Prion Protein-Specific Antibodies-Development, Modes of Action and Therapeutics Application

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Therapeutic Candidates that Modulate PrPC Expression or Accessibility to Conversion
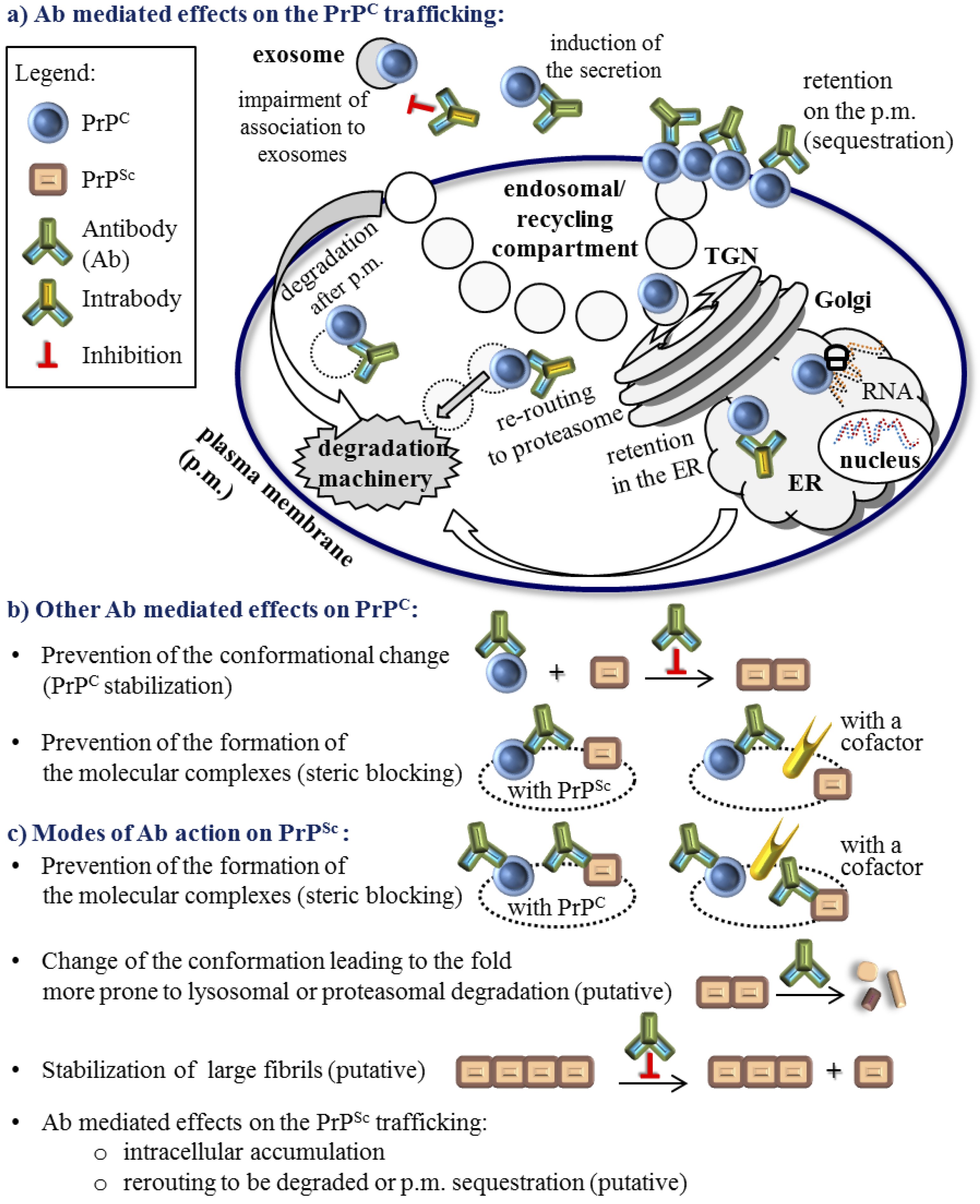
3. The Role of Antibodies in the Molecular Mechanism of the PrPC to PrPSc Conversion


4. Active and Passive Immunotherapy Approaches
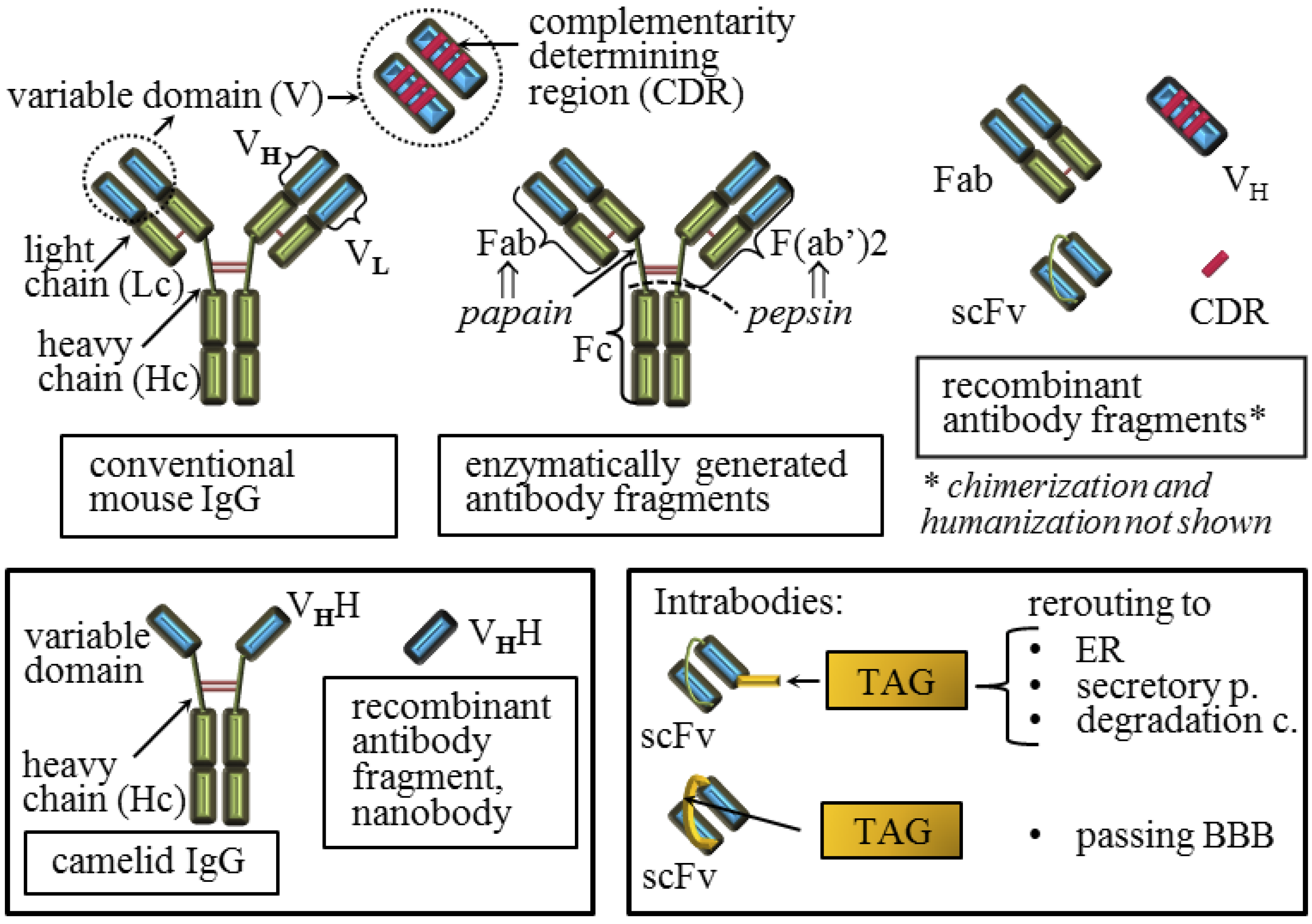
5. Molecular Parameters that Influence the Quality of the Anti-Prion Protein Antibody Effect
6. Authors’ Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar]
- Imran, M.; Mahmood, S. An overview of human prion diseases. Virol. J. 2011, 8, 559. [Google Scholar]
- Aguzzi, A.; Calella, A.M. Prions: Protein aggregation and infectious diseases. Physiol. Rev. 2009, 89, 1105–1152. [Google Scholar]
- Zou, W.Q.; Puoti, G.; Xiao, X.; Yuan, J.; Qing, L.; Cali, I.; Shimoji, M.; Langeveld, J.P.; Castellani, R.; Notari, S.; et al. Variably protease-sensitive prionopathy: A new sporadic disease of the prion protein. Ann. Neurol. 2010, 68, 162–172. [Google Scholar]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar]
- Mallucci, G.R. Prion neurodegeneration: Starts and stops at the synapse. Prion 2009, 3, 195–201. [Google Scholar]
- Grassmann, A.; Wolf, H.; Hofmann, J.; Graham, J.; Vorberg, I. Cellular aspects of prion replication in vitro. Viruses 2013, 5, 374–405. [Google Scholar]
- Gousset, K.; Zurzolo, C. Tunnelling nanotubes: A highway for prion spreading? Prion 2009, 3, 94–98. [Google Scholar]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of prp are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar]
- Mallucci, G.R.; Ratte, S.; Asante, E.A.; Linehan, J.; Gowland, I.; Jefferys, J.G.; Collinge, J. Post-natal knockout of prion protein alters hippocampal ca1 properties, but does not result in neurodegeneration. EMBO J. 2002, 21, 202–210. [Google Scholar]
- Nicoll, A.J.; Collinge, J. Preventing prion pathogenicity by targeting the cellular prion protein. Infect. Disord. Drug Targets 2009, 9, 48–57. [Google Scholar]
- Didonna, A. Prion protein and its role in signal transduction. Cell. Mol. Biol. Lett. 2013, 18, 209–230. [Google Scholar]
- Stewart, L.A.; Rydzewska, L.H.; Keogh, G.F.; Knight, R.S. Systematic review of therapeutic interventions in human prion disease. Neurology 2008, 70, 1272–1281. [Google Scholar]
- Dressel, J.; Oesper, R. The discovery of germanin by oskar dressel and richard kothe. J. Chem. Educ. 1961, 38, 620–621. [Google Scholar]
- Nunziante, M.; Kehler, C.; Maas, E.; Kassack, M.U.; Groschup, M.; Schatzl, H.M. Charged bipolar suramin derivatives induce aggregation of the prion protein at the cell surface and inhibit prpsc replication. J. Cell Sci. 2005, 118, 4959–4973. [Google Scholar]
- Kiachopoulos, S.; Heske, J.; Tatzelt, J.; Winklhofer, K.F. Misfolding of the prion protein at the plasma membrane induces endocytosis, intracellular retention and degradation. Traffic 2004, 5, 426–436. [Google Scholar]
- Taraboulos, A.; Scott, M.; Semenov, A.; Avrahami, D.; Laszlo, L.; Prusiner, S.B. Cholesterol depletion and modification of cooh-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 1995, 129, 121–132. [Google Scholar]
