
Neurodegeneration in Multiple Sclerosis: The Role of Nrf2-Dependent Pathways


 ,
, 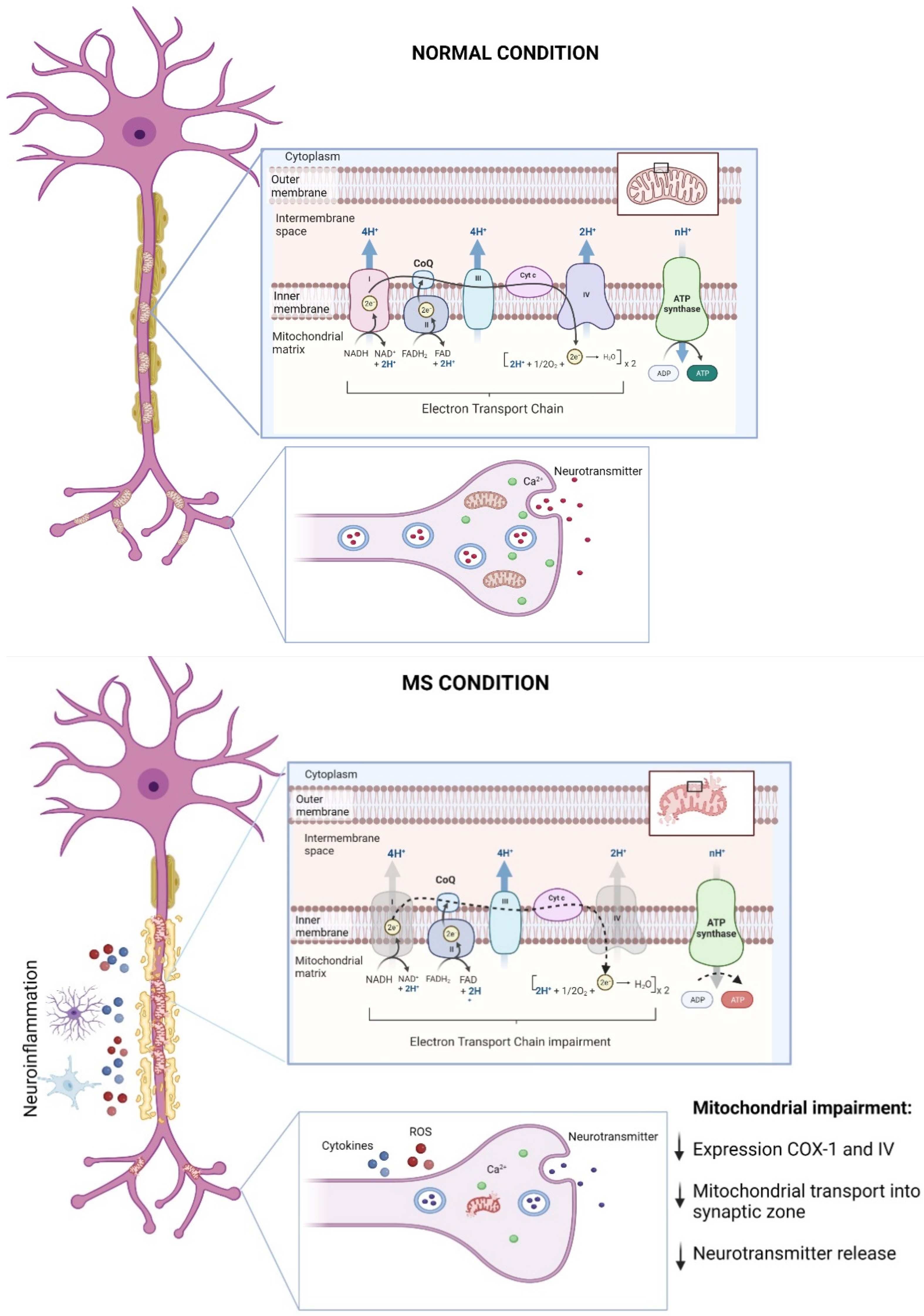{kind=link}
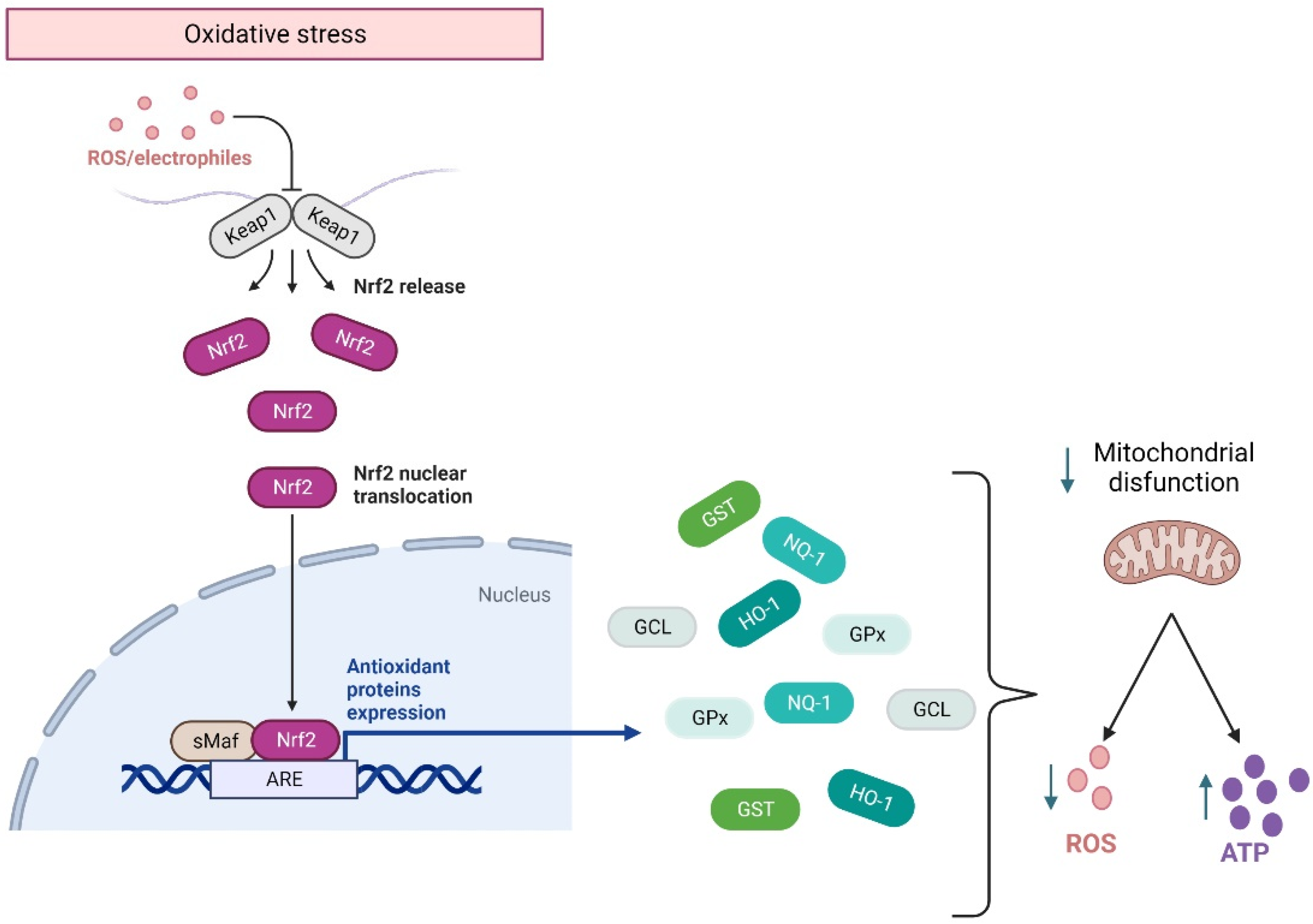{kind=link}
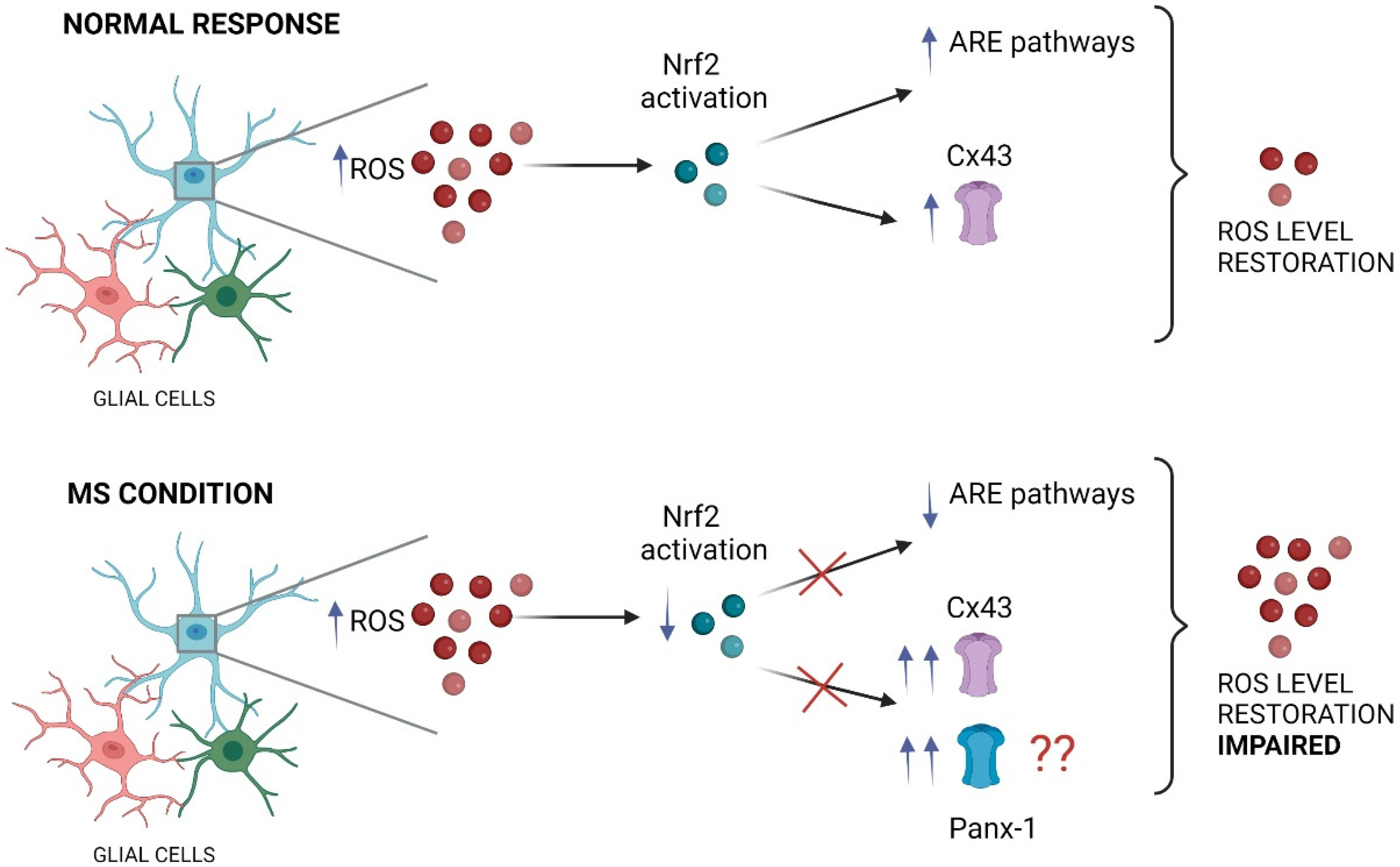{kind=link}
Abstract
:1. Introduction
2. Multiple Sclerosis
3. Glial Cell-Mediated Neuroinflammation
4. Mitochondria and Neuronal Function
5. Mitochondrial Dysfunction: A Hallmark in the Pathogenesis of MS
6. Nrf2 Signaling Network: A Key Player Combatting Oxidative Stress, Mitochondrial Dysfunction, Neuroinflammation, and Neurodegeneration in MS
7. Large-Pore Channels: A Possible Link between Glial Cell Dysfunction and Nrf2 in Multiple Sclerosis
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS Myelin—From Mechanisms to Experimental Medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The Relation between Inflammation and Neurodegeneration in Multiple Sclerosis Brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia Metabolically Support Axons and Contribute to Neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.A. Myelination and Support of Axonal Integrity by Glia. Nature 2010, 468, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C. Biology: A Degenerative Affliction. Nature 2016, 540, S2–S3. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, S.; Pontecorvo, S.; Tortorella, C.; Gasperini, C. Induction Treatment Strategy in Multiple Sclerosis: A Review of Past Experiences and Future Perspectives. Mult. Scler. Demyelinating Disord. 2018, 3, 5. [Google Scholar] [CrossRef]
- Sandi, D.; Kokas, Z.; Biernacki, T.; Bencsik, K.; Klivényi, P.; Vécsei, L. Proteomics in Multiple Sclerosis: The Perspective of the Clinician. Int. J. Mol. Sci. 2022, 23, 5162. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.M.; Ffrench-Constant, C. Remyelination in the CNS: From Biology to Therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef]
- Humphries, C. Progressive Multiple Sclerosis: The Treatment Gap. Nature 2012, 484, S10. [Google Scholar] [CrossRef] [PubMed]
- Criste, G.; Trapp, B.; Dutta, R. Axonal Loss in Multiple Sclerosis: Causes and Mechanisms. Handb. Clin. Neurol. 2014, 122, 101–113. [Google Scholar] [PubMed]
- Tiu, V.E.; Enache, I.; Panea, C.A.; Tiu, C.; Popescu, B.O. Predictive MRI Biomarkers in MS—A Critical Review. Medicina 2022, 58, 377. [Google Scholar] [CrossRef] [PubMed]
- Ziemssen, T.; Akgün, K.; Brück, W. Molecular Biomarkers in Multiple Sclerosis. J. Neuroinflammation 2019, 16, 272. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.C.; Mares, J.; Hartung, H.P. Current Therapeutic Landscape in Multiple Sclerosis: An Evolving Treatment Paradigm. Curr. Opin. Neurol. 2019, 32, 365–377. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
- Varas, R.; Ortiz, F. Neuroinflammation in Demyelinating Diseases: Oxidative Stress as a Modulator of Glial Cross-Talk. Curr. Pharm. Des. 2019, 25, 45. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Wang, J.; Yang, B.; Weng, Q.; He, Q. Targeting Microglia and Macrophages: A Potential Treatment Strategy for Multiple Sclerosis. Front. Pharmacol. 2019, 10, 286. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Miron, V.E. The Pro-Remyelination Properties of Microglia in the Central Nervous System. Nat. Rev. Neurol. 2019, 15, 447–458. [Google Scholar] [CrossRef]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple Sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Khandelwal, P.J.; Herman, A.M.; Moussa, C.E.H. Inflammation in the Early Stages of Neurodegenerative Pathology. J. Neuroimmunol. 2011, 238, 1. [Google Scholar] [CrossRef] [PubMed]
