to 2007). Genome Announc. 2016, 4, e00926-16. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; E Sanjana, N.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef]
- Wang, B.; Wang, M.; Zhang, W.; Xiao, T.; Chen, C.; Wu, A.; Wu, F.; Traugh, N.; Wang, X.; Li, Z.; et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc. 2019, 14, 756–780. [Google Scholar] [CrossRef]
- Faburay, B.; Richt, J.A. Short interfering RNA inhibits rift valley fever virus replication and degradation of protein kinase R in human cells. Front. Microbiol. 2016, 7, 1889. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Du, Z.; Li, X.; Yang, Q.; Zhang, Y.C.; Wu, M.; Li, Y.; Zhang, G. Identification of suitable reference genes for gene expression studies using quantitative polymerase chain reaction in lung cancer in vitro. Mol. Med. Rep. 2015, 11, 3767–3773. [Google Scholar] [CrossRef] [PubMed]
- Haimes, J.; Kelley, M. Demonstration of a ΔΔCq Calculation Method to Compute Relative Gene Expression from qPCR Data. Joshua GE HealthCare. 2010. Available online: https://horizondiscovery.com/-/media/Files/Horizon/resources/Technical-Manuals/delta-cq-solaris-technote.pdf (accessed on 1 March 2022).
- Lee, J.; Yu, H.; Li, Y.; Ma, J.; Lang, Y.; Duff, M.; Henningson, J.; Liu, Q.; Li, Y.; Nagy, A.; et al. Impacts of different expressions of PA-X protein on 2009 pandemic H1N1 virus replication, pathogenicity and host immune responses. Virology 2017, 504, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Penski, N.; Wagner, V.; Spiegel, M.; Överby, A.K.; Kochs, G.; Huiskonen, J.T.; Weber, F. Efficient production of Rift Valley fever virus-like particles: The antiviral protein MxA can inhibit primary transcription of bunyaviruses. Virology 2009, 385, 400–408. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hierholzer, J.C.; Killington, R.A. Virus isolation and quantitation. In Virology Methods Manual; Elsevier: Amsterdam, The Netherlands, 1996; pp. 25–46. [Google Scholar] [CrossRef]
- Savidis, G.; McDougall, W.M.; Meraner, P.; Perreira, J.M.; Portmann, J.M.; Trincucci, G.; John, S.P.; Aker, A.M.; Renzette, N.; Robbins, D.R.; et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016, 16, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Clohisey, S.M.; Chia, B.S.; Wang, B.; Cui, A.; Eisenhaure, T.; Schweitzer, L.D.; Hoover, P.; Parkinson, N.J.; Nachshon, A.; et al. Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat. Commun. 2020, 11, 164. [Google Scholar] [CrossRef]
- Merkulova, M.; Paunescu, T.G.; Azroyan, A.; Marshansky, V.; Breton, S.; Brown, D. Mapping the H+ (V)-ATPase interactome: Identification of proteins involved in trafficking, folding, assembly and phosphorylation. Sci. Rep. 2015, 5, 14827. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balaraman, V.; Indran, S.V.; Li, Y.; Meekins, D.A.; Jakkula, L.U.M.R.; Liu, H.; Hays, M.P.; Souza-Neto, J.A.; Gaudreault, N.N.; Hardwidge, P.R.; et al. Identification of Host Factors for Rift Valley Fever Phlebovirus. Viruses 2023, 15, 2251. https://0-doi-org.brum.beds.ac.uk/10.3390/v15112251
Balaraman V, Indran SV, Li Y, Meekins DA, Jakkula LUMR, Liu H, Hays MP, Souza-Neto JA, Gaudreault NN, Hardwidge PR, et al. Identification of Host Factors for Rift Valley Fever Phlebovirus. Viruses. 2023; 15(11):2251. https://0-doi-org.brum.beds.ac.uk/10.3390/v15112251
Chicago/Turabian StyleBalaraman, Velmurugan, Sabarish V. Indran, Yonghai Li, David A. Meekins, Laxmi U. M. R. Jakkula, Heidi Liu, Micheal P. Hays, Jayme A. Souza-Neto, Natasha N. Gaudreault, Philip R. Hardwidge, and et al. 2023. "Identification of Host Factors for Rift Valley Fever Phlebovirus" Viruses 15, no. 11: 2251. https://0-doi-org.brum.beds.ac.uk/10.3390/v15112251