
Acellular Human Amniotic Fluid-Derived Extracellular Vesicles as Novel Anti-Inflammatory Therapeutics against SARS-CoV-2 Infection

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. acAF Collection, Preparation, and Cytotoxicity Evaluation
2.3. Nanoparticle Tracking Analysis of acAF Preparations
2.4. AF-EV Preparation
2.5. Cytokine Array Dot Blot Analysis
2.6. In Vitro Virus Infection and acAF Treatments
2.7. In Vitro Ccytokine and Chemokine Analysis upon Stimulation with SARS-CoV-2 Structural Proteins and TLR Agonists
2.8. Mice Infection and acAF Treatments
2.9. Virus Titer Assay and Gene Expression
2.10. Immuno-Labeling and Fluorescence-Activated Cell Sorting (FACS) Assay to Assess Llung Myeloid Cells
2.11. Data Analysis
3. Results
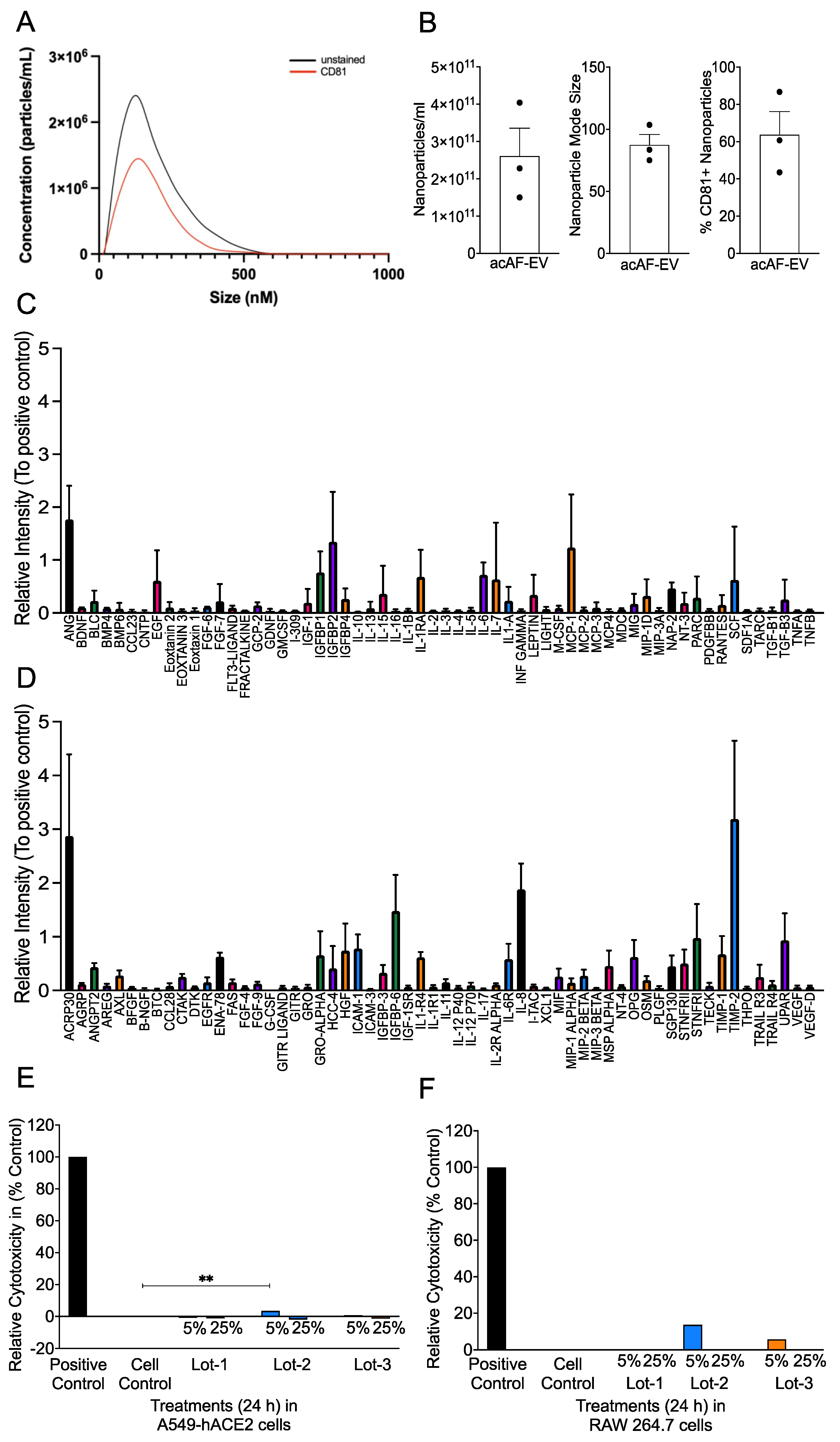
3.1. acAF Preparations Contain AF-EVs and Cytokines/Chemokines and Are a Non-Toxic Biologic

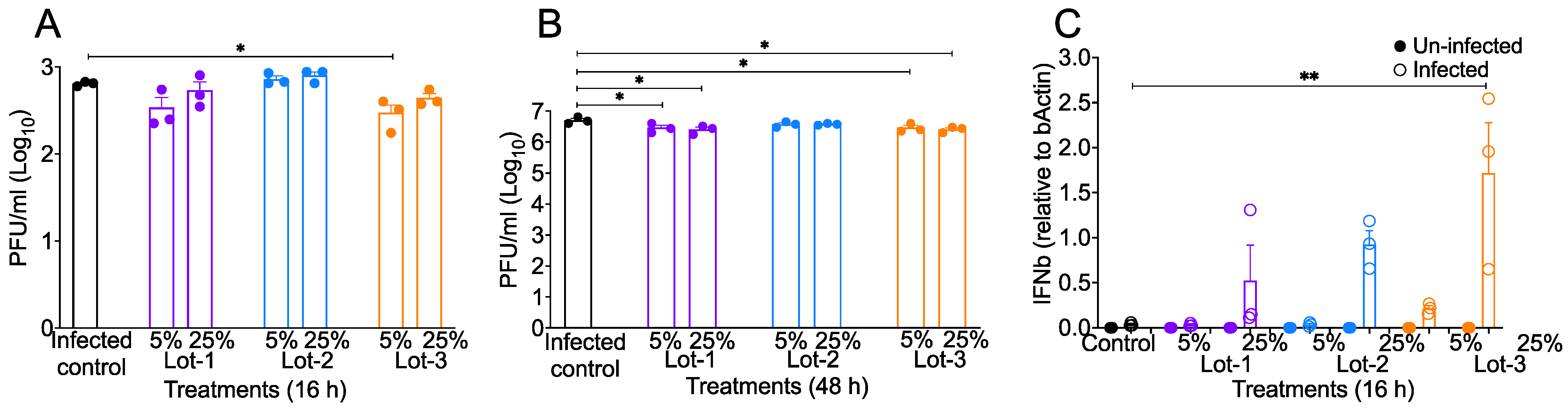
3.2. acAF Reduces Virus Titers and Promotes Anti-Viral Response to SARS-CoV-2 Infection
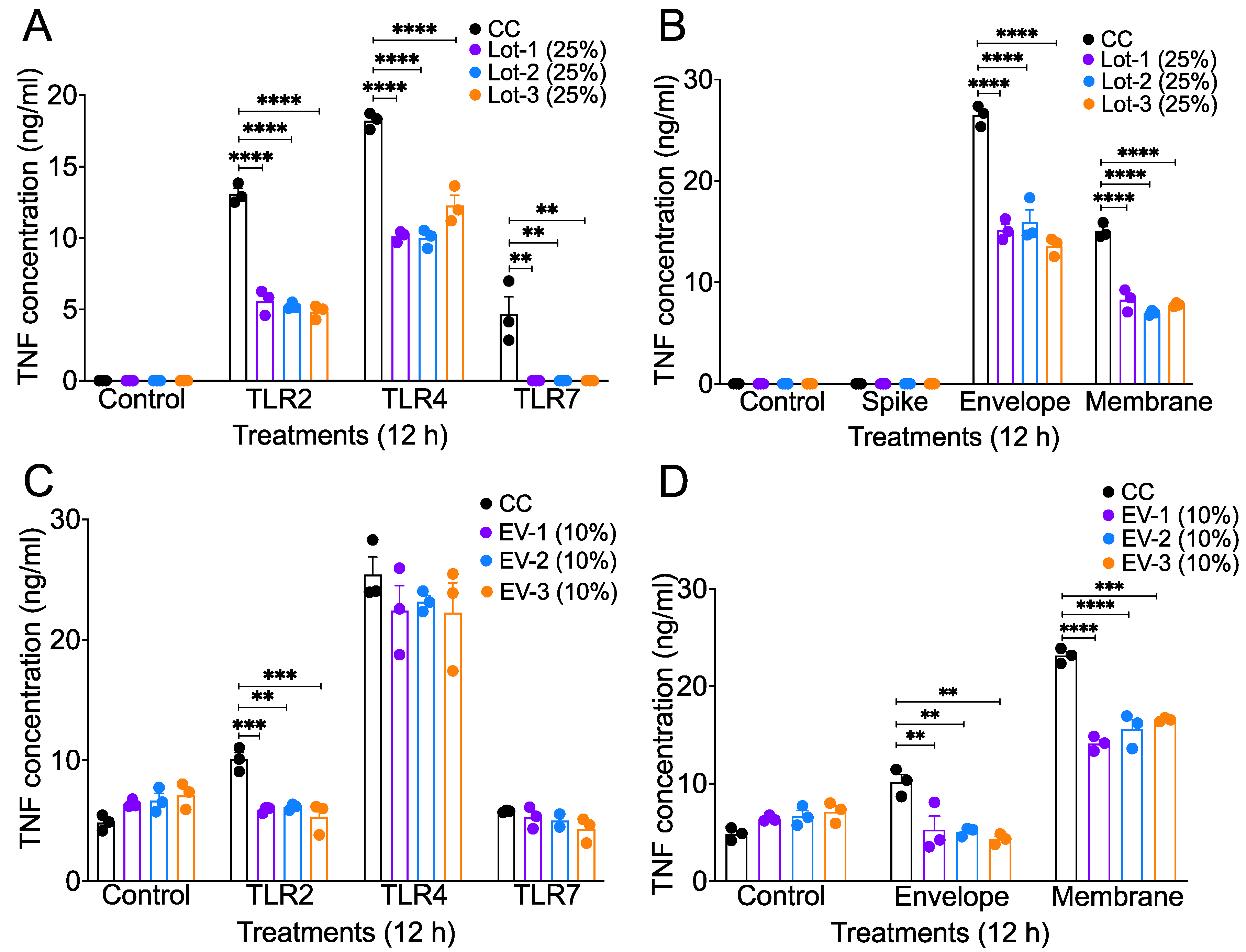
