Immunoinformatics and Structural Analysis for Identification of Immunodominant Epitopes in SARS-CoV-2 as Potential Vaccine Targets

 ,
, 
Abstract
:1. Introduction
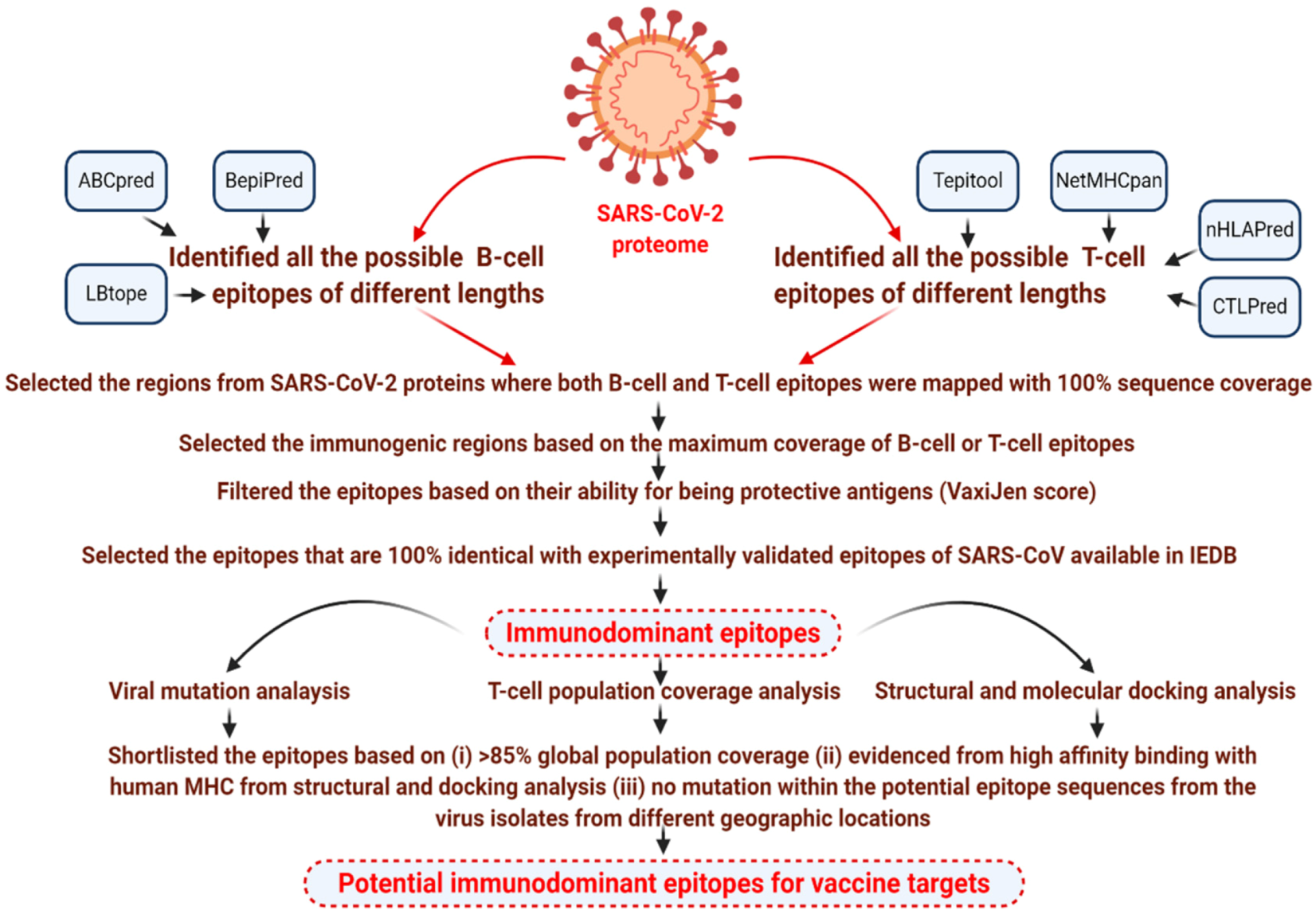
2. Materials and Methods
2.1. Immunoinformatics Analysis
2.1.1. Data Retrieval
2.1.2. Predicting Potential Linear B-cell Epitopes in SARS-CoV-2
2.1.3. Prediction of Potential T-cell Epitopes in SARS-CoV-2
2.1.4. Prediction of Protective Antigens
2.1.5. Analysis of Epitope Conservation and Population Coverage of T-cell Epitopes
2.1.6. Prediction of Allergenicity, Toxicity and Possibilities of Autoimmune Reactions
2.2. Structural Analysis
2.2.1. Data Collection for Structural Analysis
2.2.2. Modeling of Epitope MHC-bound Conformations
2.2.3. Molecular Docking
3. Results
3.1. Identification of Immunodominant Epitopes from the Proteins of SARS-CoV-2
3.2. Analysis of Viral Mutations within the Potential Epitope Regions
3.3. Population Coverage of Immunodominant Epitopes
3.4. Analysis of Allergenicity, Toxicity and Autoimmune Reactivity
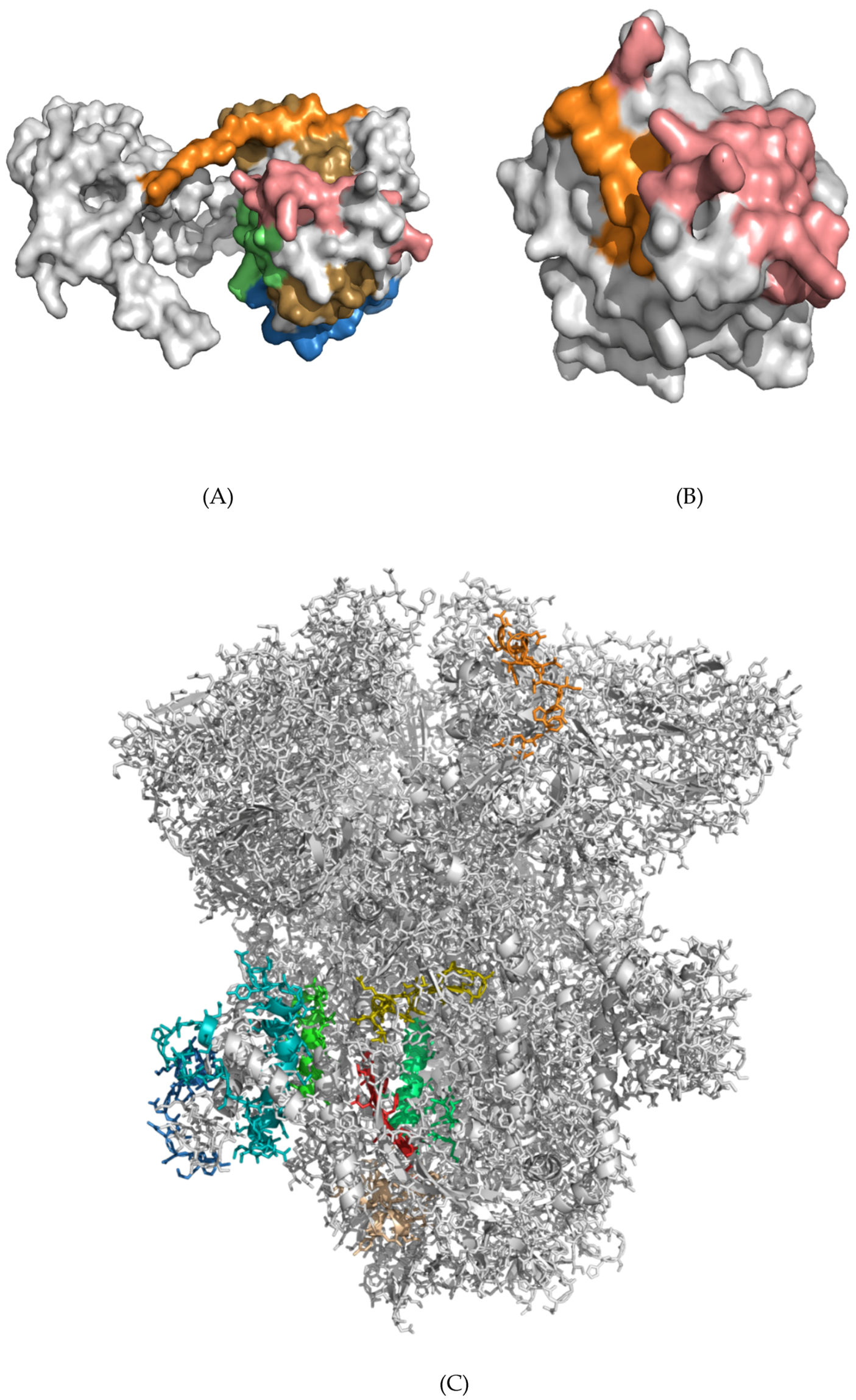
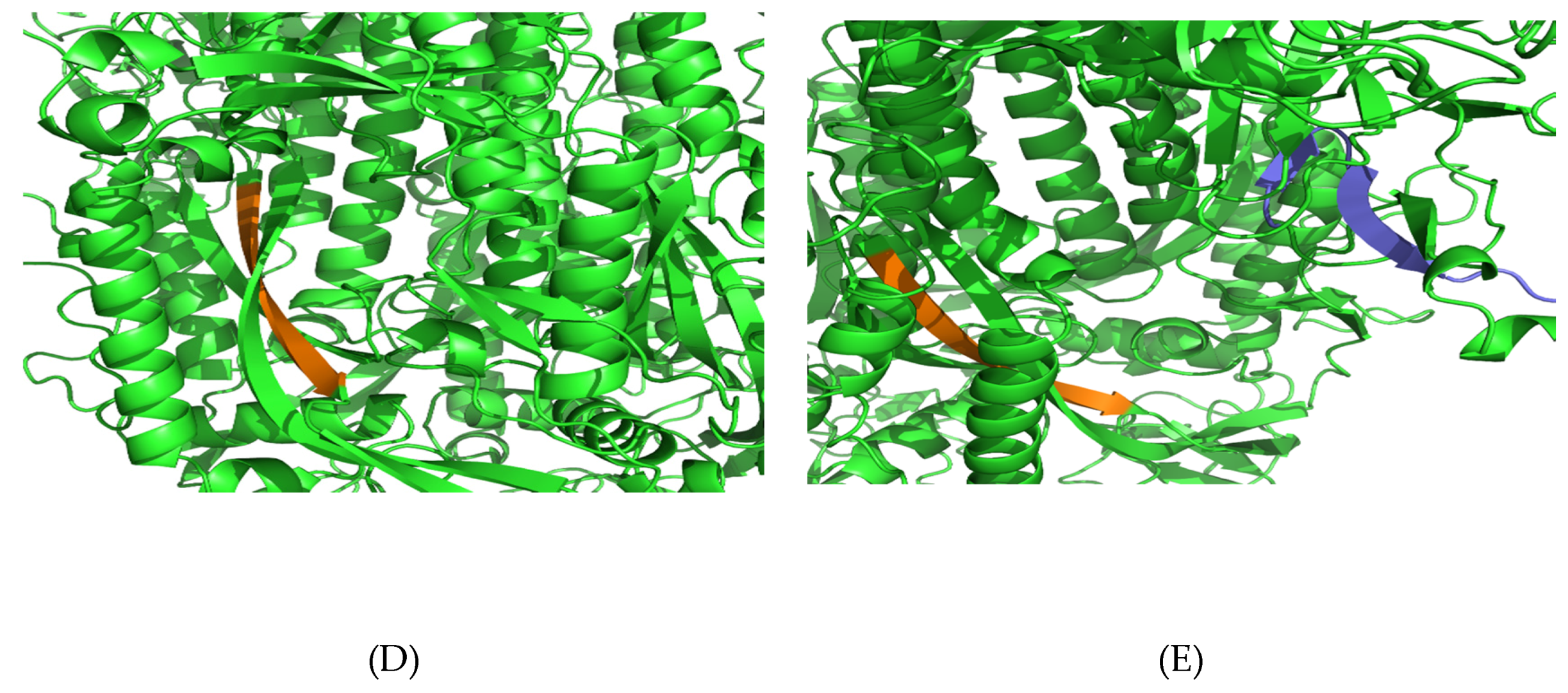
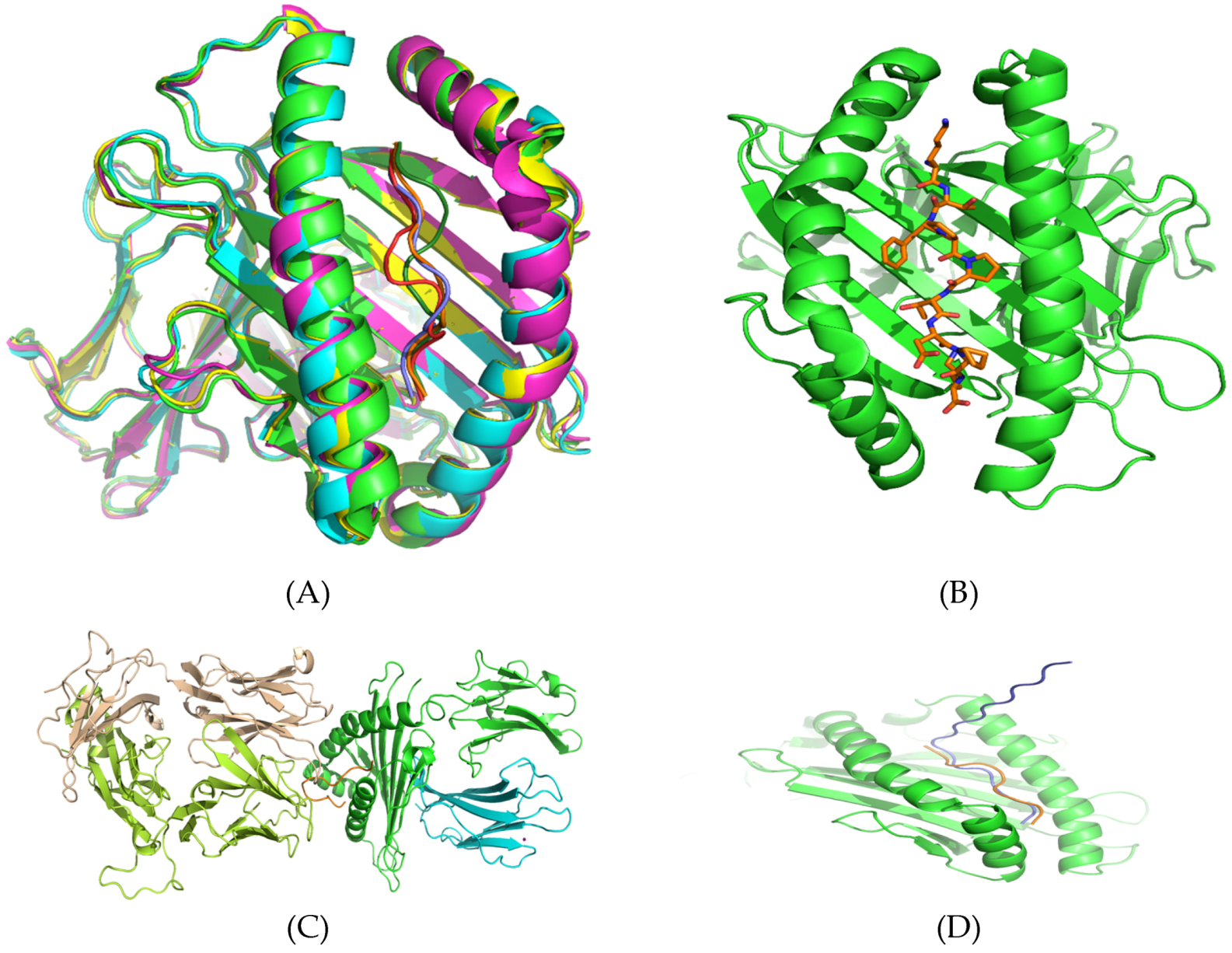
3.5. Structural Analysis and Modeling of Epitope Presentation by MHC Class I and II Systems
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, D.K.W.; Pan, Y.; Cheng, S.M.S.; Hui, K.P.Y.; Krishnan, P.; Liu, Y.; Ng, D.Y.M.; Wan, C.K.C.; Yang, P.; Wang, Q.; et al. Molecular Diagnosis of a Novel Coronavirus (2019-nCoV) Causing an Outbreak of Pneumonia. Clin. Chem. 2020, 66, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Yuan, S.; Kok, K.H.; To, K.K.W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.Y.; Poon, R.W.S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.W.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.W.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020. [Google Scholar] [CrossRef]