- Herx, L.M.; Rivest, S.; Yong, V.W. Central Nervous System-Initiated Inflammation and Neurotrophism in Trauma: IL-1β Is Required for the Production of Ciliary Neurotrophic Factor. J. Immunol. 2000, 165, 2232–2239. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.-C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I Interferons and Microbial Metabolites of Tryptophan Modulate Astrocyte Activity and Central Nervous System Inflammation via the Aryl Hydrocarbon Receptor. Nature 2016, 22, 586–600. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Ghilardi, N.; Xie, M.H.; De Sauvage, F.J.; Gurney, A.L. Interleukin-23 Promotes a Distinct CD4 T Cell Activation State Characterized by the Production of Interleukin-17. J. Biol. Chem. 2003, 278, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Rostami, A.; Ciric, B. Role of Th17 Cells in the Pathogenesis of CNS Inflammatory Demyelination. J. Neurol. Sci. 2013, 333, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative Stress in Multiple Sclerosis: Central and Peripheral Mode of Action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef]
- Li, S.; Xiong, G.J.; Huang, N.; Sheng, Z.H. The Cross-Talk of Energy Sensing and Mitochondrial Anchoring Sustains Synaptic Efficacy by Maintaining Presynaptic Metabolism. Nat. Metab. 2020, 2, 1077–1095. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Brand, M.D.; Gerencser, A.A. Mitochondrial Bioenergetics and Neuronal Survival Modelled in Primary Neuronal Culture and Isolated Nerve Terminals. J. Bioenerg. Biomembr. 2015, 47, 63–74. [Google Scholar] [CrossRef]
- Chang, D.T.W.; Reynolds, I.J. Mitochondrial Trafficking and Morphology in Healthy and Injured Neurons. Prog. Neurobiol. 2006, 80, 241–268. [Google Scholar] [CrossRef]
- Matsushima, Y.; Kaguni, L.S. Matrix Proteases in Mitochondrial DNA Function. Biochim. Biophys. Acta 2012, 1819, 1080–1087. [Google Scholar] [CrossRef]
- Cagalinec, M.; Safiulina, D.; Liiv, M.; Liiv, J.; Choubey, V.; Wareski, P.; Veksler, V.; Kaasik, A. Principles of the Mitochondrial Fusion and Fission Cycle in Neurons. J. Cell Sci. 2013, 126, 2187–2197. [Google Scholar] [CrossRef]
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and Roles of Mitochondrial Localisation and Dynamics in Neuronal Function. Neuronal Signal. 2020, 4, 20200008. [Google Scholar] [CrossRef] [PubMed]
- Ames, A. CNS Energy Metabolism as Related to Function. Brain Res. Rev. 2000, 34, 42–68. [Google Scholar] [CrossRef]
- Bergmann, F.; Keller, B.U. Impact of Mitochondrial Inhibition on Excitability and Cytosolic Ca2+ Levels in Brainstem Motoneurones from Mouse. J. Physiol. 2004, 555, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Ruthel, G.; Hollenbeck, P.J. Response of Mitochondrial Traffic to Axon Determination and Differential Branch Growth. J. Neurosci. 2003, 23, 8618–8624. [Google Scholar] [CrossRef]
- Verstreken, P.; Ly, C.V.; Venken, K.J.T.; Koh, T.W.; Zhou, Y.; Bellen, H.J. Synaptic Mitochondria Are Critical for Mobilization of Reserve Pool Vesicles at Drosophila Neuromuscular Junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef]