- Alberts, A.W.; Chen, J.; Kuron, G.; Hunt, V.; Huff, J.; Hoffman, C.; Rothrock, J.; Lopez, M.; Joshua, H.; Harris, E.; et al. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme a reductase and a cholesterol-lowering agent. Proc. Natl. Acad. Sci. USA 1980, 77, 3957–3961. [Google Scholar]
- Gilch, S.; Kehler, C.; Schatzl, H.M. The prion protein requires cholesterol for cell surface localization. Mol. Cell. Neurosci. 2006, 31, 346–353. [Google Scholar]
- Group, B.I.G.C.; Mouridsen, H.; Giobbie-Hurder, A.; Goldhirsch, A.; Thurlimann, B.; Paridaens, R.; Smith, I.; Mauriac, L.; Forbes, J.; Price, K.N.; et al. Letrozole therapy alone or in sequence with tamoxifen in women with breast cancer. N. Engl. J. Med. 2009, 361, 766–776. [Google Scholar]
- Marzo, L.; Marijanovic, Z.; Browman, D.; Chamoun, Z.; Caputo, A.; Zurzolo, C. 4-hydroxytamoxifen leads to prpsc clearance by conveying both prpc and prpsc to lysosomes independently of autophagy. J. Cell Sci. 2013, 126, 1345–1354. [Google Scholar]
- Sim, V.L.; Caughey, B. Recent advances in prion chemotherapeutics. Infect. Disord. Drug Targets 2009, 9, 81–91. [Google Scholar]
- Karapetyan, Y.E.; Sferrazza, G.F.; Zhou, M.; Ottenberg, G.; Spicer, T.; Chase, P.; Fallahi, M.; Hodder, P.; Weissmann, C.; Lasmezas, C.I. Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc. Natl. Acad. Sci. USA 2013, 110, 7044–7049. [Google Scholar]
- Trevitt, C.R.; Collinge, J. A systematic review of prion therapeutics in experimental models. Brain: J. Neurol. 2006, 129, 2241–2265. [Google Scholar]
- Gilch, S.; Schatzl, H.M. Aptamers against prion proteins and prions. Cell. Mol. Life Sci.: CMLS 2009, 66, 2445–2455. [Google Scholar]
- Peretz, D.; Williamson, R.A.; Kaneko, K.; Vergara, J.; Leclerc, E.; Schmitt-Ulms, G.; Mehlhorn, I.R.; Legname, G.; Wormald, M.R.; Rudd, P.M.; et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 2001, 412, 739–743. [Google Scholar]
- Antonyuk, S.V.; Trevitt, C.R.; Strange, R.W.; Jackson, G.S.; Sangar, D.; Batchelor, M.; Cooper, S.; Fraser, C.; Jones, S.; Georgiou, T.; et al. Crystal structure of human prion protein bound to a therapeutic antibody. Proc. Natl. Acad. Sci. USA 2009, 106, 2554–2558. [Google Scholar]
- Feraudet, C.; Morel, N.; Simon, S.; Volland, H.; Frobert, Y.; Creminon, C.; Vilette, D.; Lehmann, S.; Grassi, J. Screening of 145 anti-prp monoclonal antibodies for their capacity to inhibit prpsc replication in infected cells. J. Biol. Chem. 2005, 280, 11247–11258. [Google Scholar]
- Enari, M.; Flechsig, E.; Weissmann, C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl. Acad. Sci. USA 2001, 98, 9295–9299. [Google Scholar]
- Berry, D.B.; Lu, D.; Geva, M.; Watts, J.C.; Bhardwaj, S.; Oehler, A.; Renslo, A.R.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Drug resistance confounding prion therapeutics. Proc. Natl. Acad. Sci. USA 2013, 110, E4160–E4169. [Google Scholar]
- Ghaemmaghami, S.; May, B.C.; Renslo, A.R.; Prusiner, S.B. Discovery of 2-aminothiazoles as potent antiprion compounds. J. Virol. 2010, 84, 3408–3412. [Google Scholar]
