
Temporal Regulation of Distinct Internal Ribosome Entry Sites of the Dicistroviridae Cricket Paralysis Virus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Materials and Methods
2.1. Cell Culture, Virus Infection, and Viral Titers
2.2. Reporter Construct and CrPV2 Replicon
2.3. RNA Transfection and Luciferase Assay
2.4. Western Blotting
2.5. [35S]-Met/Cys Pulse-Labelling
2.6. Immunoprecipitation
2.7. dsRNA-Mediated Knockdown
2.8. S2 Translation Extracts and in Vitro Translation Assay
2.9. Ribosome Profiling
2.10. Calculations of Translational Efficiency
2.11. Uridine Labelling of RNA
3. Results
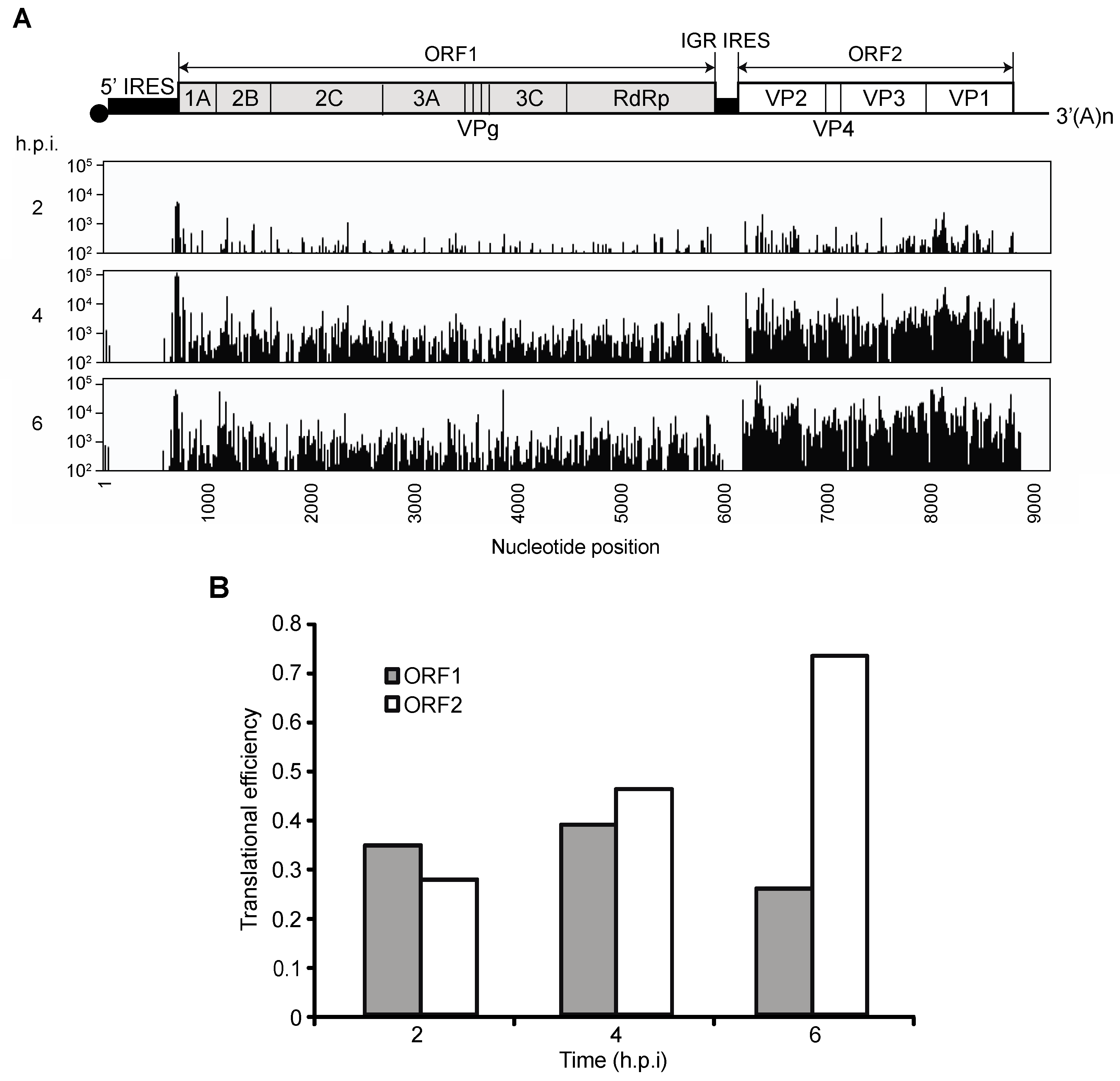
3.1. Translation of CrPV ORF1 and ORF2 during CrPV Infection
3.2. Ribosome Profiling of Viral RNA in CrPV-Infected S2 Cells

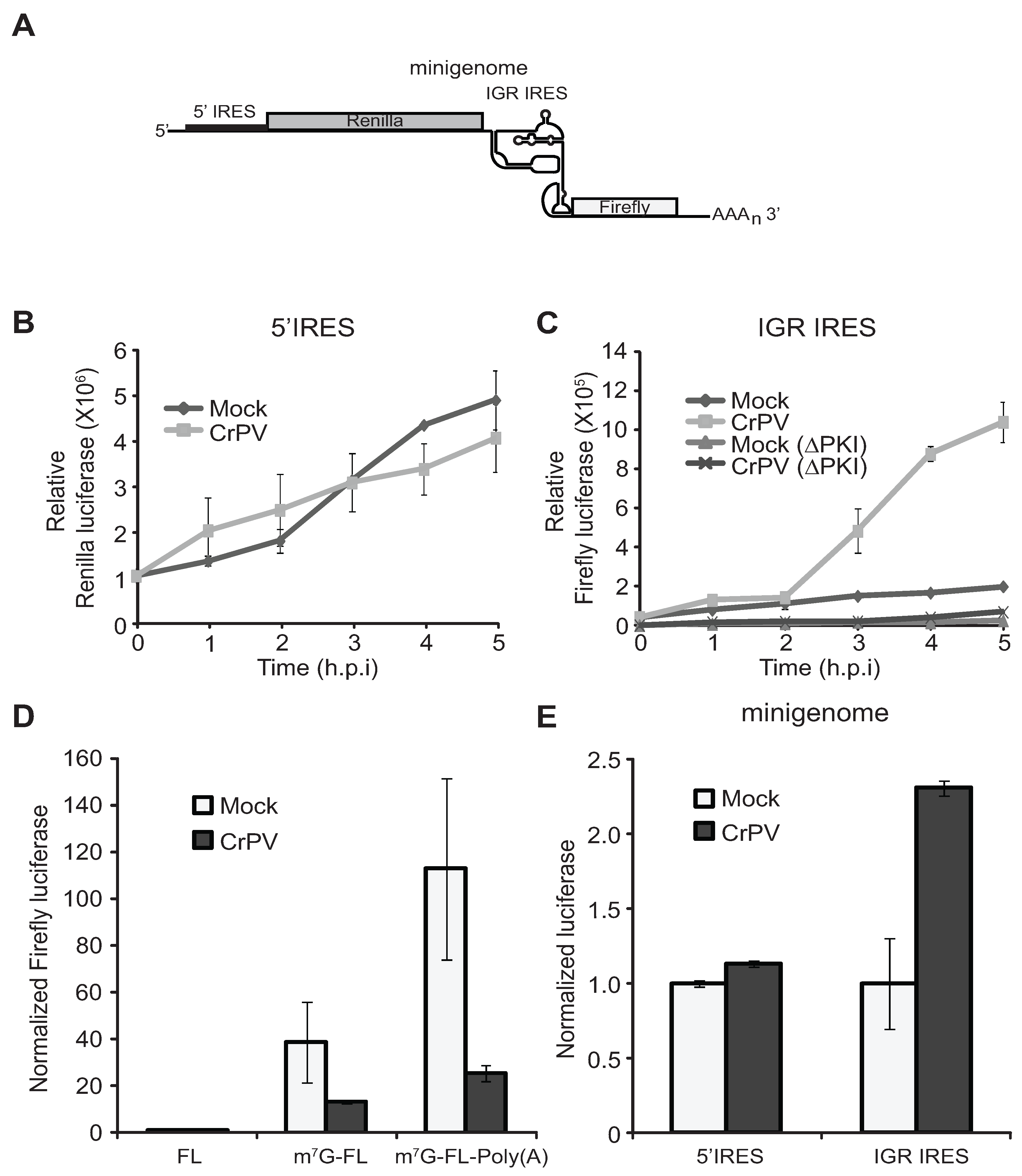
3.3. IRES Translation Using CrPV Minigenome Reporter Analysis

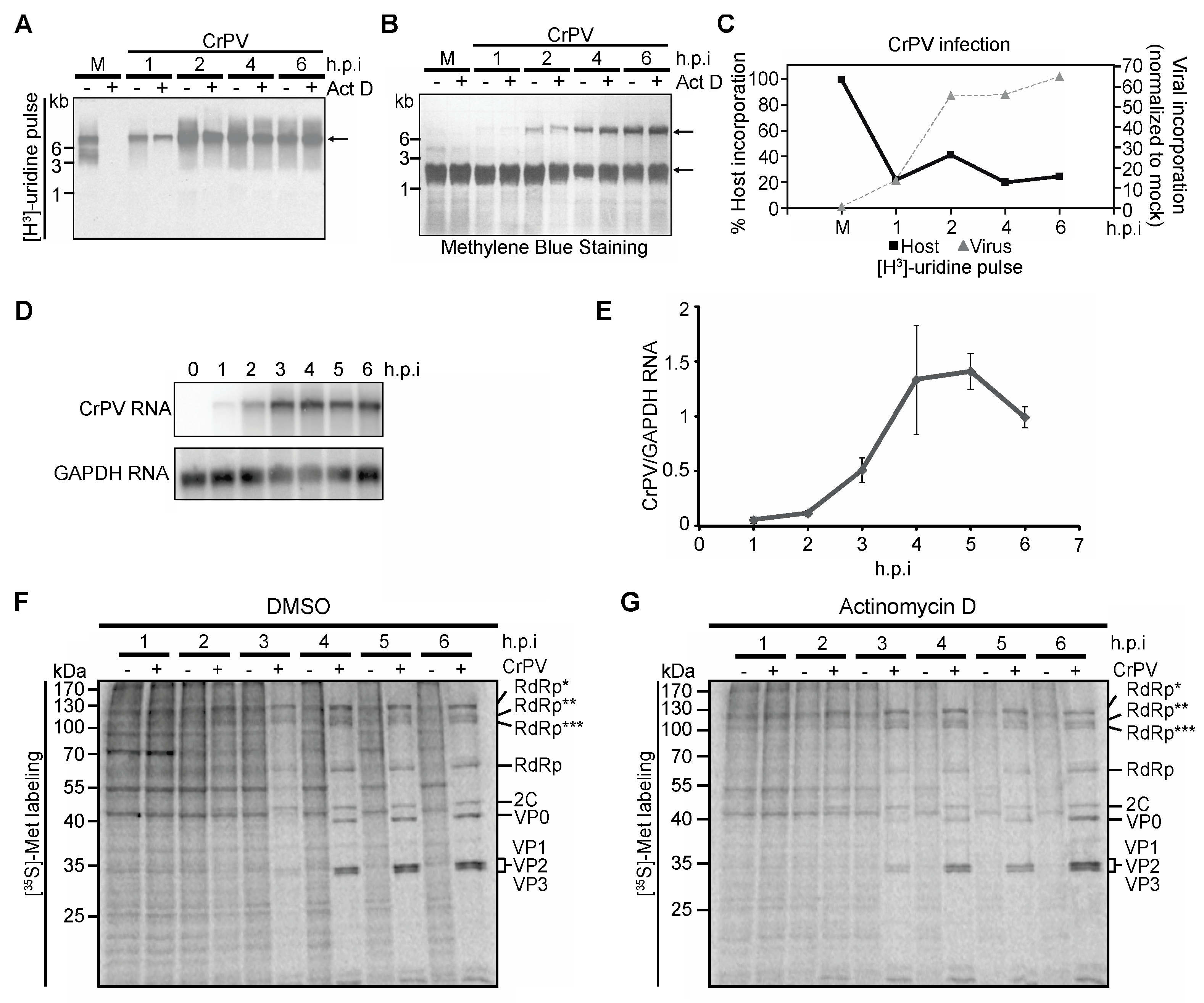
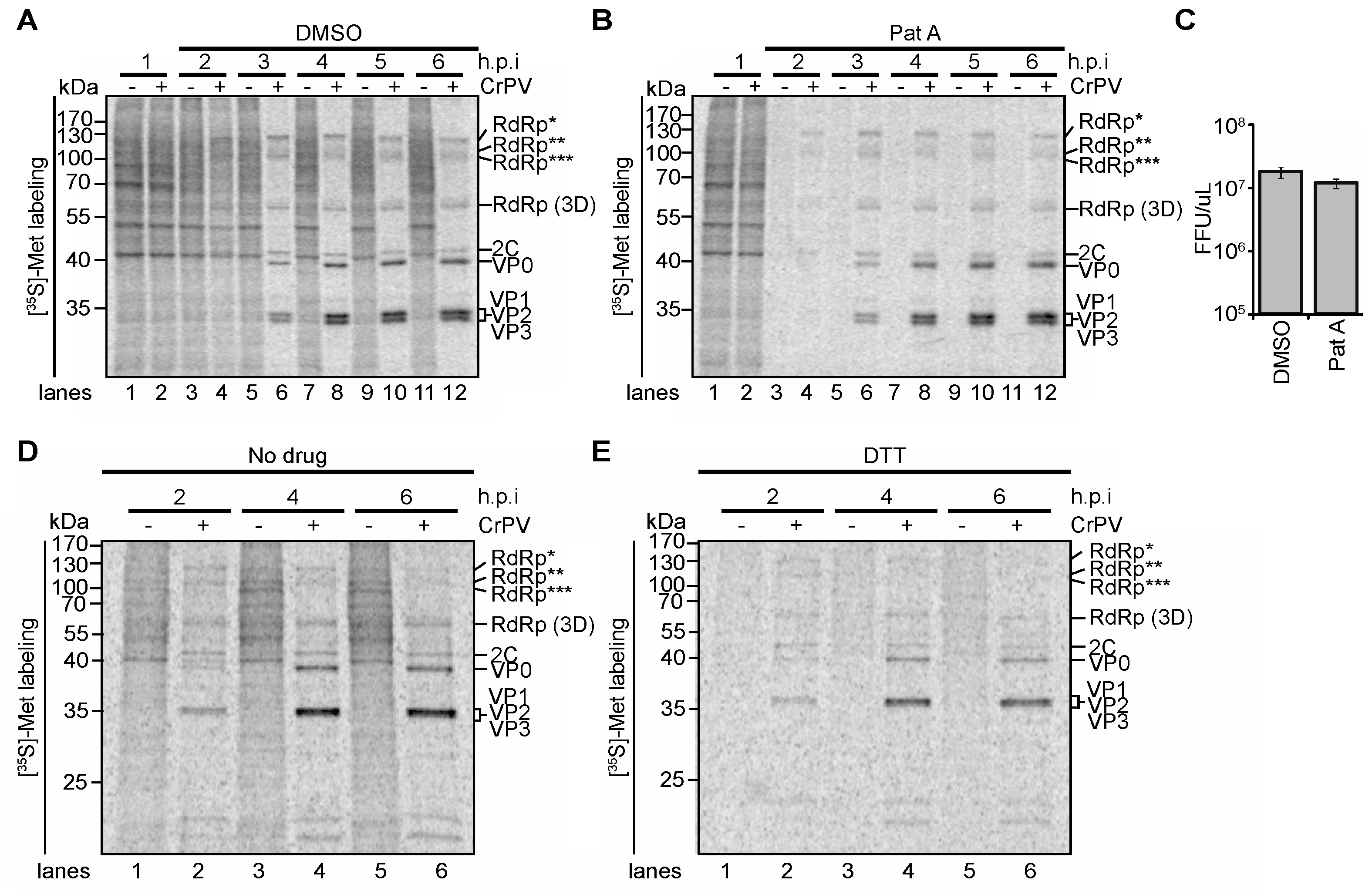
3.4. Role of Transcriptional Shutoff in Viral Protein Synthesis under CrPV Infection

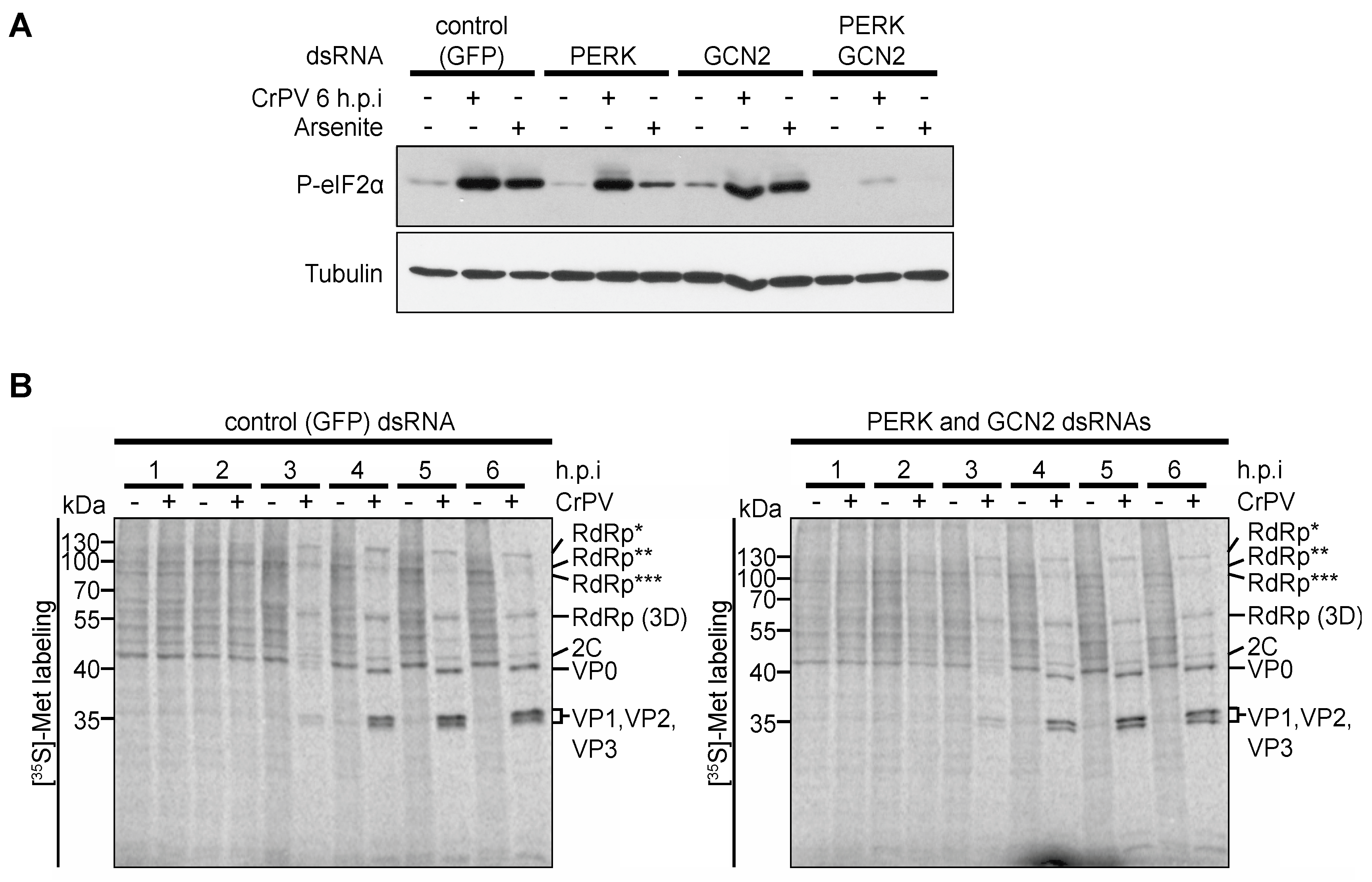
3.5. Role of eIF2α Phosphorylation in Viral Protein Synthesis under CrPV Infection

3.6. Premature Translational Shutoff and CrPV IRES Translation

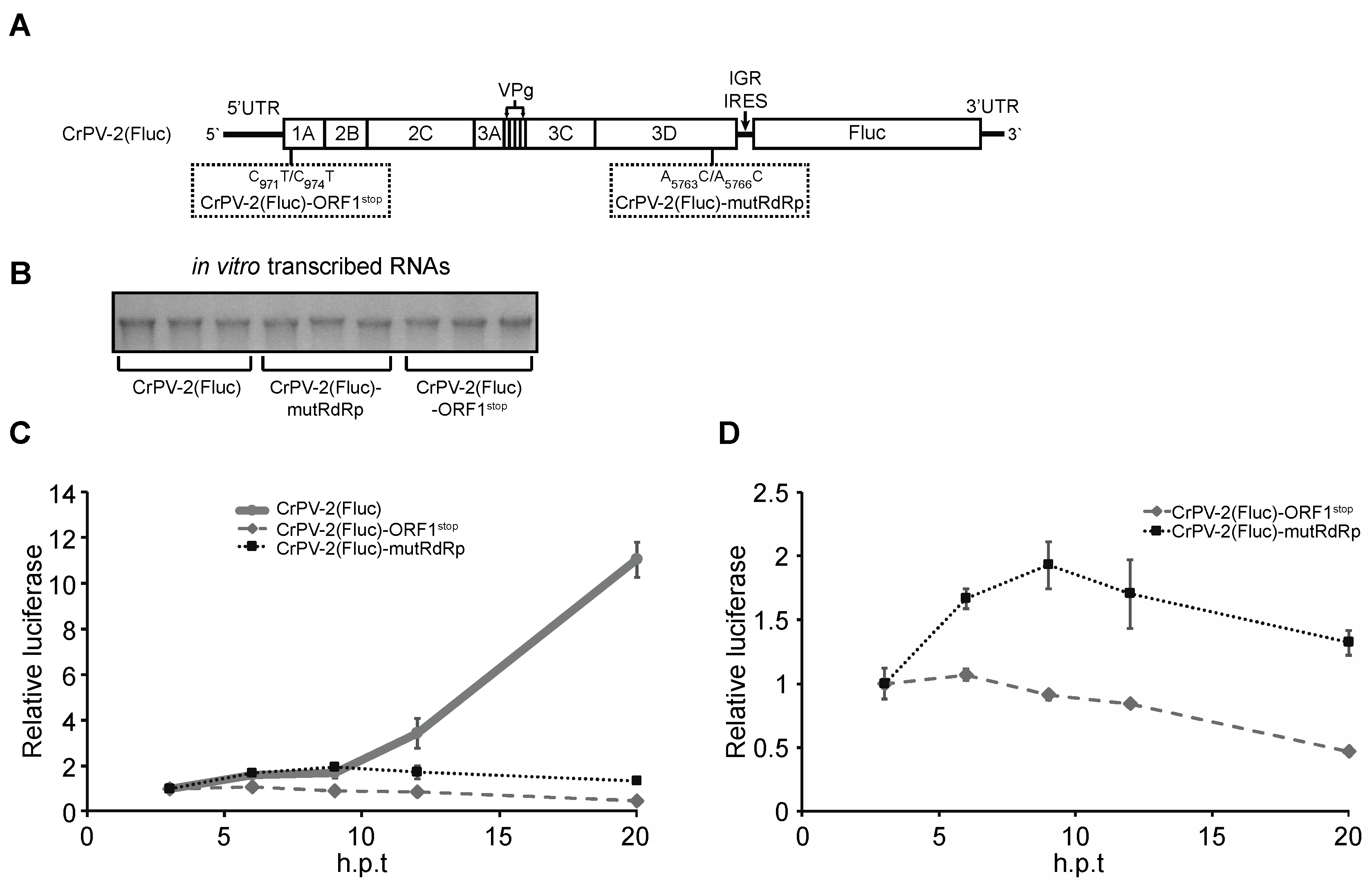
3.7. Non-Structural Viral Proteins Promote Expression of Structural Viral Proteins

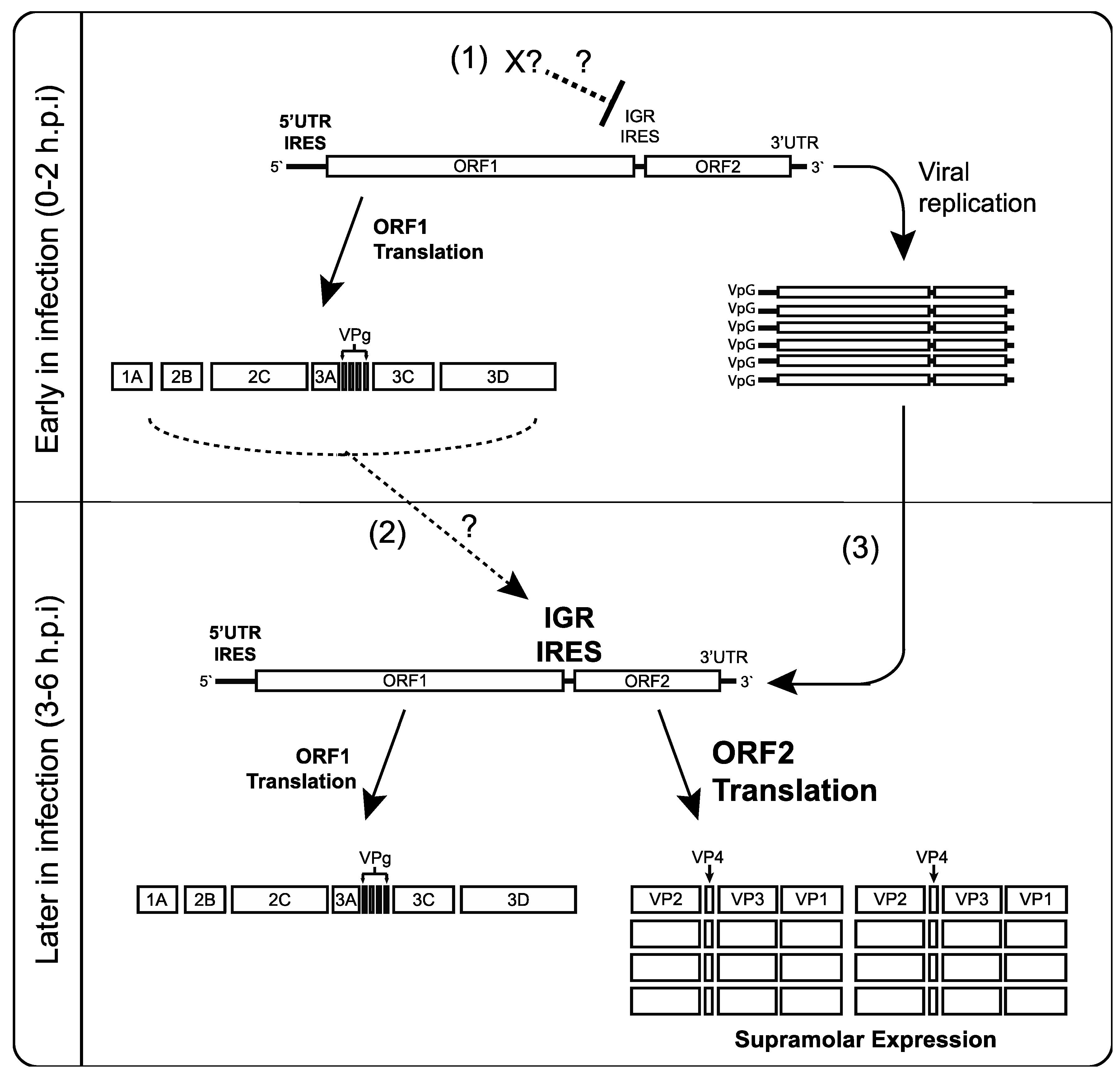
4. Discussion

5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walsh, D.; Mohr, I. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 2011, 9, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Joachims, M.; van Breugel, P.C.; Lloyd, R.E. Cleavage of poly(a)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 1999, 73, 718–727. [Google Scholar] [PubMed]