- Augustine, G.J.; Santamaria, F.; Tanaka, K. Local Calcium Signaling in Neurons. Neuron 2003, 40, 331–346. [Google Scholar] [CrossRef]
- David, G.; Barett, E.F. Mitochondrial Ca2+ Uptake Prevents Desynchronization of Quantal Release and Minimizes Depletion during Repetitive Stimulation of Mouse Motor Nerve Terminals. J. Physiol. 2003, 548, 425. [Google Scholar] [CrossRef]
- Marland, J.R.K.; Hasel, P.; Bonnycastle, K.; Cousin, M.A. Mitochondrial Calcium Uptake Modulates Synaptic Vesicle Endocytosis in Central Nerve Terminals. J. Biol. Chem. 2016, 291, 2080–2086. [Google Scholar] [CrossRef]
- Ivannikov, M.V.; Sugimori, M.; Llinás, R.R. Synaptic Vesicle Exocytosis in Hippocampal Synaptosomes Correlates Directly with Total Mitochondrial Volume. J. Mol. Neurosci. 2013, 49, 223. [Google Scholar] [CrossRef]
- Talbot, J.D.; David, G.; Barrett, E.F. Inhibition of Mitochondrial Ca2+ Uptake Affects Phasic Release from Motor Terminals Differently Depending on External [Ca2+]. J. Neurophysiol. 2003, 90, 491–502. [Google Scholar] [CrossRef]
- Lopez-Manzaneda, M.; Franco-Espin, J.; Tejero, R.; Cano, R.; Tabares, L. Calcium Is Reduced in Presynaptic Mitochondria of Motor Nerve Terminals during Neurotransmission in SMA Mice. Hum. Mol. Genet. 2021, 30, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Bourne, J.N.; Cao, G.; Chirillo, M.A.; Ostroff, L.E.; Watson, D.J.; Harris, K.M. Mitochondrial Support of Persistent Presynaptic Vesicle Mobilization with Age-Dependent Synaptic Growth after LTP. eLife 2016, 5, e15275. [Google Scholar] [CrossRef] [PubMed]
- Billups, B.; Forsythe, I.D. Presynaptic Mitochondrial Calcium Sequestration Influences Transmission at Mammalian Central Synapses. J. Neurosci. 2002, 22, 5840–5847. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.J.; Pekkurnaz, G. Powerhouse of the Mind: Mitochondrial Plasticity at the Synapse. Curr. Opin. Neurobiol. 2019, 57, 149–155. [Google Scholar] [CrossRef]
- Divakaruni, S.S.; Van Dyke, A.M.; Chandra, R.; LeGates, T.A.; Contreras, M.; Dharmasri, P.A.; Higgs, H.N.; Lobo, M.K.; Thompson, S.M.; Blanpied, T.A. Long-Term Potentiation Requires a Rapid Burst of Dendritic Mitochondrial Fission during Induction. Neuron 2018, 100, 860–875.e7. [Google Scholar] [CrossRef]
- Li, Z.; Okamoto, K.I.; Hayashi, Y.; Sheng, M. The Importance of Dendritic Mitochondria in the Morphogenesis and Plasticity of Spines and Synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef]
- Fukumitsu, K.; Hatsukano, T.; Yoshimura, A.; Heuser, J.; Fujishima, K.; Kengaku, M. Mitochondrial Fission Protein Drp1 Regulates Mitochondrial Transport and Dendritic Arborization in Cerebellar Purkinje Cells. Mol. Cell. Neurosci. 2016, 71, 56–65. [Google Scholar] [CrossRef]
- Sturm, D.; Gurevitz, S.L.; Turner, A. Multiple Sclerosis: A Review of the Disease and Treatment Options. Consult. Pharm. 2014, 29, 469–479. [Google Scholar] [CrossRef]
- Michaličková, D.; Kübra Öztürk, H.; Hroudová, J.; Ľupták, M.; Kučera, T.; Hrnčíř, T.; Kutinová Canová, N.; Šíma, M.; Slanař, O.; Michaličková, D. Edaravone Attenuates Disease Severity of Experimental Auto-Immune Encephalomyelitis and Increases Gene Expression of Nrf2 and HO-1. Physiol. Res. 2022, 71, 147–157. [Google Scholar] [CrossRef]