3.3. acAF and AF-EVs Suppress Inflammatory Cytokine Production in TLR Agonist- and SARS-CoV-2 Structural Protein-Stimulated Mouse Macrophages
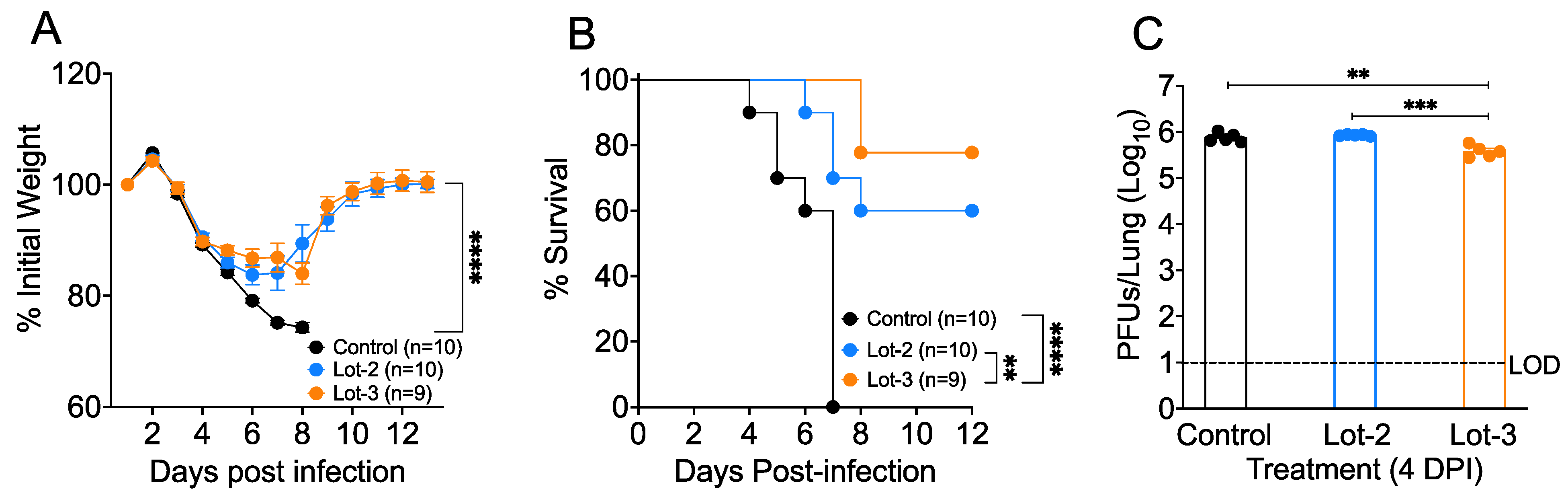
3.4. acAF Provides Protection against SARS-CoV-2-Induced Lethal Disease and Mortality
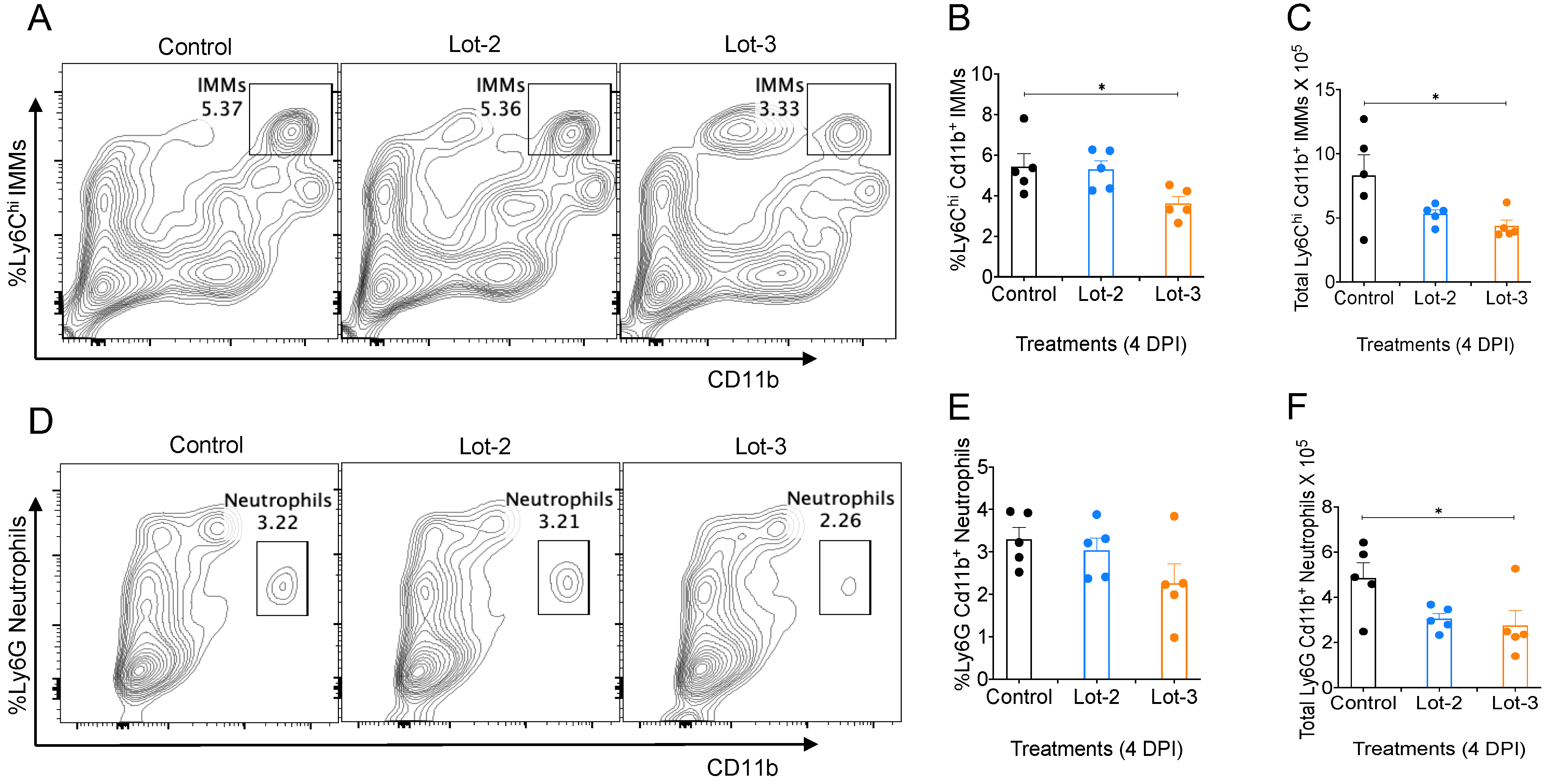
3.5. acAF Suppresses Myeloid Cell Accumulation in Mouse Lungs
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singhal, T. A Review of Coronavirus Disease-2019 (COVID-19). Indian J. Pediatr. 2020, 87, 281–286. [Google Scholar] [CrossRef] [PubMed]
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 4 October 2023).