- Etchison, D.; Milburn, S.C.; Edery, I.; Sonenberg, N.; Hershey, J.W. Inhibition of hela cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J. Biol. Chem. 1982, 257, 14806–14810. [Google Scholar] [PubMed]
- Kuyumcu-Martinez, N.M.; Joachims, M.; Lloyd, R.E. Efficient cleavage of ribosome-associated poly(A)-binding protein by enterovirus 3C protease. J. Virol. 2002, 76, 2062–2074. [Google Scholar] [CrossRef] [PubMed]
- Daijogo, S.; Semler, B.L. Mechanistic intersections between picornavirus translation and RNA replication. Adv. Virus Res. 2011, 80, 1–24. [Google Scholar] [PubMed]
- Bonning, B.C.; Miller, W.A. Dicistroviruses. Annu. Rev. Entomol. 2010, 55, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Garrey, J.L.; Lee, Y.Y.; Au, H.H.; Bushell, M.; Jan, E. Host and viral translational mechanisms during cricket paralysis virus infection. J. Virol. 2010, 84, 1124–1138. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.F.; Kearns, A.; Pullin, J.S. Characterization of cricket paralysis virus-induced polypeptides in Drosophila cells. J. Virol. 1980, 33, 1–9. [Google Scholar] [PubMed]
- Wilson, J.E.; Powell, M.J.; Hoover, S.E.; Sarnow, P. Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol. Cell. Biol. 2000, 20, 4990–4999. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.R.; Fraser, T.S.; Fraser, M.J., Jr. Examining the relative activity of several dicistrovirus intergenic internal ribosome entry site elements in uninfected insect and mammalian cell lines. J. Gen. Virol. 2008, 89, 3150–3155. [Google Scholar] [CrossRef] [PubMed]
- Hertz, M.I.; Thompson, S.R. In vivo functional analysis of the dicistroviridae intergenic region internal ribosome entry sites. Nucleic Acids Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Woolaway, K.E.; Lazaridis, K.; Belsham, G.J.; Carter, M.J.; Roberts, L.O. The 5′ untranslated region of Rhopalosiphum padi virus contains an internal ribosome entry site which functions efficiently in mammalian, plant, and insect translation systems. J. Virol. 