- Al-Kafaji, G.; Bakheit, H.F.; AlAli, F.; Fattah, M.; Alhajeri, S.; Alharbi, M.A.; Daif, A.; Alsabbagh, M.M.; Alwehaidah, M.S.; Bakhiet, M. Next-Generation Sequencing of the Whole Mitochondrial Genome Identifies Functionally Deleterious Mutations in Patients with Multiple Sclerosis. PLoS ONE 2022, 17, e0263606. [Google Scholar] [CrossRef]
- Licht-Mayer, S.; Campbell, G.R.; Canizares, M.; Mehta, A.R.; Gane, A.B.; McGill, K.; Ghosh, A.; Fullerton, A.; Menezes, N.; Dean, J.; et al. Enhanced Axonal Response of Mitochondria to Demyelination Offers Neuroprotection: Implications for Multiple Sclerosis. Acta Neuropathol. 2020, 140, 143–167. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Mahad, D.J. Mitochondrial Changes Associated with Demyelination: Consequences for Axonal Integrity. Mitochondrion 2012, 12, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Reddy, P.H. Is Multiple Sclerosis a Mitochondrial Disease? Biochim. Biophys. Acta-Mol. Basis Dis. 2010, 1802, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Andrews, H.E.; Nichols, P.P.; Bates, D.; Turnbull, D.M. Mitochondrial Dysfunction Plays a Key Role in Progressive Axonal Loss in Multiple Sclerosis. Med. Hypotheses 2005, 64, 669–677. [Google Scholar] [CrossRef] [PubMed]
- de Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial Dysfunction as a Cause of Axonal Degeneration in Multiple Sclerosis Patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial Defects in Acute Multiple Sclerosis Lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial Changes within Axons in Multiple Sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Ohno, N.; Turnbull, D.M.; Mahad, D.J. Mitochondrial Changes within Axons in Multiple Sclerosis: An Update. Curr. Opin. Neurol. 2012, 25, 221–230. [Google Scholar] [CrossRef]
- Sathornsumetee, S.; McGavern, D.B.; Ure, D.R.; Rodriguez, M. Quantitative Ultrastructural Analysis of a Single Spinal Cord Demyelinated Lesion Predicts Total Lesion Load, Axonal Loss, and Neurological Dysfunction in a Murine Model of Multiple Sclerosis. Am. J. Pathol. 2000, 157, 1365–1376. [Google Scholar] [CrossRef]
- Tobore, T.O. Oxidative/Nitroxidative Stress and Multiple Sclerosis. J. Mol. Neurosci. 2021, 71, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.; Adamczyk-Sowa, M. New Insights into the Role of Oxidative Stress Mechanisms in the Pathophysiology and Treatment of Multiple Sclerosis. Oxid. Med. Cell. Longev. 2016, 2016, 1973834. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Minghetti, L.; Sette, G.; Fieschi, C.; Levi, G. Cerebrospinal Fluid Isoprostane Shows Oxidative Stress in Patients with Multiple Sclerosis. Neurology 1999, 53, 1876–1879. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Selak, M.; O’Connor, J.; Croul, S.; Lorenzana, C.; Butunoi, C.; Kalman, B. Oxidative Damage to Mitochondrial DNA and Activity of Mitochondrial Enzymes in Chronic Active Lesions of Multiple Sclerosis. J. Neurol. Sci. 2000, 177, 95–103. [Google Scholar] [CrossRef]
- Qi, X.; Lewin, A.S.; Sun, L.; Hauswirth, W.W.; Guy, J. Mitochondrial Protein Nitration Primes Neurodegeneration in Experimental Autoimmune Encephalomyelitis. J. Biol. Chem. 2006, 281, 31950–31962. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J. Oxidative Stress and Its Impact on Neurons and Glia in Multiple Sclerosis Lesions. Biochim. Biophys. Acta-Mol. Basis Dis. 2016, 1862, 506–510. [Google Scholar] [CrossRef]