- Ferreira, N.C.; Marques, I.A.; Conceicao, W.A.; Macedo, B.; Machado, C.S.; Mascarello, A.; Chiaradia-Delatorre, L.D.; Yunes, R.A.; Nunes, R.J.; Hughson, A.G.; et al. Anti-prion activity of a panel of aromatic chemical compounds: In vitro and in silico approaches. PLoS One 2014, 9, e84531. [Google Scholar]
- Supattapone, S.; Wille, H.; Uyechi, L.; Safar, J.; Tremblay, P.; Szoka, F.C.; Cohen, F.E.; Prusiner, S.B.; Scott, M.R. Branched polyamines cure prion-infected neuroblastoma cells. J. Virol. 2001, 75, 3453–3461. [Google Scholar]
- Ertmer, A.; Gilch, S.; Yun, S.W.; Flechsig, E.; Klebl, B.; Stein-Gerlach, M.; Klein, M.A.; Schatzl, H.M. The tyrosine kinase inhibitor sti571 induces cellular clearance of prpsc in prion-infected cells. J. Biol. Chem. 2004, 279, 41918–41927. [Google Scholar]
- Caspi, S.; Halimi, M.; Yanai, A.; Sasson, S.B.; Taraboulos, A.; Gabizon, R. The anti-prion activity of congo red. Putative mechanism. J. Biol. Chem. 1998, 273, 3484–3489. [Google Scholar]
- Margalith, I.; Suter, C.; Ballmer, B.; Schwarz, P.; Tiberi, C.; Sonati, T.; Falsig, J.; Nystrom, S.; Hammarstrom, P.; Aslund, A.; et al. Polythiophenes inhibit prion propagation by stabilizing prion protein (prp) aggregates. J. Biol. Chem. 2012, 287, 18872–18887. [Google Scholar]
- Silveira, J.R.; Raymond, G.J.; Hughson, A.G.; Race, R.E.; Sim, V.L.; Hayes, S.F.; Caughey, B. The most infectious prion protein particles. Nature 2005, 437, 257–261. [Google Scholar]
- Simoneau, S.; Rezaei, H.; Sales, N.; Kaiser-Schulz, G.; Lefebvre-Roque, M.; Vidal, C.; Fournier, J.G.; Comte, J.; Wopfner, F.; Grosclaude, J.; et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog. 2007, 3, e125. [Google Scholar]
- Paramithiotis, E.; Pinard, M.; Lawton, T.; LaBoissiere, S.; Leathers, V.L.; Zou, W.Q.; Estey, L.A.; Lamontagne, J.; Lehto, M.T.; Kondejewski, L.H.; et al. A prion protein epitope selective for the pathologically misfolded conformation. Nat. Med. 2003, 9, 893–899. [Google Scholar]
- Hedlin, P.D.; Cashman, N.R.; Li, L.; Gupta, J.; Babiuk, L.A.; Potter, A.A.; Griebel, P.; Napper, S. Design and delivery of a cryptic prp(c) epitope for induction of prp(sc)-specific antibody responses. Vaccine 2010, 28, 981–988. [Google Scholar]
- Jones, M.; Wight, D.; McLoughlin, V.; Norrby, K.; Ironside, J.W.; Connolly, J.G.; Farquhar, C.F.; MacGregor, I.R.; Head, M.W. An antibody to the aggregated synthetic prion protein peptide (prp106–126) selectively recognizes disease-associated prion protein (prp) from human brain specimens. Brain Pathol. 2009, 19, 293–302. [Google Scholar]
- Horiuchi, M.; Karino, A.; Furuoka, H.; Ishiguro, N.; Kimura, K.; Shinagawa, M. Generation of monoclonal antibody that distinguishes prpsc from prpc and neutralizes prion infectivity. Virology 2009, 394, 200–207. [Google Scholar]
- Curin Serbec, V.; Bresjanac, M.; Popovic, M.; Pretnar Hartman, K.; Galvani, V.; Rupreht, R.; Cernilec, M.; Vranac, T.; Hafner, I.; Jerala, R. Monoclonal antibody against a peptide of human prion protein discriminates between creutzfeldt-jacob's disease-affected and normal brain tissue. J. Biol. Chem. 2004, 279, 3694–3698. [Google Scholar]
- Korth, C.; Stierli, B.; Streit, P.; Moser, M.; Schaller, O.; Fischer, R.; Schulz-Schaeffer, W.; Kretzschmar, H.; Raeber, A.; Braun, U.; et al. Prion (prpsc)-specific epitope defined by a monoclonal antibody. Nature 1997, 390, 74–77. [Google Scholar]