2001, 75, 10244–10249. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Nakashima, N. Characterization of the 5′ internal ribosome entry site of plautia stali intestine virus. J. Gen. Virol. 2006, 87, 3679–3686. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.E.; Pestova, T.V.; Hellen, C.U.; Sarnow, P. Initiation of protein synthesis from the a site of the ribosome. Cell 2000, 102, 511–520. [Google Scholar] [CrossRef]
- Costantino, D.A.; Pfingsten, J.S.; Rambo, R.P.; Kieft, J.S. tRNA-mRNA mimicry drives translation initiation from a viral IRES. Nat. Struct. Mol. Biol. 2008, 15, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Spahn, C.M.; Jan, E.; Mulder, A.; Grassucci, R.A.; Sarnow, P.; Frank, J. Cryo-EM visualization of a viral internal ribosome entry site bound to human ribosomes: The IRES functions as an RNA-based translation factor. Cell 2004, 118, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Pestova, T.V.; Hellen, C.U. Translation elongation after assembly of ribosomes on the cricket paralysis virus internal ribosomal entry site without initiation factors or initiator tRNA. Genes Dev. 2003, 17, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Jan, E.; Kinzy, T.G.; Sarnow, P. Divergent tRNA-like element supports initiation, elongation, and termination of protein biosynthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 15410–15415. [Google Scholar] [CrossRef] [PubMed]
- Jan, E.; Sarnow, P. Factorless ribosome assembly on the internal ribosome entry site of cricket paralysis virus. J. Mol. Biol. 2002, 324, 889–902. [Google Scholar] [CrossRef]