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, S.; Xia, H.; Shi, D.; Chen, Y.; Zheng, S.; Chen, Y.; Gao, H.; Guo, F.; Ji, Z.; et al. Cytokine Signature Associated with Disease Severity in COVID-19. Front. Immunol. 2021, 12, 681516. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 2020, 27, 883–890.e2. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Mann, E.R.; Menon, M.; Knight, S.B.; Konkel, J.E.; Jagger, C.; Shaw, T.N.; Krishnan, S.; Rattray, M.; Ustianowski, A.; Bakerly, N.D.; et al. Longitudinal immune profiling reveals key myeloid signatures associated with COVID-19. Sci. Immunol. 2020, 5, eabd6197. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Kizaki, T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon 2021, 7, e06187. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; To, R.K.; Hanna, J.; Spector, S.A. SARS-CoV-2, SARS-CoV-1, and HIV-1 derived ssRNA sequences activate the NLRP3 inflammasome in human macrophages through a non-classical pathway. iScience 2021, 24, 102295. [Google Scholar] [CrossRef] [PubMed]
- Kouwaki, T.; Nishimura, T.; Wang, G.; Oshiumi, H. RIG-I-Like Receptor-Mediated Recognition of Viral Genomic RNA of Severe Acute Respiratory Syndrome Coronavirus-2 and Viral Escape From the Host Innate Immune Responses. Front. Immunol. 2021, 12, 700926. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Riva, L.; Pu, Y.; Martin-Sancho, L.; Kanamune, J.; Yamamoto, Y.; Sakai, K.; Gotoh, S.; Miorin, L.; De Jesus, P.D.; et al. MDA5 Governs the Innate Immune Response to SARS-CoV-2 in Lung Epithelial Cells. Cell Rep. 2021, 34, 108628. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Eutimio, M.A.; López-Macías, C.; Pastelin-Palacios, R. Bioinformatic analysis and identification of single-stranded RNA sequences recognized by TLR7/8 in the SARS-CoV-2, SARS-CoV, and MERS-CoV genomes. Microbes Infect. 2020, 22, 226–229. [Google Scholar] [CrossRef]
- Rebendenne, A.; Chaves Valadão, A.L.; Tauziet, M.; Maarifi, G.; Bonaventure, B.; McKellar, J.; Planès, R.; Nisole, S.; Arnaud-Arnould, M.; Moncorgé, O.; et al. SARS-CoV-2 Triggers an MDA-5-Dependent Interferon Response Which Is Unable to Control Replication in Lung Epithelial Cells. J. Virol. 2021, 95, 20. [Google Scholar] [CrossRef]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. eLife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Planès, R.; Bert, J.-B.; Tairi, S.; BenMohamed, L.; Bahraoui, E. SARS-CoV-2 Envelope (E) Protein Binds and Activates TLR2 Pathway: A Novel Molecular Target for COVID-19 Interventions. Viruses 2022, 14, 999. [Google Scholar] [CrossRef]
- Bortolotti, D.; Gentili, V.; Rizzo, S.; Schiuma, G.; Beltrami, S.; Strazzabosco, G.; Fernandez, M.; Caccuri, F.; Caruso, A.; Rizzo, R. TLR3 and TLR7 RNA Sensor Activation during SARS-CoV-2 Infection. Microorganisms 2021, 9, 1820. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. 2021, 31, 818–820. [Google Scholar] [CrossRef]
- Ran, X.-H.; Zhu, J.-W.; Chen, Y.-Y.; Ni, R.-Z.; Mu, D. Papain-like protease of SARS-CoV-2 inhibits RLR signaling in a deubiquitination-dependent and deubiquitination-independent manner. Front. Immunol. 2022, 13, 947272. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y.; et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664. [Google Scholar] [CrossRef] [PubMed]
- Yalcinkaya, M.; Liu, W.; Islam, M.N.; Kotini, A.G.; Gusarova, G.A.; Fidler, T.P.; Papapetrou, E.P.; Bhattacharya, J.; Wang, N.; Tall, A.R. Modulation of the NLRP3 inflammasome by Sars-CoV-2 Envelope protein. Sci. Rep. 2021, 11, 24432. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Akinyemi, I.A.; Chitre, S.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology 2022, 568, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Lee, J.S.; Park, S.; Jeong, H.W.; Ahn, J.Y.; Choi, S.J.; Lee, H.; Choi, B.; Nam, S.K.; Sa, M.; Kwon, J.-S.; et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020, 5, eabd1554. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Merck Sharp & Dohme LLC. A Phase 2/3, Randomized, Placebo-Controlled, Double-Blind Clinical Study to Evaluate the Efficacy, Safety, and Pharmacokinetics of MK-4482 in Non-Hospitalized Adults with COVID-19. Clinicaltrials.Gov, Clinical Trial Registration NCT04575597. 2023. Available online: https://clinicaltrials.gov/study/NCT04575597 (accessed on 31 December 2022).
- Ridgeback Biotherapeutics. A Phase IIa Randomized, Double-Blind, Placebo-Controlled Trial to Evaluate the Safety, Tolerability and Efficacy of EIDD-2801 to Eliminate SARS-CoV-2RNA Detection in Persons with COVID-19. Clinicaltrials.Gov, Clinical Trial Registration NCT04405570. 2022. Available online: https://clinicaltrials.gov/study/NCT04405570 (accessed on 31 December 2022).
- Venter, P.F. A Randomised, Multi-Centre, Double-blind, Phase 3 Study to Observe the Effectiveness, Safety and Tolerability of Molnupiravir Compared to Placebo Administered Orally to High-Risk Adult Outpatients with Mild COVID-19 Receiving Local Standard of Care in South Africa (CoTeT). Clinicaltrials.Gov, Clinical Trial Registration NCT05459532. 2022. Available online: https://clinicaltrials.gov/study/NCT05459532 (accessed on 31 December 2022).
- Pinto, A. Canadian Adaptive Platform Trial of Treatments for COVID-19 in Community Settings. Clinicaltrials.Gov, Clinical trial Registration NCT05614349. 2023. Available online: https://clinicaltrials.gov/study/NCT05614349 (accessed on 31 December 2022).
- Pfizer. General Investigation for PAXLOVID PAC. Clinicaltrials.Gov, Clinical Trial Registration NCT05263908. 2023. Available online: https://clinicaltrials.gov/study/NCT05263908 (accessed on 31 December 2022).
- Early Treatment with Pegylated Interferon Lambda for COVID-19|NEJM. Available online: https://www.nejm.org/doi/full/10.1056/NEJMoa2209760 (accessed on 9 November 2023).
- Bosi, E. Randomized, Controlled, Open Label, Phase 2 Clinical Trial of Interferon-β-1a (IFNβ-1a) in COVID-19 Patients. Clinicaltrials.Gov, Clinical Trial Registration NCT04449380. 2021. Available online: https://clinicaltrials.gov/study/NCT04449380 (accessed on 31 December 2022).
- Etzion, O. Pegylated Interferon Lambda for Treatment of COVID-19 Infection—A Randomized Open Label Pilot Trial. Clinicaltrials.Gov, Clinical Trial Registration NCT04534673. 2023. Available online: https://clinicaltrials.gov/study/NCT04534673 (accessed on 31 December 2022).
- Feld, J.J.; Kandel, C.; Biondi, M.J.; Kozak, R.A.; Zahoor, M.A.; Lemieux, C.; Borgia, S.M.; Boggild, A.K.; Powis, J.; McCready, J.; et al. Peginterferon lambda for the treatment of outpatients with COVID-19: A phase 2, placebo-controlled randomised trial. Lancet Respir. Med. 2021, 9, 498–510. [Google Scholar] [CrossRef]
- “Interferons Clinical Data|COVID-19,” COVID-19 Treatment Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov/tables/interferons-data/ (accessed on 9 November 2023).
- “Interferons”, COVID-19 Treatment Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov/therapies/antivirals-including-antibody-products/interferons/ (accessed on 9 November 2023).