- Petsch, B.; Muller-Schiffmann, A.; Lehle, A.; Zirdum, E.; Prikulis, I.; Kuhn, F.; Raeber, A.J.; Ironside, J.W.; Korth, C.; Stitz, L. Biological effects and use of prpsc- and prp-specific antibodies generated by immunization with purified full-length native mouse prions. J. Virol. 2011, 85, 4538–4546. [Google Scholar]
- Beringue, V.; Vilette, D.; Mallinson, G.; Archer, F.; Kaisar, M.; Tayebi, M.; Jackson, G.S.; Clarke, A.R.; Laude, H.; Collinge, J.; et al. Prpsc binding antibodies are potent inhibitors of prion replication in cell lines. J. Biol. Chem. 2004, 279, 39671–39676. [Google Scholar]
- White, A.R.; Enever, P.; Tayebi, M.; Mushens, R.; Linehan, J.; Brandner, S.; Anstee, D.; Collinge, J.; Hawke, S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003, 422, 80–83. [Google Scholar]
- Kubota, T.; Hamazoe, Y.; Hashiguchi, S.; Ishibashi, D.; Akasaka, K.; Nishida, N.; Katamine, S.; Sakaguchi, S.; Kuroki, R.; Nakashima, T.; et al. Direct evidence of generation and accumulation of beta-sheet-rich prion protein in scrapie-infected neuroblastoma cells with human igg1 antibody specific for beta-form prion protein. J. Biol. Chem. 2012, 287, 14023–14039. [Google Scholar]
- Roettger, Y.; Du, Y.; Bacher, M.; Zerr, I.; Dodel, R.; Bach, J.P. Immunotherapy in prion disease. Nat. Rev. Neurol. 2013, 9, 98–105. [Google Scholar]
- Bade, S.; Frey, A. Potential of active and passive immunizations for the prevention and therapy of transmissible spongiform encephalopathies. Exp. Rev. Vacc. 2007, 6, 153–168. [Google Scholar]
- Gilch, S.; Wopfner, F.; Renner-Muller, I.; Kremmer, E.; Bauer, C.; Wolf, E.; Brem, G.; Groschup, M.H.; Schatzl, H.M. Polyclonal anti-prp auto-antibodies induced with dimeric prp interfere efficiently with prpsc propagation in prion-infected cells. J. Biol. Chem. 2003, 278, 18524–18531. [Google Scholar]
- Pankiewicz, J.; Prelli, F.; Sy, M.S.; Kascsak, R.J.; Kascsak, R.B.; Spinner, D.S.; Carp, R.I.; Meeker, H.C.; Sadowski, M.; Wisniewski, T. Clearance and prevention of prion infection in cell culture by anti-prp antibodies. Eur. J. Neurosci. 2006, 23, 2635–2647. [Google Scholar]
- Chang, B.; Petersen, R.; Wisniewski, T.; Rubenstein, R. Influence of mabs on prp(sc) formation using in vitro and cell-free systems. PLoS One 2012, 7, e41626. [Google Scholar]
- Kim, C.L.; Karino, A.; Ishiguro, N.; Shinagawa, M.; Sato, M.; Horiuchi, M. Cell-surface retention of prpc by anti-prp antibody prevents protease-resistant prp formation. J. Gener. Virol. 2004, 85, 3473–3482. [Google Scholar]
- Sonati, T.; Reimann, R.R.; Falsig, J.; Baral, P.K.; O'Connor, T.; Hornemann, S.; Yaganoglu, S.; Li, B.; Herrmann, U.S.; Wieland, B.; et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013, 501, 102–106. [Google Scholar]
- Caughey, B.; Raymond, G.J.; Ernst, D.; Race, R.E. N-terminal truncation of the scrapie-associated form of prp by lysosomal protease(s): Implications regarding the site of conversion of prp to the protease-resistant state. J. Virol. 1991, 65, 6597–6603. [Google Scholar]