- De Gregorio, E.; Rappuoli, R. Vaccines for the future: Learning from human immunology. Microb. Biotechnol. 2012, 5, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Backert, L.; Kohlbacher, O. Immunoinformatics and epitope prediction in the age of genomic medicine. Genome Med. 2015, 7, 119. [Google Scholar] [CrossRef] [Green Version]
- Tahir Ul Qamar, M.; Saleem, S.; Ashfaq, U.A.; Bari, A.; Anwar, F.; Alqahtani, S. Epitope-based peptide vaccine design and target site depiction against Middle East Respiratory Syndrome Coronavirus: An immune-informatics study. J. Transl. Med. 2019, 17, 362. [Google Scholar] [CrossRef]
- Khan, A.; Junaid, M.; Kaushik, A.C.; Ali, A.; Ali, S.S.; Mehmood, A.; Wei, D.Q. Computational identification, characterization and validation of potential antigenic peptide vaccines from hrHPVs E6 proteins using immunoinformatics and computational systems biology approaches. PLoS ONE 2018, 13, e0196484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahir Ul Qamar, M.; Bari, A.; Adeel, M.M.; Maryam, A.; Ashfaq, U.A.; Du, X.; Muneer, I.; Ahmad, H.I.; Wang, J. Peptide vaccine against chikungunya virus: Immuno-informatics combined with molecular docking approach. J. Transl. Med. 2018, 16, 298. [Google Scholar] [CrossRef]
- Ahmad, B.; Ashfaq, U.A.; Rahman, M.U.; Masoud, M.S.; Yousaf, M.Z. Conserved B and T cell epitopes prediction of ebola virus glycoprotein for vaccine development: An immuno-informatics approach. Microb. Pathog. 2019, 132, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Mourosi, J.; Khan, M.; Hosen, M. Prediction of Epitope-Based Peptide Vaccine Against the Chikungunya Virus by Immuno-informatics Approach. Curr. Pharm. Biotechnol. 2019, 21, 325–340. [Google Scholar] [CrossRef]