- Alavi Darazam, I.; Hatami, F.; Mahdi Rabiei, M.; Amin Pourhoseingholi, M.; Shabani, M.; Shokouhi, S.; Mardani, M.; Moradi, O.; Javandoust Gharehbagh, F.; Mirtalaee, N.; et al. An investigation into the beneficial effects of high-dose interferon beta 1-a, compared to low-dose interferon beta 1-a in severe COVID-19: The COVIFERON II randomized controlled trial. Int. Immunopharmacol. 2021, 99, 107916. [Google Scholar] [CrossRef]
- Titanji, B.K.; Farley, M.M.; Mehta, A.; Connor-Schuler, R.; Moanna, A.; Cribbs, S.K.; O’Shea, J.; DeSilva, K.; Chan, B.; Edwards, A.; et al. Use of Baricitinib in Patients with Moderate to Severe Coronavirus Disease 2019. Clin. Infect. Dis. 2021, 72, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Liu, Y.; Qiu, L.; Liu, X.; Liu, D.; Li, J. Tocilizumab treatment in COVID-19: A single center experience. J. Med. Virol. 2020, 92, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Huet, T.; Beaussier, H.; Voisin, O.; Jouveshomme, S.; Dauriat, G.; Lazareth, I.; Sacco, E.; Naccache, J.-M.; Bézie, Y.; Laplanche, S.; et al. Anakinra for severe forms of COVID-19: A cohort study. Lancet Rheumatol. 2020, 2, e393–e400. [Google Scholar] [CrossRef] [PubMed]
- Issak, E.R. Timing of Corticosteroids in COVID-19, II. Post COVID-19 Follow-up. Clinicaltrials.Gov, Clinical Trial Registration NCT04530409. 2023. Available online: https://clinicaltrials.gov/study/NCT04530409 (accessed on 31 December 2022).
- Tang, X.; Feng, Y.-M.; Ni, J.-X.; Zhang, J.-Y.; Liu, L.-M.; Hu, K.; Wu, X.-Z.; Zhang, J.-X.; Chen, J.-W.; Zhang, J.-C.; et al. Early Use of Corticosteroid May Prolong SARS-CoV-2 Shedding in Non-Intensive Care Unit Patients with COVID-19 Pneumonia: A Multicenter, Single-Blind, Randomized Control Trial. Respiration 2021, 100, 116–126. [Google Scholar] [CrossRef] [PubMed]
- The RECOVERY Collaborative Group. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Abani, O.; Abbas, A.; Abbas, F.; Abbas, J.; Abbas, K.; Abbas, M.; Abbasi, S.; Abbass, H.; Abbott, A.; Abbott, A.; et al. Higher dose corticosteroids in patients admitted to hospital with COVID-19 who are hypoxic but not requiring ventilatory support (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2023, 401, 1499–1507. [Google Scholar] [CrossRef]
- Hermine, O.; Mariette, X.; Tharaux, P.-L.; Resche-Rigon, M.; Porcher, R.; Ravaud, P. CORIMUNO-19 Collaborative Group Effect of Tocilizumab vs Usual Care in Adults Hospitalized With COVID-19 and Moderate or Severe Pneumonia: A Randomized Clinical Trial. JAMA Intern. Med. 2021, 181, 32–40. [Google Scholar] [CrossRef]
- Bellio, M.A.; Young, K.C.; Milberg, J.; Santos, I.; Abdullah, Z.; Stewart, D.; Arango, A.; Chen, P.; Huang, J.; Williams, K.; et al. Amniotic fluid-derived extracellular vesicles: Characterization and therapeutic efficacy in an experimental model of bronchopulmonary dysplasia. Cytotherapy 2021, 23, 1097–1107. [Google Scholar] [CrossRef]
- Takov, K.; He, Z.; Johnston, H.E.; Timms, J.F.; Guillot, P.V.; Yellon, D.M.; Davidson, S.M. Small extracellular vesicles secreted from human amniotic fluid mesenchymal stromal cells possess cardioprotective and promigratory potential. Basic Res. Cardiol. 2020, 115, 26. [Google Scholar] [CrossRef]
- Corcelli, M.; Hawkins, K.; Vlahova, F.; Hunjan, A.; Dowding, K.; De Coppi, P.; David, A.L.; Peebles, D.; Gressens, P.; Hagberg, H.; et al. Neuroprotection of the hypoxic-ischemic mouse brain by human CD117+CD90+CD105+ amniotic fluid stem cells. Sci. Rep. 2018, 8, 2425. [Google Scholar] [CrossRef] [PubMed]
- Balbi, C.; Piccoli, M.; Barile, L.; Papait, A.; Armirotti, A.; Principi, E.; Reverberi, D.; Pascucci, L.; Becherini, P.; Varesio, L.; et al. First Characterization of Human Amniotic Fluid Stem Cell Extracellular Vesicles as a Powerful Paracrine Tool Endowed with Regenerative Potential. Stem Cells Transl. Med. 2017, 6, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Fauré, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell. Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G.; Shair, K.H.Y.; Marquitz, A.R.; Kung, C.-P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Kühne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef]
- Murphy, D.E.; de Jong, O.G.; Evers, M.J.W.; Nurazizah, M.; Schiffelers, R.M.; Vader, P. Natural or Synthetic RNA Delivery: A Stoichiometric Comparison of Extracellular Vesicles and Synthetic Nanoparticles. Nano Lett. 2021, 21, 1888–1895. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Fliervoet, L.A.L.; van der Meel, R.; Fens, M.H.A.M.; Heijnen, H.F.G.; van Bergen en Henegouwen, P.M.P.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J. Control. Release 2016, 224, 77–85. [Google Scholar] [CrossRef]
- McBride, J.D.; Rodriguez-Menocal, L.; Guzman, W.; Candanedo, A.; Garcia-Contreras, M.; Badiavas, E.V. Bone Marrow Mesenchymal Stem Cell-Derived CD63+ Exosomes Transport Wnt3a Exteriorly and Enhance Dermal Fibroblast Proliferation, Migration, and Angiogenesis In Vitro. Stem Cells Dev. 2017, 26, 1384–1398. [Google Scholar] [CrossRef]
- Jun, E.K.; Zhang, Q.; Yoon, B.S.; Moon, J.-H.; Lee, G.; Park, G.; Kang, P.J.; Lee, J.H.; Kim, A.; You, S. Hypoxic Conditioned Medium from Human Amniotic Fluid-Derived Mesenchymal Stem Cells Accelerates Skin Wound Healing through TGF-β/SMAD2 and PI3K/Akt Pathways. Int. J. Mol. Sci. 2014, 15, 605–628. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Stenqvist, A.-C.; Nagaeva, O.; Kjellberg, L.; Wulff, M.; Baranov, V.; Mincheva-Nilsson, L. Human Placenta Expresses and Secretes NKG2D Ligands via Exosomes that Down-Modulate the Cognate Receptor Expression: Evidence for Immunosuppressive Function1. J. Immunol. 2009, 183, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Silachev, D.N.; Goryunov, K.V.; Shpilyuk, M.A.; Beznoschenko, O.S.; Morozova, N.Y.; Kraevaya, E.E.; Popkov, V.A.; Pevzner, I.B.; Zorova, L.D.; Evtushenko, E.A.; et al. Effect of MSCs and MSC-Derived Extracellular Vesicles on Human Blood Coagulation. Cells 2019, 8, 258. [Google Scholar] [CrossRef] [PubMed]