- Caughey, B.; Raymond, G.J. The scrapie-associated form of prp is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J. Biol. Chem. 1991, 266, 18217–18223. [Google Scholar]
- Taraboulos, A.; Raeber, A.J.; Borchelt, D.R.; Serban, D.; Prusiner, S.B. Synthesis and trafficking of prion proteins in cultured cells. Mol. Biol. Cell 1992, 3, 851–863. [Google Scholar]
- Borchelt, D.R.; Taraboulos, A.; Prusiner, S.B. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 1992, 267, 16188–16199. [Google Scholar]
- Shyng, S.L.; Huber, M.T.; Harris, D.A. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J. Biol. Chem. 1993, 268, 15922–15928. [Google Scholar]
- Marijanovic, Z.; Caputo, A.; Campana, V.; Zurzolo, C. Identification of an intracellular site of prion conversion. PLoS Pathog. 2009, 5, e1000426. [Google Scholar]
- Yamasaki, T.; Baron, G.S.; Suzuki, A.; Hasebe, R.; Horiuchi, M. Characterization of intracellular dynamics of inoculated prp-res and newly generated prp(sc) during early stage prion infection in neuro2a cells. Virology 2014, 450–451, 324–335. [Google Scholar]
- Goold, R.; McKinnon, C.; Rabbanian, S.; Collinge, J.; Schiavo, G.; Tabrizi, S.J. Alternative fates of newly formed prpsc upon prion conversion on the plasma membrane. J. Cell Sci. 2013, 126, 3552–3562. [Google Scholar]
- Rouvinski, A.; Karniely, S.; Kounin, M.; Moussa, S.; Goldberg, M.D.; Warburg, G.; Lyakhovetsky, R.; Papy-Garcia, D.; Kutzsche, J.; Korth, C.; et al. Live imaging of prions reveals nascent prpsc in cell-surface, raft-associated amyloid strings and webs. J. Cell Biol. 2014, 204, 423–441. [Google Scholar]
- Uchiyama, K.; Muramatsu, N.; Yano, M.; Usui, T.; Miyata, H.; Sakaguchi, S. Prions disturb post-golgi trafficking of membrane proteins. Nat. Commun. 2013, 4, 1846. [Google Scholar]
- Polymenidou, M.; Heppner, F.L.; Pellicioli, E.C.; Urich, E.; Miele, G.; Braun, N.; Wopfner, F.; Schatzl, H.M.; Becher, B.; Aguzzi, A. Humoral immune response to native eukaryotic prion protein correlates with anti-prion protection. Proc. Natl. Acad. Sci. USA 2004, 101, 14670–14676. [Google Scholar] [Green Version]
- Xanthopoulos, K.; Lagoudaki, R.; Kontana, A.; Kyratsous, C.; Panagiotidis, C.; Grigoriadis, N.; Yiangou, M.; Sklaviadis, T. Immunization with recombinant prion protein leads to partial protection in a murine model of tses through a novel mechanism. PLoS One 2013, 8, e59143. [Google Scholar]
- Shimizu, Y.; Kaku-Ushiki, Y.; Iwamaru, Y.; Muramoto, T.; Kitamoto, T.; Yokoyama, T.; Mohri, S.; Tagawa, Y. A novel anti-prion protein monoclonal antibody and its single-chain fragment variable derivative with ability to inhibit abnormal prion protein accumulation in cultured cells. Microbiol. Immunol. 2010, 54, 112–121. [Google Scholar]
- David, M.A.; Jones, D.R.; Tayebi, M. Potential candidate camelid antibodies for the treatment of protein-misfolding diseases. J. Neuroimmunol. 2014, 272, 76–85. [Google Scholar]
- Perrier, V.; Solassol, J.; Crozet, C.; Frobert, Y.; Mourton-Gilles, C.; Grassi, J.; Lehmann, S. Anti-prp antibodies block prpsc replication in prion-infected cell cultures by accelerating prpc degradation. J. Neurochem. 2004, 89, 454–463. [Google Scholar]