- Shahid, F.; Ashfaq, U.A.; Javaid, A.; Khalid, H. Immunoinformatics guided rational design of a next generation multi epitope based peptide (MEBP) vaccine by exploring Zika virus proteome. Infect. Genet. Evol. 2020, 80, 104199. [Google Scholar] [CrossRef]
- Ikram, A.; Zaheer, T.; Awan, F.M.; Obaid, A.; Naz, A.; Hanif, R.; Paracha, R.Z.; Ali, A.; Naveed, A.K.; Janjua, H.A. Exploring NS3/4A, NS5A and NS5B proteins to design conserved subunit multi-epitope vaccine against HCV utilizing immunoinformatics approaches. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, B. The contribution of vaccination to global health: Past, present and future. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130433. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-Z.; Holmes, E.C. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 2020. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [Green Version]
- Van Regenmortel, M.H.V. What is a b-cell epitope? In Epitope Mapping Protocols; Humana Press: Totowa, NJ, USA, 2009; pp. 3–20. [Google Scholar]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Genet. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Singh, H.; Ansari, H.R.; Raghava, G.P.S. Improved Method for Linear B-Cell Epitope Prediction Using Antigen’s Primary Sequence. PLoS ONE 2013, 8, e62216. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988, 334, 395–402. [Google Scholar] [CrossRef]
- Patronov, A.; Doytchinova, I. T-cell epitope vaccine design by immunoinformatics. Open Biol. 2013, 3, 120139. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Sidney, J.; Sette, A.; Peters, B. TepiTool: A pipeline for computational prediction of T cell epitope candidates. Curr. Protoc. Immunol. 2016, 114, 18–19. [Google Scholar] [CrossRef]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Weiskopf, D.; Angelo, M.A.; Sidney, J.; Peters, B.; Sette, A. HLA Class I Alleles Are Associated with Peptide-Binding Repertoires of Different Size, Affinity, and Immunogenicity. J. Immunol. 