- Berckmans, R.J.; Lacroix, R.; Hau, C.M.; Sturk, A.; Nieuwland, R. Extracellular vesicles and coagulation in blood from healthy humans revisited. J. Extracell. Vesicles 2019, 8, 1688936. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.; Dukes, A.; Drewry, M.; Helwa, I.; Johnson, M.H.; Isales, C.M.; Hill, W.D.; Liu, Y.; Shi, X.; Fulzele, S.; et al. MicroRNA-183-5p Increases with Age in Bone-Derived Extracellular Vesicles, Suppresses Bone Marrow Stromal (Stem) Cell Proliferation, and Induces Stem Cell Senescence. Tissue Eng. Part. A 2017, 23, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-Associated Exosome Release from Human Prostate Cancer Cells. Cancer Res. 2008, 68, 7864–7871. [Google Scholar] [CrossRef] [PubMed]
- Kadota, T.; Yoshioka, Y.; Fujita, Y.; Araya, J.; Minagawa, S.; Hara, H.; Miyamoto, A.; Suzuki, S.; Fujimori, S.; Kohno, T.; et al. Extracellular Vesicles from Fibroblasts Induce Epithelial-Cell Senescence in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 63, 623–636. [Google Scholar] [CrossRef]
- Castelli, V.; Antonucci, I.; d’Angelo, M.; Tessitore, A.; Zelli, V.; Benedetti, E.; Ferri, C.; Desideri, G.; Borlongan, C.; Stuppia, L.; et al. Neuroprotective effects of human amniotic fluid stem cells-derived secretome in an ischemia/reperfusion model. Stem Cells Transl. Med. 2021, 10, 251–266. [Google Scholar] [CrossRef]
- Bellio, M.A.; Bennett, C.; Arango, A.; Khan, A.; Xu, X.; Barrera, C.; Friedewald, V.; Mitrani, M.I. Proof-of-concept trial of an amniotic fluid-derived extracellular vesicle biologic for treating high risk patients with mild-to-moderate acute COVID-19 infection. Biomater. Biosyst. 2021, 4, 100031. [Google Scholar] [CrossRef]
- Channappanavar, R.; Perlman, S. Age-related susceptibility to coronavirus infections: Role of impaired and dysregulated host immunity. J. Clin. Investig. 2020, 130, 6204–6213. [Google Scholar] [CrossRef]
- Wong, L.-Y.R.; Zheng, J.; Wilhelmsen, K.; Li, K.; Ortiz, M.E.; Schnicker, N.J.; Thurman, A.; Pezzulo, A.A.; Szachowicz, P.J.; Li, P.; et al. Eicosanoid signalling blockade protects middle-aged mice from severe COVID-19. Nature 2022, 605, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Del Rivero, T.; Milberg, J.; Bennett, C.; Mitrani, M.I.; Bellio, M.A. Human amniotic fluid derived extracellular vesicles attenuate T cell immune response. Front. Immunol. 2022, 13, 977809. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Selvaraj, M.; More, S.; Perlman, S. Alveolar macrophages protect mice from MERS-CoV-induced pneumonia and severe disease. Vet. Pathol. 2022, 59, 627–638. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Perlman, S. Evaluation of Activation and Inflammatory Activity of Myeloid Cells During Pathogenic Human Coronavirus Infection. In MERS Coronavirus; Vijay, R., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2020; Volume 2099, pp. 195–204. ISBN 978-1-07-160210-2. [Google Scholar]
- White, I.A. The Potential of Amniotic Fluid-Derived Extracellular Vesicles to Treat Severe Acute Respiratory Syndrome Coronavirus 2 Infection Versus Hydroxychloroquine in Human Patients. Epidemiol. Open J. 2020, 5, 8–12. [Google Scholar] [CrossRef]
- Enayatkhani, M.; Hasaniazad, M.; Faezi, S.; Gouklani, H.; Davoodian, P.; Ahmadi, N.; Einakian, M.A.; Karmostaji, A.; Ahmadi, K. Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: An in silico study. J. Biomol. Struct. Dyn. 2021, 39, 2857–2872. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Omueti-Ayoade, K.; Mutha, S.K.; Hergenrother, P.J.; Tapping, R.I. Identification of Novel Synthetic Toll-like Receptor 2 Agonists by High Throughput Screening. J. Biol. Chem. 2010, 285, 23755–23762. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Funderburg, N.T.; Jadlowsky, J.K.; Lederman, M.M.; Feng, Z.; Weinberg, A.; Sieg, S.F. The Toll-like receptor 1/2 agonists Pam3CSK4 and human β-defensin-3 differentially induce interleukin-10 and nuclear factor-κB signalling patterns in human monocytes. Immunology 2011, 134, 151–160. [Google Scholar] [CrossRef]
- Li, Z.J.; Sohn, K.-C.; Choi, D.-K.; Shi, G.; Hong, D.; Lee, H.-E.; Whang, K.U.; Lee, Y.H.; Im, M.; Lee, Y.; et al. Roles of TLR7 in Activation of NF-κB Signaling of Keratinocytes by Imiquimod. PLoS ONE 2013, 8, e77159. [Google Scholar] [CrossRef]
- Sohn, K.M.; Lee, S.-G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 Patients Upregulate Toll-like Receptor 4-mediated Inflammatory Signaling That Mimics Bacterial Sepsis. J. Korean Med. Sci. 2020, 35, e343. [Google Scholar] [CrossRef]
- Melms, J.C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A.F.; Amin, A.D.; Schapiro, D.; et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 2021, 595, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Koike-Soko, C.; Sugimoto, J.; Yoshida, T.; Okabe, M.; Nikaido, T. Human Amnion-Derived Stem Cells Have Immunosuppressive Properties on NK Cells and Monocytes. Cell Transplant. 2015, 24, 2065–2076. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.L.; Chan, S.T.; Wallace, E.M.; Lim, R. Human Amnion Epithelial Cells Mediate Lung Repair by Directly Modulating Macrophage Recruitment and Polarization. Cell Transplant. 2014, 23, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Niederkorn, J.Y.; Neelam, S.; Mayhew, E.; Word, R.A.; McCulley, J.P.; Alizadeh, H. Immunosuppressive Factors Secreted by Human Amniotic Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 900. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061. [Google Scholar] [CrossRef] [PubMed]