- Heppner, F.L.; Musahl, C.; Arrighi, I.; Klein, M.A.; Rulicke, T.; Oesch, B.; Zinkernagel, R.M.; Kalinke, U.; Aguzzi, A. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science 2001, 294, 178–182. [Google Scholar]
- Jones, D.R.; Taylor, W.A.; Bate, C.; David, M.; Tayebi, M. A camelid anti-prp antibody abrogates prp replication in prion-permissive neuroblastoma cell lines. PLoS One 2010, 5, e9804. [Google Scholar]
- Cardinale, A.; Biocca, S. Gene-based antibody strategies for prion diseases. Int. J. Cell Biol. 2013, 2013, 710406. [Google Scholar]
- Gilch, S.; Nunziante, M.; Ertmer, A.; Schatzl, H.M. Strategies for eliminating prp(c) as substrate for prion conversion and for enhancing prp(sc) degradation. Vet. Microbiol. 2007, 123, 377–386. [Google Scholar]
- Wei, X.; Roettger, Y.; Tan, B.; He, Y.; Dodel, R.; Hampel, H.; Wei, G.; Haney, J.; Gu, H.; Johnstone, B.H.; et al. Human anti-prion antibodies block prion peptide fibril formation and neurotoxicity. J. Biol. Chem. 2012, 287, 12858–12866. [Google Scholar]
- Madampage, C.A.; Maattanen, P.; Marciniuk, K.; Brownlie, R.; Andrievskaia, O.; Potter, A.; Cashman, N.R.; Lee, J.S.; Napper, S. Binding of bovine t194a prp(c) by prp(sc)-specific antibodies: Potential implications for immunotherapy of familial prion diseases. Prion 2013, 7, 301–311. [Google Scholar]
- Goni, F.; Knudsen, E.; Schreiber, F.; Scholtzova, H.; Pankiewicz, J.; Carp, R.; Meeker, H.C.; Rubenstein, R.; Brown, D.R.; Sy, M.S.; et al. Mucosal vaccination delays or prevents prion infection via an oral route. Neuroscience 2005, 133, 413–421. [Google Scholar]
- Bade, S.; Baier, M.; Boetel, T.; Frey, A. Intranasal immunization of balb/c mice against prion protein attenuates orally acquired transmissible spongiform encephalopathy. Vaccine 2006, 24, 1242–1253. [Google Scholar]
- Sigurdsson, E.M.; Sy, M.S.; Li, R.; Scholtzova, H.; Kascsak, R.J.; Kascsak, R.; Carp, R.; Meeker, H.C.; Frangione, B.; Wisniewski, T. Anti-prion antibodies for prophylaxis following prion exposure in mice. Neurosci. Lett. 2003, 336, 185–187. [Google Scholar]
- Song, C.H.; Furuoka, H.; Kim, C.L.; Ogino, M.; Suzuki, A.; Hasebe, R.; Horiuchi, M. Effect of intraventricular infusion of anti-prion protein monoclonal antibodies on disease progression in prion-infected mice. J. Gener. Virol. 2008, 89, 1533–1544. [Google Scholar]
- Ohsawa, N.; Song, C.H.; Suzuki, A.; Furuoka, H.; Hasebe, R.; Horiuchi, M. Therapeutic effect of peripheral administration of an anti-prion protein antibody on mice infected with prions. Microbiol. Immunol. 2013, 57, 288–297. [Google Scholar]
- Wuertzer, C.A.; Sullivan, M.A.; Qiu, X.; Federoff, H.J. Cns delivery of vectored prion-specific single-chain antibodies delays disease onset. Mol. Ther. 2008, 16, 481–486. [Google Scholar]
- Moda, F.; Vimercati, C.; Campagnani, I.; Ruggerone, M.; Giaccone, G.; Morbin, M.; Zentilin, L.; Giacca, M.; Zucca, I.; Legname, G.; et al. Brain delivery of aav9 expressing an anti-prp monovalent antibody delays prion disease in mice. Prion 2012, 6, 383–390. [Google Scholar]
- Campana, V.; Zentilin, L.; Mirabile, I.; Kranjc, A.; Casanova, P.; Giacca, M.; Prusiner, S.B.; Legname, G.; Zurzolo, C. Development of antibody fragments for immunotherapy of prion diseases. Biochem. J. 2009, 418, 507–515. [Google Scholar]
