
First Crystal Structure of Bacterial Oligopeptidase B in an Intermediate State: The Roles of the Hinge Region Modification and Spermine

 ,
,
Abstract
:Simple Summary
Abstract
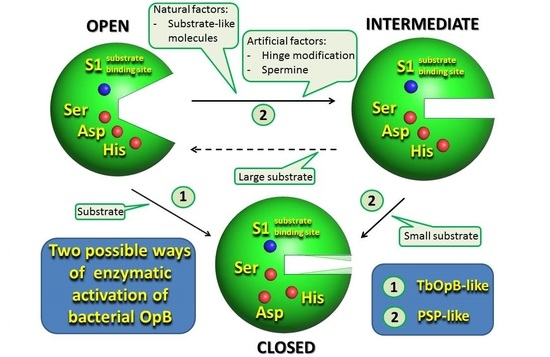
1. Introduction
2. Materials and Methods
2.1. Mutagenesis
2.2. Recombinant Proteins Purification and Characterization
2.3. Enzymatic Study
2.4. Far-UV Circular Dichroism Spectroscopy
2.5. Differential Scanning Calorimetry
2.6. Protein Crystallization, Data Collection, Processing, Structure Refinement and Analysis
2.7. Data Bank Accession Numbers
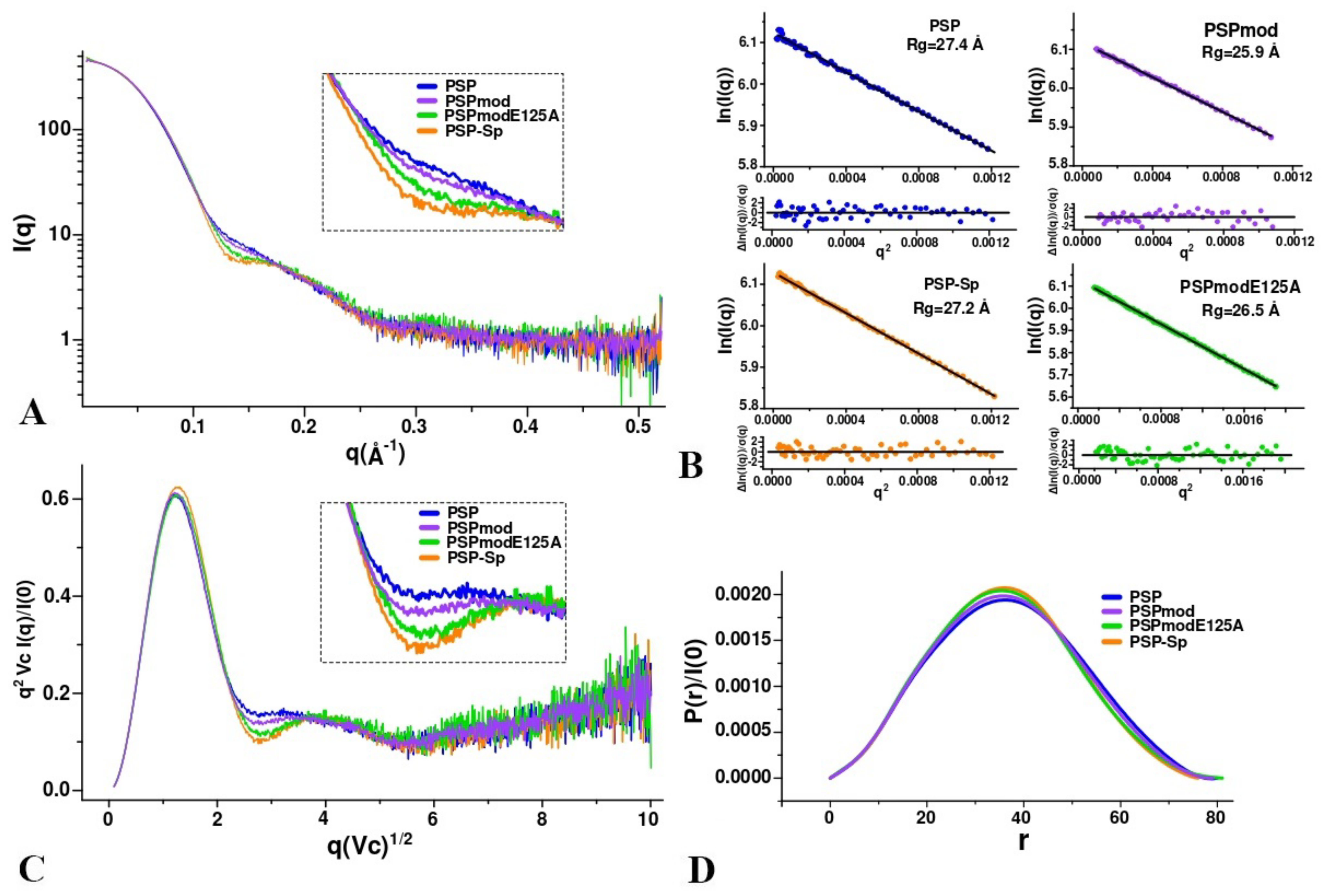
2.8. SAXS Measurement
3. Results and Discussion
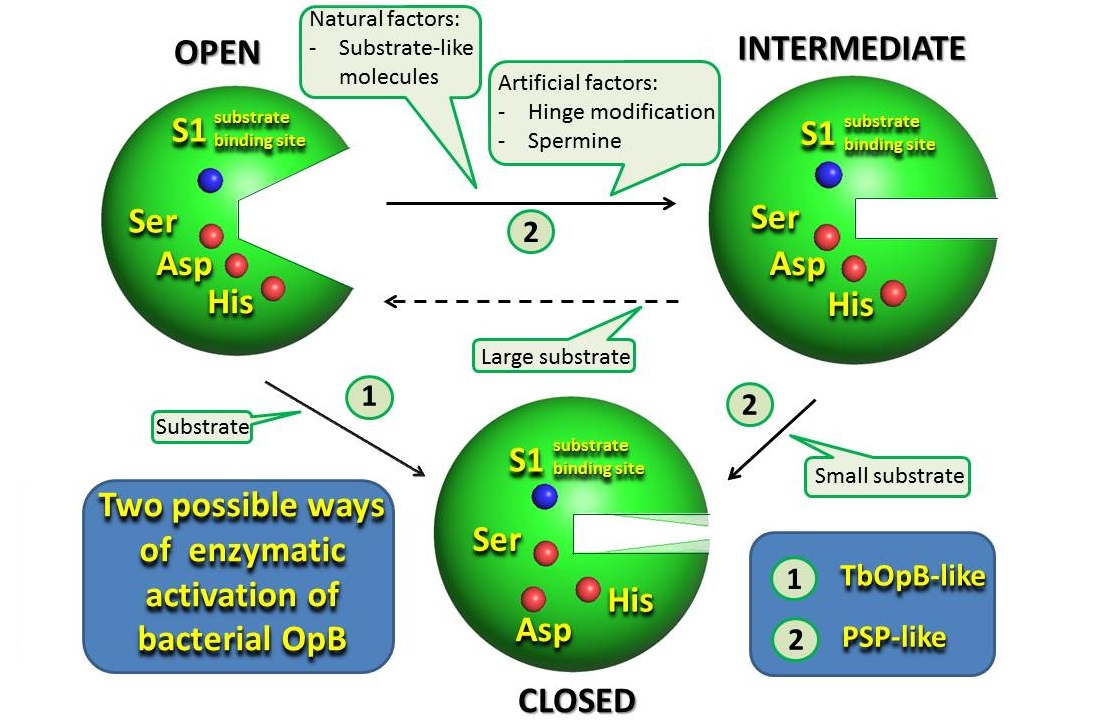
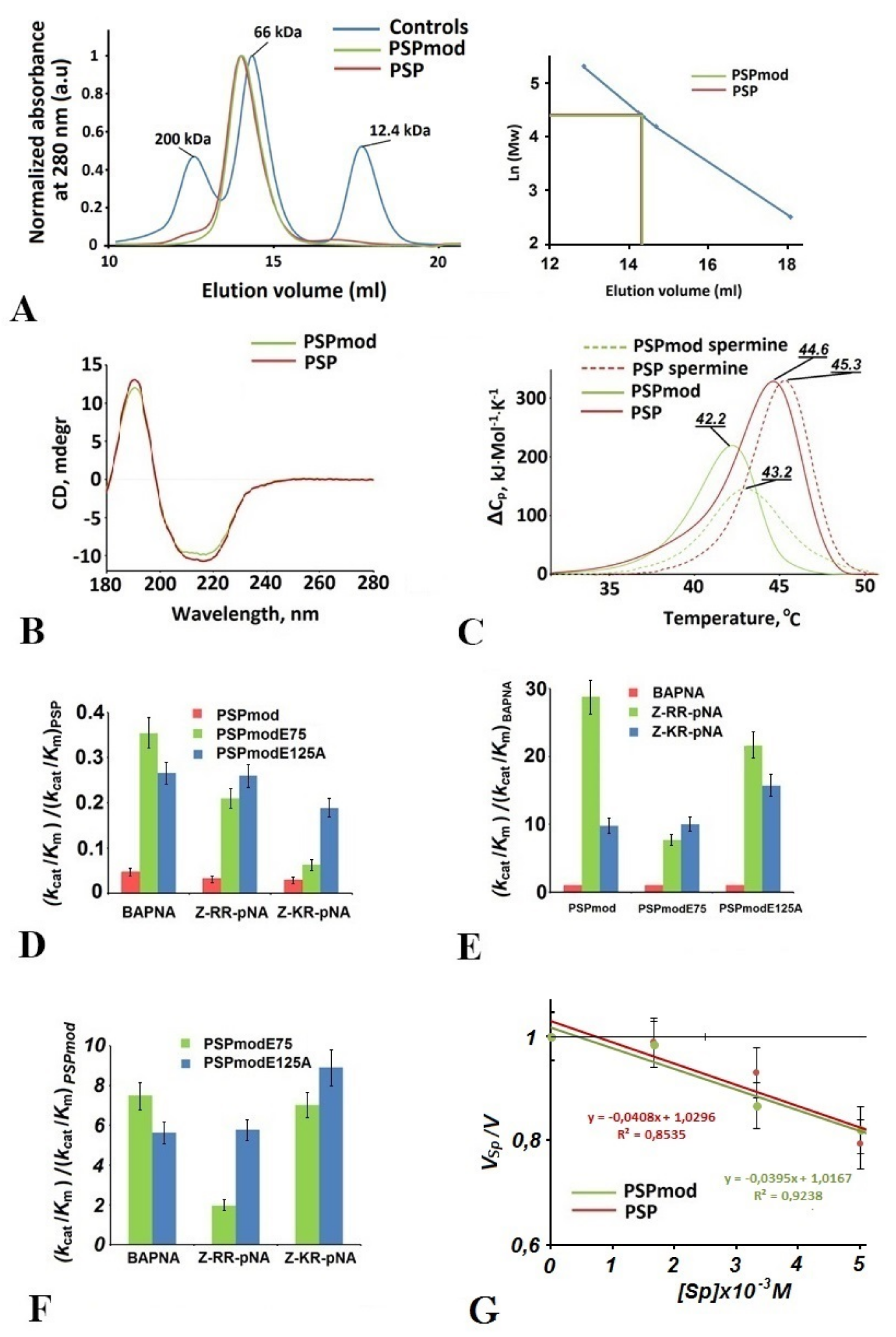
3.1. Comparative Analysis of Physicochemical Features and Enzymatic Activity of PSPmod