- Wendisch, D.; Dietrich, O.; Mari, T.; von Stillfried, S.; Ibarra, I.L.; Mittermaier, M.; Mache, C.; Chua, R.L.; Knoll, R.; Timm, S.; et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell 2021, 184, 6243–6261.e27. [Google Scholar] [CrossRef] [PubMed]
- Dennis, A.; Cuthbertson, D.J.; Wootton, D.; Crooks, M.; Gabbay, M.; Eichert, N.; Mouchti, S.; Pansini, M.; Roca-Fernandez, A.; Thomaides-Brears, H.; et al. Multi-organ impairment and long COVID: A 1-year prospective, longitudinal cohort study. J. R. Soc. Med. 2023, 116, 97–112. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Brown, J.Q.; Heide, R.S.V. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Yao, X.H.; Li, T.Y.; He, Z.C.; Ping, Y.F.; Liu, H.W.; Yu, S.C.; Mou, H.M.; Wang, L.H.; Zhang, H.R.; Fu, W.J.; et al. A pathological report of three COVID-19 cases by minimal invasive autopsies. Zhonghua Bing Li Xue Za Zhi 2020, 49, 411–417. [Google Scholar] [CrossRef]
- Dennison, D.; Khabori, M.A.; Mamari, S.A.; Aurelio, A.; Hinai, H.A.; Maamari, K.A.; Alshekaili, J.; Khadouri, G.A. Circulating activated neutrophils in COVID-19: An independent predictor for mechanical ventilation and death. Int. J. Infect. Dis. 2021, 106, 155–159. [Google Scholar] [CrossRef]
- Sánchez-Cerrillo, I.; Landete, P.; Aldave, B.; Sánchez-Alonso, S.; Sánchez-Azofra, A.; Marcos-Jiménez, A.; Ávalos, E.; Alcaraz-Serna, A.; de los Santos, I.; Mateu-Albero, T.; et al. COVID-19 severity associates with pulmonary redistribution of CD1c+ DCs and inflammatory transitional and nonclassical monocytes. J. Clin. Investig. 2020, 130, 6290–6300. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Willms, E.; Johansson, H.J.; Mäger, I.; Lee, Y.; Blomberg, K.E.M.; Sadik, M.; Alaarg, A.; Smith, C.I.E.; Lehtiö, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef] [PubMed]
- Lamparski, H.G.; Metha-Damani, A.; Yao, J.-Y.; Patel, S.; Hsu, D.-H.; Ruegg, C.; Le Pecq, J.-B. Production and characterization of clinical grade exosomes derived from dendritic cells. J. Immunol. Methods 2002, 270, 211–226. [Google Scholar] [CrossRef]
- Hao, S.; Liu, Y.; Yuan, J.; Zhang, X.; He, T.; Wu, X.; Wei, Y.; Sun, D.; Xiang, J. Novel Exosome-Targeted CD4+ T Cell Vaccine Counteracting CD4+25+ Regulatory T Cell-Mediated Immune Suppression and Stimulating Efficient Central Memory CD8+ CTL Responses1. J. Immunol. 2007, 179, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Bedford, J.G.; Infusini, G.; Dagley, L.F.; Villalon-Letelier, F.; Zheng, M.Z.M.; Bennett-Wood, V.; Reading, P.C.; Wakim, L.M. Airway Exosomes Released During Influenza Virus Infection Serve as a Key Component of the Antiviral Innate Immune Response. Front. Immunol. 2020, 11, 887. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00887 (accessed on 9 November 2023). [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, 887. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harris, S.L.; Levine, A.J. The Regulation of Exosome Secretion: A Novel Function of the p53 Protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Keller, S.; Ridinger, J.; Rupp, A.-K.; Janssen, J.W.; Altevogt, P. Body fluid derived exosomes as a novel template for clinical diagnostics. J. Transl. Med. 2011, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; Van Eijndhoven, M.A.J.; Hopmans, E.S.; Lindenberg, J.L.; De Gruijl, T.D.; Würdinger, T.; Middeldorp, J.M. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.S.; Apcher, G.S.; Comber, J.D.; Eisenlohr, L.C. Exosome-Driven Antigen Transfer for MHC Class II Presentation Facilitated by the Receptor Binding Activity of Influenza Hemagglutinin. J. Immunol. 2010, 185, 6608–6616. [Google Scholar] [CrossRef]
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes Derived from HIV-1-infected Cells Contain Trans-activation Response Element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotič, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitaš, A.; Peterlin, B.M. HIV Nef is Secreted in Exosomes and Triggers Apoptosis in Bystander CD4+ T Cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; De Ruiter, P.E.; Willemsen, R.; Demmers, J.A.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [PubMed]
- Alenquer, M.; Amorim, M. Exosome Biogenesis, Regulation, and Function in Viral Infection. Viruses 2015, 7, 5066–5083. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhan, Y.; Zhu, L.; Hou, Z.; Liu, F.; Song, P.; Qiu, F.; Wang, X.; Zou, X.; Wan, D.; et al. Retrospective Multicenter Cohort Study Shows Early Interferon Therapy Is Associated with Favorable Clinical Responses in COVID-19 Patients. Cell Host Microbe 2020, 28, 455–464.e2. [Google Scholar] [CrossRef]
- Chen, J.S.; Alfajaro, M.M.; Chow, R.D.; Wei, J.; Filler, R.B.; Eisenbarth, S.C.; Wilen, C.B. Nonsteroidal Anti-inflammatory Drugs Dampen the Cytokine and Antibody Response to SARS-CoV-2 Infection. J. Virol. 2021, 95, 21. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhao, S.; Gan, L.; Wang, Z.; Peng, S.; Li, Q.; Liu, H.; Liu, X.; Wang, Z.; Shi, Q.; et al. Use of non-steroidal anti-inflammatory drugs and adverse outcomes during the COVID-19 pandemic: A systematic review and meta-analysis. eClinicalMedicine 2022, 46, 101373. [Google Scholar] [CrossRef]