- Pfingsten, J.S.; Costantino, D.A.; Kieft, J.S. Structural basis for ribosome recruitment and manipulation by a viral IRES RNA. Science 2006, 314, 1450–1454. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Connell, S.R.; Lescoute, A.; Giesebrecht, J.; Dabrowski, M.; Schroeer, B.; Mielke, T.; Penczek, P.A.; Westhof, E.; Spahn, C.M. Structure of the ribosome-bound cricket paralysis virus IRES RNA. Nat. Struct. Mol. Biol. 2006, 13, 1092–1096. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.S.; Bai, X.C.; Murshudov, G.; Scheres, S.H.; Ramakrishnan, V. Initiation of translation by cricket paralysis virus IRES requires its translocation in the ribosome. Cell 2014, 157, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Muhs, M.; Hilal, T.; Mielke, T.; Skabkin, M.A.; Sanbonmatsu, K.Y.; Pestova, T.V.; Spahn, C.M. Cryo-EM of ribosomal 80S complexes with termination factors reveals the translocated cricket paralysis virus IRES. Mol. Cell 2015, 57, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Terenin, I.M.; Dmitriev, S.E.; Andreev, D.E.; Royall, E.; Belsham, G.J.; Roberts, L.O.; Shatsky, I.N. A cross-kingdom internal ribosome entry site reveals a simplified mode of internal ribosome entry. Mol. Cell. Biol. 2005, 25, 7879–7888. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.S.; Jan, E. Switch from cap- to factorless IRES-dependent 0 and +1 frame translation during cellular stress and dicistrovirus infection. PLoS ONE 2014, 9, e103601. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.A.; Koev, G. Synthesis of subgenomic RNAs by positive-strand RNA viruses. Virology 2000, 273, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kerr, C.H.; Wang, Q.S.; Keatings, K.; Khong, A.; Allan, D.; Yip, C.K.; Foster, L.J.; Jan, E. The 5′ untranslated region of a novel infectious molecular clone of the dicistrovirus cricket paralysis virus modulates infection. J. Virol. 2015, 89, 5919–5934. [Google Scholar] [CrossRef] [PubMed]
- Rasband, W.S. Image J, version 1.8.0; National Institutes of Health: Bethesda, MD, USA, 1997–2015.