- Su, K.; Bourdette, D.; Forte, M. Mitochondrial Dysfunction and Neurodegeneration in Multiple Sclerosis. Front. Physiol. 2013, 4, 169. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.; Yamamoto, M. Nrf2-MafG Heterodimers Contribute Globally to Antioxidant and Metabolic Networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The Emerging Role of Nrf2 in Mitochondrial Function. Free Radic. Biol. Med. 2015, 88, 179. [Google Scholar] [CrossRef]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-Mediated Neuroprotection in the MPTP Mouse Model of Parkinson’s Disease: Critical Role for the Astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef]
- Branca, C.; Ferreira, E.; Nguyen, T.V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic Reduction of Nrf2 Exacerbates Cognitive Deficits in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef] [PubMed]
- Michaličková, D.; Hrnčíř, T.; Canová, N.K.; Slanař, O. Targeting Keap1/Nrf2/ARE Signaling Pathway in Multiple Sclerosis. Eur. J. Pharmacol. 2020, 873, 172973. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 Regulates Microglial Dynamics and Neuroinflammation in Experimental Parkinson’s Disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Rosito, M.; Testi, C.; Parisi, G.; Cortese, B.; Baiocco, P.; Di Angelantonio, S. Exploring the Use of Dimethyl Fumarate as Microglia Modulator for Neurodegenerative Diseases Treatment. Antioxidants 2020, 9, 700. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.; Sagarra, M.; Cuadrado, A. GSK-3beta down-Regulates the Transcription Factor Nrf2 after Oxidant Damage: Relevance to Exposure of Neuronal Cells to Oxidative Stress. J. Neurochem. 2008, 105, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased Nuclear DNA Oxidation in the Brain in Alzheimer’s Disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef] [PubMed]
- Min, K.J.; Yang, M.S.; Kim, S.U.; Jou, I.; Joe, E.H. Astrocytes Induce Hemeoxygenase-1 Expression in Microglia: A Feasible Mechanism for Preventing Excessive Brain Inflammation. J. Neurosci. 2006, 26, 1880–1887. [Google Scholar] [CrossRef]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-ҚB Interplay in Cerebrovascular and Neurodegenerative Disorders: Molecular Mechanisms and Possible Therapeutic Approaches. Redox Biol. 2019, 21. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription Factors NRF2 and NF-ΚB Are Coordinated Effectors of the Rho Family, GTP-Binding Protein RAC1 during Inflammation *. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting Molecular Cross-Talk between Nrf2 and NF-ΚB Response Pathways. Biochem. Soc. Trans. 2015, 43, 621. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.; Johnson, J. The Nrf2-ARE Cytoprotective Pathway in Astrocytes. Expert Rev. Mol. Med. 2009, 11. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, S.; Ardizzone, A.; Paterniti, I.; Esposito, E.; Campolo, M. Antioxidant and Anti-Inflammatory Effect of Nrf2 Inducer Dimethyl Fumarate in Neurodegenerative Diseases. Antioxidants 2020, 9, 630. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-Synuclein Expression and Nrf2 Deficiency Cooperate to Aggravate Protein Aggregation, Neuronal Death and Inflammation in Early-Stage Parkinson’s Disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Johnson, J. Nrf2--a Therapeutic Target for the Treatment of Neurodegenerative Diseases. Free Radic. Biol. Med. 2015, 88, 253–267. [Google Scholar] [CrossRef]