2013, 191, 5831–5839. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Sidney, J.; Dow, C.; Mothé, B.; Sette, A.; Peters, B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 2008, 4, e1000048. [Google Scholar] [CrossRef] [Green Version]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Lata, S.; Bhasin, M.; Raghava, G.P.S. Application of machine learning techniques in predicting MHC binders. In Immunoinformatics; Humana Press: Totowa, NJ, USA, 2007; pp. 201–215. [Google Scholar]
- Bhasin, M.; Raghava, G.P.S. Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine 2004, 22, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v.2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. AlgPred: Prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F. BLAST Algorithm. In Encyclopedia of Life Sciences; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Mahajan, S.; Yan, Z.; Jespersen, M.C.; Jensen, K.K.; Marcatili, P.; Nielsen, M.; Sette, A.; Peters, B. Benchmark datasets of immune receptor-epitope structural complexes. BMC Bioinform. 2019, 20, 1–7. [Google Scholar] [CrossRef] [Green Version]
- London, N.; Movshovitz-Attias, D.; Schueler-Furman, O. The Structural Basis of Peptide-Protein Binding Strategies. Structure 2010, 18, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Canutescu, A.A.; Dunbrack, R.L. SCWRL and MolIDE: Computer programs for side-chain conformation prediction and homology modeling. Nat. Protoc. 2008, 3, 1832. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Morley, C.; Hutchison, G.R. Pybel: A Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J. 2008, 2, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grifoni, A.; Sidney, J.; Zhang, Y.; Scheuermann, R.H.; Peters, B.; Sette, A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe 2020. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Karrer, U.; Lucas, A.; Klenerman, P. Viral escape mechanisms—Escapology taught by viruses. Int. J. Exp. Pathol. 2001, 82, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, J.M.; Jamieson, S.E.; Burgner, D. HLA and infectious diseases. Clin. Microbiol. Rev. 2009, 22, 370–385. [Google Scholar] [CrossRef] [Green Version]
- Vanderlugt, C.J.; Miller, S.D. Epitope spreading. Curr. Opin. Immunol. 1996, 8, 831. [Google Scholar] [CrossRef]
- Powell, A.M.; Black, M.M. Epitope spreading: Protection from pathogens, but propagation of autoimmunity? Clin. Exp. Dermatol. 2001, 26, 427–433. [Google Scholar] [CrossRef]
- Ehreth, J. The global value of vaccination. Vaccine 2003, 21, 596–600. [Google Scholar] [CrossRef]
- Scarselli, M.; Giuliani, M.M.; Adu-Bobie, J.; Pizza, M.; Rappuoli, R. The impact of genomics on vaccine design. Trends Biotechnol. 2005, 23, 84–91. [Google Scholar] [CrossRef]
- Soria-Guerra, R.E.; Nieto-Gomez, R.; Govea-Alonso, D.O.; Rosales-Mendoza, S. An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. J. Biomed. Inform. 2015, 53, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asai, A.; Konno, M.; Ozaki, M.; Otsuka, C.; Vecchione, A.; Arai, T.; Kitagawa, T.; Ofusa, K.; Yabumoto, M.; Hirotsu, T.; et al. COVID-19 drug discovery using intensive approaches. Int. J. Mol. Sci. 2020, 21, 2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanh Le, T.; Andreadakis, Z.; Kumar, A.; Gómez Román, R.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef]
- Prompetchara, E.; Ketloy, C.; Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pacific J. Allergy Immunol. 2020, 38, 1–9. [Google Scholar]
- Xu, J.; Zhao, S.; Teng, T.; Abdalla, A.E.; Zhu, W.; Xie, L.; Wang, Y.; Guo, X. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 2020, 12, 244. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary identification of potential vaccine targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [Green Version]