- Cherry, S.; Doukas, T.; Armknecht, S.; Whelan, S.; Wang, H.; Sarnow, P.; Perrimon, N. Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes Dev. 2005, 19, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Snapdragon. DRSC website, Harvard Medical School: Boston, MA, USA.
- Brasey, A.; Lopez-Lastra, M.; Ohlmann, T.; Beerens, N.; Berkhout, B.; Darlix, J.L.; Sonenberg, N. The leader of human immunodeficiency virus type 1 genomic RNA harbors an internal ribosome entry segment that is active during the G2/M phase of the cell cycle. J. Virol. 2003, 77, 3939–3949. [Google Scholar] [CrossRef] [PubMed]
- Roy, G.; Miron, M.; Khaleghpour, K.; Lasko, P.; Sonenberg, N. The Drosophila poly(A) binding protein-interacting protein, dPaip2, is a novel effector of cell growth. Mol. Cell. Biol. 2004, 24, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Lareau, L.F.; Weissman, J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 2011, 147, 789–802. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Available online: www.ncbi.nlm.nih.gov/nuccore/NC_003924.1 (accessed on 1 Janurary 2012).
- Merkling, S.H.; Overheul, G.J.; van Mierlo, J.T.; Arends, D.; Gilissen, C.; van Rij, R.P. The heat shock response restricts virus infection in Drosophila. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, C.; Mueller, S.; Goto, A.; Barbier, V.; Paro, S.; Bonnay, F.; Dostert, C.; Troxler, L.; Hetru, C.; Meignin, C.; et al. Broad RNA interference-mediated antiviral immunity and virus-specific inducible responses in Drosophila. J. Immunol. 2013, 190, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Jouanguy, E.; Irving, P.; Troxler, L.; Galiana-Arnoux, D.; Hetru, C.; Hoffmann, J.A.; Imler, J.L. The JaK-STAT signaling pathway is required but not sufficient for the antiviral response of Drosophila. Nat. Immunol. 