- Cuadrado, A.; Kügler, S.; Lastres-Becker, I. Pharmacological Targeting of GSK-3 and NRF2 Provides Neuroprotection in a Preclinical Model of Tauopathy. Redox Biol. 2018, 14, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, G.; Jasoliya, M.; Sahdeo, S.; Saccà, F.; Pane, C.; Filla, A.; Marsili, A.; Puorro, G.; Lanzillo, R.; Brescia Morra, V.; et al. Dimethyl Fumarate Mediates Nrf2-Dependent Mitochondrial Biogenesis in Mice and Humans. Hum. Mol. Genet. 2017, 26, 2864–2873. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.A.; Ogrodnik, M.A.; Plave, A.; Mao-Draayer, Y. Emerging Understanding of the Mechanism of Action for Dimethyl Fumarate in the Treatment of Multiple Sclerosis. Front. Neurol. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Mikulskis, A.; Gold, R.; Fox, R.J.; Dawson, K.T.; Amaravadi, L. Evidence of Activation of the Nrf2 Pathway in Multiple Sclerosis Patients Treated with Delayed-Release Dimethyl Fumarate in the Phase 3 DEFINE and CONFIRM Studies. Mult. Scler. 2017, 23, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Hammer, A.; Waschbisch, A.; Kuhbandner, K.; Bayas, A.; Lee, D.H.; Duscha, A.; Haghikia, A.; Gold, R.; Linker, R.A. The NRF2 Pathway as Potential Biomarker for Dimethyl Fumarate Treatment in Multiple Sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Bomprezzi, R. Dimethyl Fumarate in the Treatment of Relapsing-Remitting Multiple Sclerosis: An Overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef] [PubMed]
- van der Star, B.J.; Vogel, D.Y.S.; Kipp, M.; Puentes, F.; Baker, D.; Amor, S. In Vitro and in Vivo Models of Multiple Sclerosis. CNS Neurol. Disord. Drug Targets 2012, 11, 570–588. [Google Scholar] [CrossRef] [PubMed]
- Draheim, T.; Liessem, A.; Scheld, M.; Wilms, F.; Weißflog, M.; Denecke, B.; Kensler, T.W.; Zendedel, A.; Beyer, C.; Kipp, M.; et al. Activation of the Astrocytic Nrf2/ARE System Ameliorates the Formation of Demyelinating Lesions in a Multiple Sclerosis Animal Model. Glia 2016, 64, 2219–2230. [Google Scholar] [CrossRef]
- Nellessen, A.; Nyamoya, S.; Zendedel, A.; Slowik, A.; Wruck, C.; Beyer, C.; Fragoulis, A.; Clarner, T. Nrf2 Deficiency Increases Oligodendrocyte Loss, Demyelination, Neuroinflammation and Axonal Damage in an MS Animal Model. Metab. Brain Dis. 2020, 35, 353–362. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.H.; Ryan, S.; Van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric Acid Esters Exert Neuroprotective Effects in Neuroinflammation via Activation of the Nrf2 Antioxidant Pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef]
- Mela, V.; Gaban, A.S.; O’neill, E.; Bechet, S.; Walsh, A.; Lynch, M.A. The Modulatory Effects of DMF on Microglia in Aged Mice Are Sex-Specific. Cells 2022, 11, 729. [Google Scholar] [CrossRef]
- Abudara, V.; Retamal, M.A.; Del Rio, R.; Orellana, J.A. Synaptic Functions of Hemichannels and Pannexons: A Double-Edged Sword. Front. Mol. Neurosci. 2018, 11, 435. [Google Scholar] [CrossRef]
- Cheung, G.; Chever, O.; Rouach, N. Connexons and Pannexons: Newcomers in Neurophysiology. Front. Cell. Neurosci. 2014, 8, 348. [Google Scholar] [CrossRef]