- Yamey, G.; Schäferhoff, M.; Hatchett, R.; Pate, M.; Zhao, F.; McDade, K.K. Ensuring global access to COVID-19 vaccines. Lancet 2020, 395, 1405–1406. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential Immunogenic Regions from Proteins of SARS-CoV-2, Isolated in Wuhan-Hu-1 (NC_045512.2) | The Number of Epitopes Mapped | Potential Immunodominant Epitopes |
|---|---|---|
| Membrane glycoprotein (61–70) | 1 | TLACFVLAAV |
| Membrane glycoprotein (157–187) | 3 | GRCDIKDLPKEITVATSR PKEITVATSRTLSYYKL TSRTLSYYKLGASQRV |
| Nucleocapsid phosphoprotein (176–191) | 1 | SRGGSQASSRSSSRSR |
| Nucleocapsid phosphoprotein (240–264) | 2 | QQQGQTVTKKSAAEASKK KKSAAEASKKPRQKRTA |
| Nucleocapsid phosphoprotein (268–286) | 1 | YNVTQAFGRRGPEQTQGNF |
| Nucleocapsid phosphoprotein (292–330) | 3 | IRQGTDYKHWPQIAQFA QFAPSASAFFGMSRIGM FFGMSRIGMEVTPSGTW |
| Nucleocapsid phosphoprotein (360–375) | 1 | YKTFPPTEPKKDKKKK |
| Spike glycoprotein (327–343) | 1 | VRFPNITNLCPFGEVFN |
| Spike glycoprotein (663–680) | 1 | DIPIGAGICASYHTVSLL |
| Spike glycoprotein (817–833) | 1 | FIEDLLFNKVTLADAGF |
| Spike glycoprotein (891–918) | 3 | GAALQIPFAMQMAYRFN PFAMQMAYRFNGIGVTQ MAYRFNGIGVTQNVLYE |
| Spike glycoprotein (1019–1041) | 2 | RASANLAATKMSECVLG AATKMSECVLGQSKRVD |
| Spike glycoprotein (1060–1068) | 1 | VVFLHVTYV |
| Spike glycoprotein (1157–1209) | 3 | KNHTSPDVDLGDISGIN DLGDISGINASVVNIQK EIDRLNEVAKNLNESLIDLQELGKYEQY |
| Spike glycoprotein (1254–1273) | 1 | CKFDEDDSEPVLKGVKLHYT |
| Epitopes | Epitope Location | World Population Coverage (%) | Predicted HLA Locus |
|---|---|---|---|
| PKEITVATSRTLSYYKL | Membrane glycoprotein: 165–181 | 95.82% | HLA-A, HLA-B, HLA-DRB1, HLA-DRB3, HLA-DRB4, HLA-DRB5, HLA-DQA1, HLA-DQB1 |