2005, 6, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Chtarbanova, S.; Lamiable, O.; Lee, K.Z.; Galiana, D.; Troxler, L.; Meignin, C.; Hetru, C.; Hoffmann, J.A.; Daeffler, L.; Imler, J.L. Drosophila C virus systemic infection leads to intestinal obstruction. J. Virol. 2014, 88, 14057–14069. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.R. Demonstration of intact 26S ribosomal RNA molecules in Drosophila cells. J. Mol. Biol. 1975, 98, 277–280. [Google Scholar] [CrossRef]
- Roberts, L.O.; Groppelli, E. An atypical IRES within the 5′ UTR of a dicistrovirus genome. Virus Res. 2009, 139, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.; Yaman, I.; Sarnow, P.; Snider, M.D.; Hatzoglou, M. Regulation of internal ribosomal entry site-mediated translation by phosphorylation of the translation initiation factor eIF2α. J. Biol. Chem. 2002, 277, 19198–19205. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.R.; Gulyas, K.D.; Sarnow, P. Internal initiation in saccharomyces cerevisiae mediated by an initiator tRNA/eIF2-independent internal ribosome entry site element. Proc. Natl. Acad. Sci. USA 2001, 98, 12972–12977. [Google Scholar] [CrossRef] [PubMed]
- Farny, N.G.; Kedersha, N.L.; Silver, P.A. Metazoan stress granule assembly is mediated by p-eIF2α-dependent and -independent mechanisms. RNA 2009, 15, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, M.E.; Matthews, J.; Wojnar, J.M.; Lindqvist, L.; Novac, O.; Jankowsky, E.; Sonenberg, N.; Northcote, P.; Teesdale-Spittle, P.; Pelletier, J. Stimulation of mammalian translation initiation factor eIF4α activity by a small molecule inhibitor of eukaryotic translation. Proc. Natl. Acad. Sci. USA 2005, 102, 10460–10465. [Google Scholar] [CrossRef] [PubMed]
- Joyce, C.M.; Steitz, T.A. Polymerase structures and function: Variations on a theme? J. Bacteriol. 1995, 177, 6321–6329. [Google Scholar] [PubMed]