- Dahl, G. The Pannexin1 Membrane Channel: Distinct Conformations and Functions. FEBS Lett. 2018, 592, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Avendano, B.C.; Montero, T.D. Role of Connexins and Pannexins in Ischemic Stroke. Curr. Med. Chem. 2014, 21, 2165–2182. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor Necrosis Factor-Alpha Induces Neurotoxicity via Glutamate Release from Hemichannels of Activated Microglia in an Autocrine Manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef] [PubMed]
- Vejar, S.; Oyarzún, J.E.; Retamal, M.A.; Ortiz, F.C.; Orellana, J.A. Connexin and Pannexin-Based Channels in Oligodendrocytes: Implications in Brain Health and Disease. Front. Cell. Neurosci. 2019, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Arias, J.C.; Wicki-Stordeur, L.E.; Swayne, L.A. Perspectives on the Role of Pannexin 1 in Neural Precursor Cell Biology. Neural Regen. Res. 2016, 11, 1540. [Google Scholar] [CrossRef] [PubMed]
- Ardiles, A.O.; Flores-Muñoz, C.; Toro-Ayala, G.; Cárdenas, A.M.; Palacios, A.G.; Muñoz, P.; Fuenzalida, M.; Sáez, J.C.; Martínez, A.D. Pannexin 1 Regulates Bidirectional Hippocampal Synaptic Plasticity in Adult Mice. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- Chever, O.; Lee, C.Y.; Rouach, N. Astroglial Connexin43 Hemichannels Tune Basal Excitatory Synaptic Transmission. J. Neurosci. 2014, 34, 11228–11232. [Google Scholar] [CrossRef]
- Meunier, C.; Wang, N.; Yi, C.; Dallerac, G.; Ezan, P.; Koulakoff, A.; Leybaert, L.; Giaume, C. Contribution of Astroglial Cx43 Hemichannels to the Modulation of Glutamatergic Currents by D-Serine in the Mouse Prefrontal Cortex. J. Neurosci. 2017, 37, 9064–9075. [Google Scholar] [CrossRef]
- Stehberg, J.; Moraga-Amaro, R.; Salazar, C.; Becerra, A.; Echeverría, C.; Orellana, J.A.; Bultynck, G.; Ponsaerts, R.; Leybaert, L.; Simon, F.; et al. Release of Gliotransmitters through Astroglial Connexin 43 Hemichannels Is Necessary for Fear Memory Consolidation in the Basolateral Amygdala. FASEB J. 2012, 26, 3649–3657. [Google Scholar] [CrossRef]
- Walrave, L.; Vinken, M.; Albertini, G.; de Bundel, D.; Leybaert, L.; Smolders, I.J. Inhibition of Connexin43 Hemichannels Impairs Spatial Short-Term Memory without Affecting Spatial Working Memory. Front. Cell. Neurosci. 2016, 10, 288. [Google Scholar] [CrossRef]
- Sargiannidou, I.; Vavlitou, N.; Aristodemou, S.; Hadjisavvas, A.; Kyriacou, K.; Scherer, S.S.; Kleopa, K.A. Connexin32 Mutations Cause Loss of Function in Schwann Cells and Oligodendrocytes Leading to PNS and CNS Myelination Defects. J. Neurosci. 2009, 29, 4736. [Google Scholar] [CrossRef] [PubMed]
- Papaneophytou, C.; Georgiou, E.; Kleopa, K.A. The Role of Oligodendrocyte Gap Junctions in Neuroinflammation. Channels 2019, 13, 247. [Google Scholar] [CrossRef] [PubMed]
- Díaz, E.F.; Labra, V.C.; Alvear, T.F.; Mellado, L.A.; Inostroza, C.A.; Oyarzún, J.E.; Salgado, N.; Quintanilla, R.A.; Orellana, J.A. Connexin 43 Hemichannels and Pannexin-1 Channels Contribute to the α-Synuclein-Induced Dysfunction and Death of Astrocytes. Glia 2019, 67, 1598–1619. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.L.; Ebihara, L.; Takemoto, L.J.; Swenson, K.I.; Goodenough, D.A. Connexin46, a Novel Lens Gap Junction Protein, Induces Voltage-Gated Currents in Nonjunctional Plasma Membrane of Xenopus Oocytes. J. Cell Biol. 1991, 115, 1077–1089. [Google Scholar] [CrossRef]