| QFAPSASAFFGMSRIGM | Nucleocapsid phosphoprotein: 306–322 | 92.81% | HLA-A, HLA-B, HLA-DRB1, HLA-DRB5, HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1 |
| FFGMSRIGMEVTPSGTW | Nucleocapsid phosphoprotein: 314–330 | 87.42% | HLA-A, HLA-B, HLA-DRB1, HLA-DRB4, HLA-DRB5, HLA-DQA1, HLA-DQB1 |
| FIEDLLFNKVTLADAGF | Spike glycoprotein: 817–833 | 94.26% | HLA-A, HLA-B, HLA-DRB1, HLA-DRB3, HLA-DRB4, HLA-DRB5, HLA-DQA1, HLA-DQB1, HLA-DPA1, HLA-DPB1 |
| GAALQIPFAMQMAYRFN | Spike glycoprotein: 891–907 | 97.46% | HLA-A, HLA-B, HLA-DRB1, HLA-DRB4, HLA-DRB5, HLA-DQA1, HLA-DQB1, HLA-DPA1, HLA-DPB1 |
| PFAMQMAYRFNGIGVTQ | Spike glycoprotein: 897–913 | 92.52% | HLA-A, HLA-B, HLA-DRB1, HLA-DRB3, HLA-DRB4, HLA-DRB5, HLA-DQA1, HLA-DQB1, HLA-DPA1 |
| EIDRLNEVAKNLNESLIDLQELGKYEQY | Spike glycoprotein:1182–1209 | 88.57% | HLA-A, HLA-B, HLA-DRB1, HLA-DRB3, HLA-DQA1, HLA-DQB1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, S.; Tworowski, D.; Detroja, R.; Mukherjee, S.B.; Frenkel-Morgenstern, M. Immunoinformatics and Structural Analysis for Identification of Immunodominant Epitopes in SARS-CoV-2 as Potential Vaccine Targets. Vaccines 2020, 8, 290. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines8020290
Mukherjee S, Tworowski D, Detroja R, Mukherjee SB, Frenkel-Morgenstern M. Immunoinformatics and Structural Analysis for Identification of Immunodominant Epitopes in SARS-CoV-2 as Potential Vaccine Targets. Vaccines. 2020; 8(2):290. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines8020290
Chicago/Turabian StyleMukherjee, Sumit, Dmitry Tworowski, Rajesh Detroja, Sunanda Biswas Mukherjee, and Milana Frenkel-Morgenstern. 2020. "Immunoinformatics and Structural Analysis for Identification of Immunodominant Epitopes in SARS-CoV-2 as Potential Vaccine Targets" Vaccines 8, no. 2: 290. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines8020290