- Back, S.H.; Kim, Y.K.; Kim, W.J.; Cho, S.; Oh, H.R.; Kim, J.E.; Jang, S.K. Translation of polioviral mRNA is inhibited by cleavage of polypyrimidine tract-binding proteins executed by polioviral 3Cpro. J. Virol. 2002, 76, 2529–2542. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.; Daijogo, S.; Walter, B.L.; Nguyen, J.H.; Semler, B.L. Cellular protein modification by poliovirus: The two faces of poly(rc)-binding protein. J. Virol. 2007, 81, 8919–8932. [Google Scholar] [CrossRef] [PubMed]
- Bonderoff, J.M.; Larey, J.L.; Lloyd, R.E. Cleavage of poly(A)-binding protein by poliovirus 3C proteinase inhibits viral internal ribosome entry site-mediated translation. J. Virol. 2008, 82, 9389–9399. [Google Scholar] [CrossRef] [PubMed]
- Hertz, M.I.; Landry, D.M.; Willis, A.E.; Luo, G.; Thompson, S.R. Ribosomal protein S25 dependency reveals a common mechanism for diverse internal ribosome entry sites and ribosome shunting. Mol. Cell. Biol. 2013, 33, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Jack, K.; Bellodi, C.; Landry, D.M.; Niederer, R.O.; Meskauskas, A.; Musalgaonkar, S.; Kopmar, N.; Krasnykh, O.; Dean, A.M.; Thompson, S.R.; et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol. Cell 2011, 44, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Yoon, A.; Peng, G.; Brandenburger, Y.; Zollo, O.; Xu, W.; Rego, E.; Ruggero, D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 2006, 312, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Majzoub, K.; Hafirassou, M.L.; Meignin, C.; Goto, A.; Marzi, S.; Fedorova, A.; Verdier, Y.; Vinh, J.; Hoffmann, J.A.; Martin, F.; et al. RACK1 controls IRES-mediated translation of viruses. Cell 2014, 159, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, K.A.; Bushell, M.; Mitchell, S.A.; Willis, A.E. Internal ribosome entry segment-mediated translation during apoptosis: The role of IRES-trans-acting factors. Cell Death Differ. 