- Solforosi, L.; Criado, J.R.; McGavern, D.B.; Wirz, S.; Sanchez-Alavez, M.; Sugama, S.; DeGiorgio, L.A.; Volpe, B.T.; Wiseman, E.; Abalos, G.; et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 2004, 303, 1514–1516. [Google Scholar]
- Klohn, P.C.; Farmer, M.; Linehan, J.M.; O’Malley, C.; Fernandez de Marco, M.; Taylor, W.; Farrow, M.; Khalili-Shirazi, A.; Brandner, S.; Collinge, J. Prp antibodies do not trigger mouse hippocampal neuron apoptosis. Science 2012, 335, 52. [Google Scholar]
- Kellett, K.A.; Hooper, N.M. Prion protein and alzheimer disease. Prion 2009, 3, 190–194. [Google Scholar]
- Brody, D.L.; Holtzman, D.M. Active and passive immunotherapy for neurodegenerative disorders. Annu. Rev. Neurosc. 2008, 31, 175–193. [Google Scholar]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate alzheimer's disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar]
- Klyubin, I.; Nicoll, A.J.; Khalili-Shirazi, A.; Farmer, M.; Canning, S.; Mably, A.; Linehan, J.; Brown, A.; Wakeling, M.; Brandner, S.; et al. Peripheral administration of a humanized anti-prp antibody blocks alzheimer's disease abeta synaptotoxicity. J. Neurosci. 2014, 34, 6140–6145. [Google Scholar]
- Bendheim, P.E.; Barry, R.A.; DeArmond, S.J.; Stites, D.P.; Prusiner, S.B. Antibodies to a scrapie prion protein. Nature 1984, 310, 418–421. [Google Scholar]
- Williamson, R.A.; Peretz, D.; Smorodinsky, N.; Bastidas, R.; Serban, H.; Mehlhorn, I.; DeArmond, S.J.; Prusiner, S.B.; Burton, D.R. Circumventing tolerance to generate autologous monoclonal antibodies to the prion protein. Proc. Natl. Acad. Sci. USA 1996, 93, 7279–7282. [Google Scholar]
- McCutcheon, S.; Langeveld, J.P.; Tan, B.C.; Gill, A.C.; de Wolf, C.; Martin, S.; Gonzalez, L.; Alibhai, J.; Blanco, A.R.; Campbell, L.; et al. Prion protein-specific antibodies that detect multiple tse agents with high sensitivity. PLoS One 2014, 9, e91143. [Google Scholar]
- Stanker, L.H.; Serban, A.V.; Cleveland, E.; Hnasko, R.; Lemus, A.; Safar, J.; DeArmond, S.J.; Prusiner, S.B. Conformation-dependent high-affinity monoclonal antibodies to prion proteins. J. Immunol. 2010, 185, 729–737. [Google Scholar]
- Polymenidou, M.; Moos, R.; Scott, M.; Sigurdson, C.; Shi, Y.Z.; Yajima, B.; Hafner-Bratkovic, I.; Jerala, R.; Hornemann, S.; Wuthrich, K.; et al. The pom monoclonals: A comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS One 2008, 3, e3872. [Google Scholar]
- Abskharon, R.N.; Giachin, G.; Wohlkonig, A.; Soror, S.H.; Pardon, E.; Legname, G.; Steyaert, J. Probing the N-terminal beta-sheet conversion in the crystal structure of the human prion protein bound to a nanobody. J. Am. Chem. Soc. 2014, 136, 937–944. [Google Scholar]
- Feraudet-Tarisse, C.; Andreoletti, O.; Morel, N.; Simon, S.; Lacroux, C.; Mathey, J.; Lamourette, P.; Relano, A.; Torres, J.M.; Creminon, C.; et al. Immunotherapeutic effect of anti-prp monoclonal antibodies in transmissible spongiform encephalopathy mouse models: Pharmacokinetic and pharmacodynamic analysis. J. Gener. Virol. 2010, 91, 1635–1645. [Google Scholar]
- Alexandrenne, C.; Hanoux, V.; Dkhissi, F.; Boquet, D.; Couraud, J.Y.; Wijkhuisen, A. Curative properties of antibodies against prion protein: A comparative in vitro study of monovalent fragments and divalent antibodies. J. Neuroimmunol. 2009, 209, 50–56. [Google Scholar]