- Shijie, J.; Takeuchi, H.; Yawata, I.; Harada, Y.; Sonobe, Y.; Doi, Y.; Liang, J.; Hua, L.; Yasuoka, S.; Zhou, Y.; et al. Blockade of Glutamate Release from Microglia Attenuates Experimental Autoimmune Encephalomyelitis in Mice. Tohoku J. Exp. Med. 2009, 217, 87–92. [Google Scholar] [CrossRef]
- Zhou, L.; Ao, L.; Yan, Y.; Li, C.; Li, W.; Ye, A.; Liu, J.; Hu, Y.; Fang, W.; Li, Y. Levo-Corydalmine Attenuates Vincristine-Induced Neuropathic Pain in Mice by Upregulating the Nrf2/HO-1/CO Pathway to Inhibit Connexin 43 Expression. Neurotherapeutics 2020, 17, 340–355. [Google Scholar] [CrossRef]
- Chen, Z.; Xie, X.; Huang, J.; Gong, W.; Zhu, X.; Chen, Q.; Huang, J.; Huang, H. Connexin43 Regulates High Glucose-Induced Expression of Fibronectin, ICAM-1 and TGF-Β1 via Nrf2/ARE Pathway in Glomerular Mesangial Cells. Free Radic. Biol. Med. 2017, 102, 77–86. [Google Scholar] [CrossRef]
- Yang, Y.; Li, J.; Zhang, L.; Lin, Z.; Xiao, H.; Sun, X.; Zhang, M.; Liu, P.; Huang, H. CKIP-1 Acts Downstream to Cx43 on the Activation of Nrf2 Signaling Pathway to Protect from Renal Fibrosis in Diabetes. Pharmacol. Res. 2021, 163. [Google Scholar] [CrossRef]
- Chen, X.; Liang, H.; Xi, Z.; Yang, Y.; Shan, H.; Wang, B.; Zhong, Z.; Xu, C.; Yang, G.Y.; Sun, Q.; et al. BM-MSC Transplantation Alleviates Intracerebral Hemorrhage-Induced Brain Injury, Promotes Astrocytes Vimentin Expression, and Enhances Astrocytes Antioxidation via the Cx43/Nrf2/HO-1 Axis. Front. Cell Dev. Biol. 2020, 8, 302. [Google Scholar] [CrossRef]
- Negoro, H.; Lutz, S.E.; Liou, L.S.; Kanematsu, A.; Ogawa, O.; Scemes, E.; Suadicani, S.O. Pannexin 1 Involvement in Bladder Dysfunction in a Multiple Sclerosis Model. Sci. Rep. 2013, 3, 2152. [Google Scholar] [CrossRef]
- Hainz, N.; Becker, P.; Rapp, D.; Wagenpfeil, S.; Wonnenberg, B.; Beisswenger, C.; Tschernig, T.; Meier, C. Probenecid-Treatment Reduces Demyelination Induced by Cuprizone Feeding. J. Chem. Neuroanat. 2017, 85, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Hainz, N.; Wolf, S.; Beck, A.; Wagenpfeil, S.; Tschernig, T.; Meier, C. Probenecid Arrests the Progression of Pronounced Clinical Symptoms in a Mouse Model of Multiple Sclerosis. Sci. Rep. 2017, 7, 17214. [Google Scholar] [CrossRef] [PubMed]
- Hainz, N.; Wolf, S.; Tschernig, T.; Meier, C. Probenecid Application Prevents Clinical Symptoms and Inflammation in Experimental Autoimmune Encephalomyelitis. Inflammation 2016, 39, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, F.; Puebla, C. Pannexin 1-Based Channels Activity as a Novel Regulator of Multiple Sclerosis Progression. Neural Regen. Res. 2020, 15, 65–66. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maldonado, P.P.; Guevara, C.; Olesen, M.A.; Orellana, J.A.; Quintanilla, R.A.; Ortiz, F.C. Neurodegeneration in Multiple Sclerosis: The Role of Nrf2-Dependent Pathways. Antioxidants 2022, 11, 1146. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11061146
Maldonado PP, Guevara C, Olesen MA, Orellana JA, Quintanilla RA, Ortiz FC. Neurodegeneration in Multiple Sclerosis: The Role of Nrf2-Dependent Pathways. Antioxidants. 2022; 11(6):1146. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11061146
Chicago/Turabian StyleMaldonado, Paloma P., Coram Guevara, Margrethe A. Olesen, Juan Andres Orellana, Rodrigo A. Quintanilla, and Fernando C. Ortiz. 2022. "Neurodegeneration in Multiple Sclerosis: The Role of Nrf2-Dependent Pathways" Antioxidants 11, no. 6: 1146. https://0-doi-org.brum.beds.ac.uk/10.3390/antiox11061146