- Campochiaro, C.; Della-Torre, E.; Cavalli, G.; De Luca, G.; Ripa, M.; Boffini, N.; Tomelleri, A.; Baldissera, E.; Rovere-Querini, P.; Ruggeri, A.; et al. Efficacy and safety of tocilizumab in severe COVID-19 patients: A single-centre retrospective cohort study. Eur. J. Intern. Med. 2020, 76, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.H.; Kim, M.H.; Sohn, Y.; Cho, Y.; Baek, Y.J.; Kim, J.H.; Ahn, J.Y.; Choi, J.Y.; Yeom, J.S.; Ahn, M.Y.; et al. Effects of early corticosteroid use in patients with severe coronavirus disease 2019. BMC Infect. Dis. 2021, 21, 506. [Google Scholar] [CrossRef] [PubMed]
- Mitrani, M.I.; Bellio, M.A.; Sagel, A.; Saylor, M.; Kapp, W.; VanOsdol, K.; Haskell, G.; Stewart, D.; Abdullah, Z.; Santos, I.; et al. Case Report: Administration of Amniotic Fluid-Derived Nanoparticles in Three Severely Ill COVID-19 Patients. Front. Med. 2021, 8, 583842. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.J.; Peltan, I.D.; Jensen, P.; Hoda, D.; Hunter, B.; Silver, A.; Starr, N.; Buckel, W.; Grisel, N.; Hummel, E.; et al. Clinical criteria for COVID-19-associated hyperinflammatory syndrome: A cohort study. Lancet Rheumatol. 2020, 2, e754–e763. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, N.; Inatomi, T.; Sotozono, C.; Fullwood, N.J.; Quantock, A.J.; Kinoshita, S. Growth factor mRNA and protein in preserved human amniotic membrane. Curr. Eye Res. 2000, 20, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Han, J.; Chen, S.; Xie, R.; Yang, J.; Zhou, T.; Zhang, Q.; Xia, R. MicroLet-7b Regulates Neutrophil Function and Dampens Neutrophilic Inflammation by Suppressing the Canonical TLR4/NF-κB Pathway. Front. Immunol. 2021, 12, 653344. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2021.653344 (accessed on 19 January 2024). [CrossRef] [PubMed]
- Xie, C.; Chen, Y.; Luo, D.; Zhuang, Z.; Jin, H.; Zhou, H.; Li, X.; Lin, H.; Zheng, X.; Zhang, J.; et al. Therapeutic potential of C1632 by inhibition of SARS-CoV-2 replication and viral-induced inflammation through upregulating let-7. Signal Transduct. Target. Ther. 2021, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, C.; Xia, C.; Liu, S.; Mei, Q. MicroRNA let-7 Suppresses Influenza A Virus Infection by Targeting RPS16 and Enhancing Type I Interferon Response. Front. Cell Infect. Microbiol. 2022, 12, 904775. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Zhang, L.; Sun, H.-X.; Zhang, Z.; Xu, J.; Xu, Y.; Lin, Y.; Zhu, A.; Luo, Y.; et al. Plasma cell-free RNA characteristics in COVID-19 patients. Genome Res. 2022, 32, 228–241. [Google Scholar] [CrossRef]
- Bernardo, M.M.; Fridman, R. TIMP-2 (tissue inhibitor of metalloproteinase-2) regulates MMP-2 (matrix metalloproteinase-2) activity in the extracellular environment after pro-MMP-2 activation by MT1 (membrane type 1)-MMP. Biochem. J. 2003, 374, 739–745. [Google Scholar] [CrossRef]
- Costanzo, L.; Soto, B.; Meier, R.; Geraghty, P. The Biology and Function of Tissue Inhibitor of Metalloproteinase 2 in the Lungs. Pulm. Med. 2022, 2022, 3632764. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.J.; Seaton, T.; Mitchell, J.; Howe, A.; Blackburn, K.; Burkhart, W.; Moyer, M.; Patel, I.; Waitt, G.M.; Becherer, J.D.; et al. The Τumor Necrosis Factor-α Converting Enzyme (TACE): A Unique Metalloproteinase with Highly Defined Substrate Selectivity. Biochemistry 2002, 41, 9462–9469. [Google Scholar] [CrossRef] [PubMed]
- Schönbeck, U.; Mach, F.; Libby, P. Generation of Biologically Active IL-1β by Matrix Metalloproteinases: A Novel Caspase-1-Independent Pathway of IL-1β Processing1. J. Immunol. 1998, 161, 3340–3346. [Google Scholar] [CrossRef] [PubMed]
- Manicone, A.M.; McGuire, J.K. Matrix Metalloproteinases as Modulators of Inflammation. Semin. Cell Dev. Biol. 2008, 19, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Hermanowski-Vosatka, A.; Mundt, S.S.; Ayala, J.M.; Goyal, S.; Hanlon, W.A.; Czerwinski, R.M.; Wright, S.D.; Whitman, C.P. Enzymatically Inactive Macrophage Migration Inhibitory Factor Inhibits Monocyte Chemotaxis and Random Migration. Biochemistry 1999, 38, 12841–12849. [Google Scholar] [CrossRef] [PubMed]
- Ietta, F.; Todros, T.; Ticconi, C.; Piccoli, E.; Zicari, A.; Piccione, E.; Paulesu, L. Macrophage Migration Inhibitory Factor in Human Pregnancy and Labor: MIF IN PREGNANCY AND LABOR. Am. J. Reprod. Immunol. 2002, 48, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Vestad, B.; Llorente, A.; Neurauter, A.; Phuyal, S.; Kierulf, B.; Kierulf, P.; Skotland, T.; Sandvig, K.; Haug, K.B.F.; Øvstebø, R. Size and concentration analyses of extracellular vesicles by nanoparticle tracking analysis: A variation study. J. Extracell. Vesicles 2017, 6, 1344087. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chanda, D.; Del Rivero, T.; Ghimire, R.; More, S.; Mitrani, M.I.; Bellio, M.A.; Channappanavar, R. Acellular Human Amniotic Fluid-Derived Extracellular Vesicles as Novel Anti-Inflammatory Therapeutics against SARS-CoV-2 Infection. Viruses 2024, 16, 273. https://0-doi-org.brum.beds.ac.uk/10.3390/v16020273
Chanda D, Del Rivero T, Ghimire R, More S, Mitrani MI, Bellio MA, Channappanavar R. Acellular Human Amniotic Fluid-Derived Extracellular Vesicles as Novel Anti-Inflammatory Therapeutics against SARS-CoV-2 Infection. Viruses. 2024; 16(2):273. https://0-doi-org.brum.beds.ac.uk/10.3390/v16020273
Chicago/Turabian StyleChanda, Debarati, Tania Del Rivero, Roshan Ghimire, Sunil More, Maria Ines Mitrani, Michael A. Bellio, and Rudragouda Channappanavar. 2024. "Acellular Human Amniotic Fluid-Derived Extracellular Vesicles as Novel Anti-Inflammatory Therapeutics against SARS-CoV-2 Infection" Viruses 16, no. 2: 273. https://0-doi-org.brum.beds.ac.uk/10.3390/v16020273