- Weir, A.N.; Nesbitt, A.; Chapman, A.P.; Popplewell, A.G.; Antoniw, P.; Lawson, A.D. Formatting antibody fragments to mediate specific therapeutic functions. Biochem. Soc. Trans. 2002, 30, 512–516. [Google Scholar]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar]
- Huang, L.; Su, X.; Federoff, H.J. Single-chain fragment variable passive immunotherapies for neurodegenerative diseases. Int. J. Mol. Sci. 2013, 14, 19109–19127. [Google Scholar]
- Donofrio, G.; Heppner, F.L.; Polymenidou, M.; Musahl, C.; Aguzzi, A. Paracrine inhibition of prion propagation by anti-prp single-chain fv miniantibodies. J. Virol. 2005, 79, 8330–8338. [Google Scholar]
- Fujita, K.; Yamaguchi, Y.; Mori, T.; Muramatsu, N.; Miyamoto, T.; Yano, M.; Miyata, H.; Ootsuyama, A.; Sawada, M.; Matsuda, H.; et al. Effects of a brain-engraftable microglial cell line expressing anti-prion scfv antibodies on survival times of mice infected with scrapie prions. Cell. Mol. Neurobiol. 2011, 31, 999–1008. [Google Scholar]
- Muller-Schiffmann, A.; Petsch, B.; Leliveld, S.R.; Muyrers, J.; Salwierz, A.; Mangels, C.; Schwarzinger, S.; Riesner, D.; Stitz, L.; Korth, C. Complementarity determining regions of an anti-prion protein scfv fragment orchestrate conformation specificity and antiprion activity. Mol. Immunol. 2009, 46, 532–540. [Google Scholar]
- Skrlj, N.; Vranac, T.; Popovic, M.; Curin Serbec, V.; Dolinar, M. Specific binding of the pathogenic prion isoform: Development and characterization of a humanized single-chain variable antibody fragment. PLoS One 2011, 6, e15783. [Google Scholar]
- Skrlj, N.; Drevensek, G.; Hudoklin, S.; Romih, R.; Curin Serbec, V.; Dolinar, M. Recombinant single-chain antibody with the trojan peptide penetratin positioned in the linker region enables cargo transfer across the blood-brain barrier. Appl. Biochem. Biotechnol. 2013, 169, 159–169. [Google Scholar]
- Prusiner, S.B.; Groth, D.; Serban, A.; Koehler, R.; Foster, D.; Torchia, M.; Burton, D.; Yang, S.L.; DeArmond, S.J. Ablation of the prion protein (prp) gene in mice prevents scrapie and facilitates production of anti-prp antibodies. Proc. Natl. Acad. Sci. USA 1993, 90, 10608–10612. [Google Scholar]
- Vorberg, I.; Raines, A.; Story, B.; Priola, S.A. Susceptibility of common fibroblast cell lines to transmissible spongiform encephalopathy agents. J. Infect. Dis. 2004, 189, 431–439. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rovis, T.L.; Legname, G. Prion Protein-Specific Antibodies-Development, Modes of Action and Therapeutics Application. Viruses 2014, 6, 3719-3737. https://0-doi-org.brum.beds.ac.uk/10.3390/v6103719
Rovis TL, Legname G. Prion Protein-Specific Antibodies-Development, Modes of Action and Therapeutics Application. Viruses. 2014; 6(10):3719-3737. https://0-doi-org.brum.beds.ac.uk/10.3390/v6103719
Chicago/Turabian StyleRovis, Tihana Lenac, and Giuseppe Legname. 2014. "Prion Protein-Specific Antibodies-Development, Modes of Action and Therapeutics Application" Viruses 6, no. 10: 3719-3737. https://0-doi-org.brum.beds.ac.uk/10.3390/v6103719