2005, 12, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Li, M.L.; Shih, S.R. Far upstream element binding protein 2 interacts with enterovirus 71 internal ribosomal entry site and negatively regulates viral translation. Nucleic Acids Res. 2009, 37, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Whitlow, Z.W.; Connor, J.H.; Lyles, D.S. New mRNAs are preferentially translated during vesicular stomatitis virus infection. J. Virol. 2008, 82, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; He, W.T.; Tian, S.; Meng, D.; Li, Y.; Chen, W.; Li, L.; Tian, L.; Zhong, C.Q.; Han, F.; et al. pelo Is required for high efficiency viral replication. PLoS Pathog. 2014, 10, e1004034. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, T.; Kuroha, K.; Kudo, K.; Makino, S.; Inoue, E.; Kashima, I.; Inada, T. Dom34:hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3′ end of aberrant mRNA. Mol. Cell 2012, 46, 518–529. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khong, A.; Bonderoff, J.M.; Spriggs, R.V.; Tammpere, E.; Kerr, C.H.; Jackson, T.J.; Willis, A.E.; Jan, E. Temporal Regulation of Distinct Internal Ribosome Entry Sites of the Dicistroviridae Cricket Paralysis Virus. Viruses 2016, 8, 25. https://0-doi-org.brum.beds.ac.uk/10.3390/v8010025
Khong A, Bonderoff JM, Spriggs RV, Tammpere E, Kerr CH, Jackson TJ, Willis AE, Jan E. Temporal Regulation of Distinct Internal Ribosome Entry Sites of the Dicistroviridae Cricket Paralysis Virus. Viruses. 2016; 8(1):25. https://0-doi-org.brum.beds.ac.uk/10.3390/v8010025
Chicago/Turabian StyleKhong, Anthony, Jennifer M. Bonderoff, Ruth V. Spriggs, Erik Tammpere, Craig H. Kerr, Thomas J. Jackson, Anne E. Willis, and Eric Jan. 2016. "Temporal Regulation of Distinct Internal Ribosome Entry Sites of the Dicistroviridae Cricket Paralysis Virus" Viruses 8, no. 1: 25. https://0-doi-org.brum.beds.ac.uk/10.3390/v8010025