3.2. Intermediate States Were Observed in the Crystal Structures of PSPmod and Its Derivatives
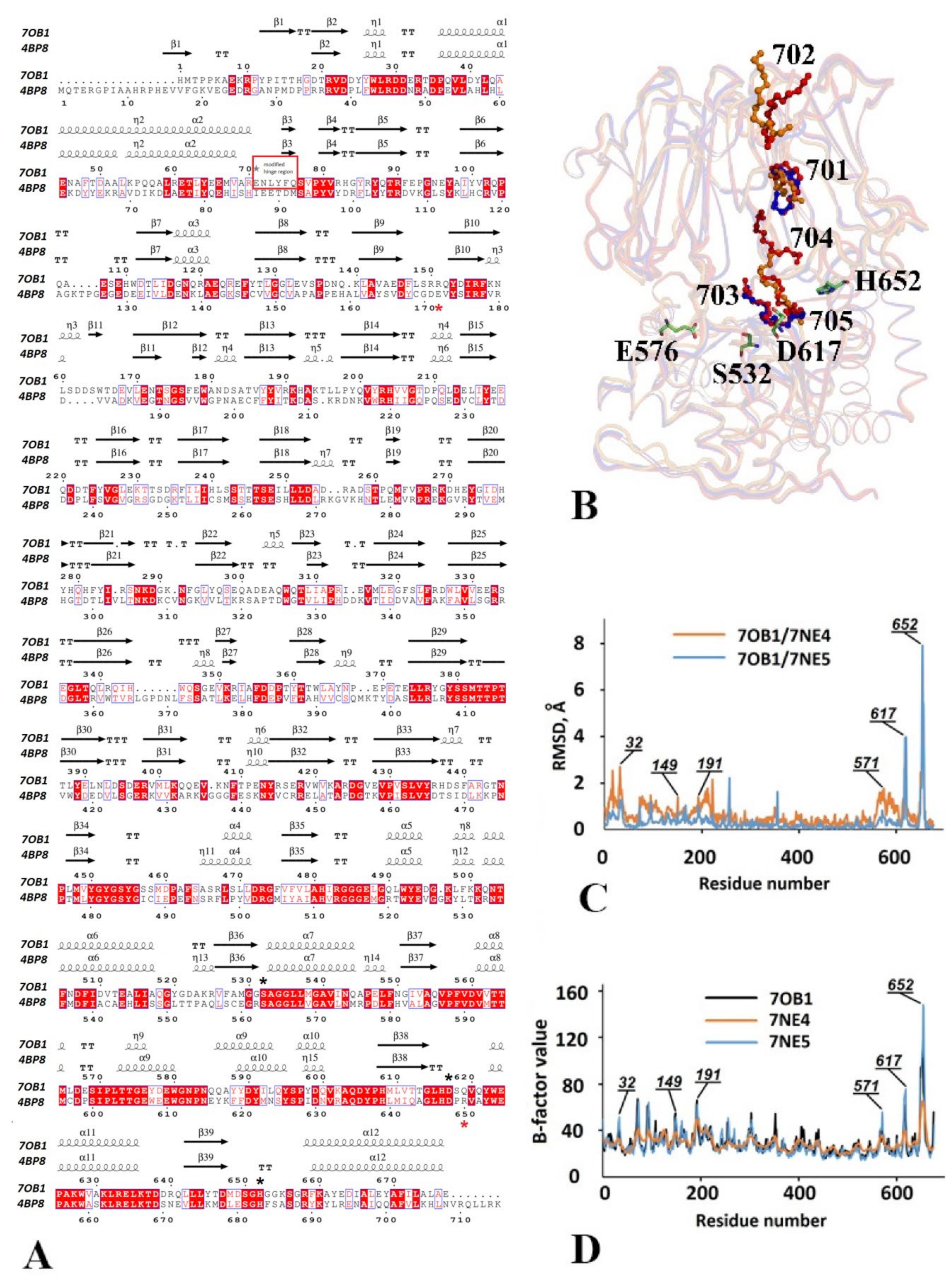
3.2.1. Structural Overview
3.2.2. The Catalytic Triad Arrangement
3.2.3. Comparative Analysis of the Domain Positioning
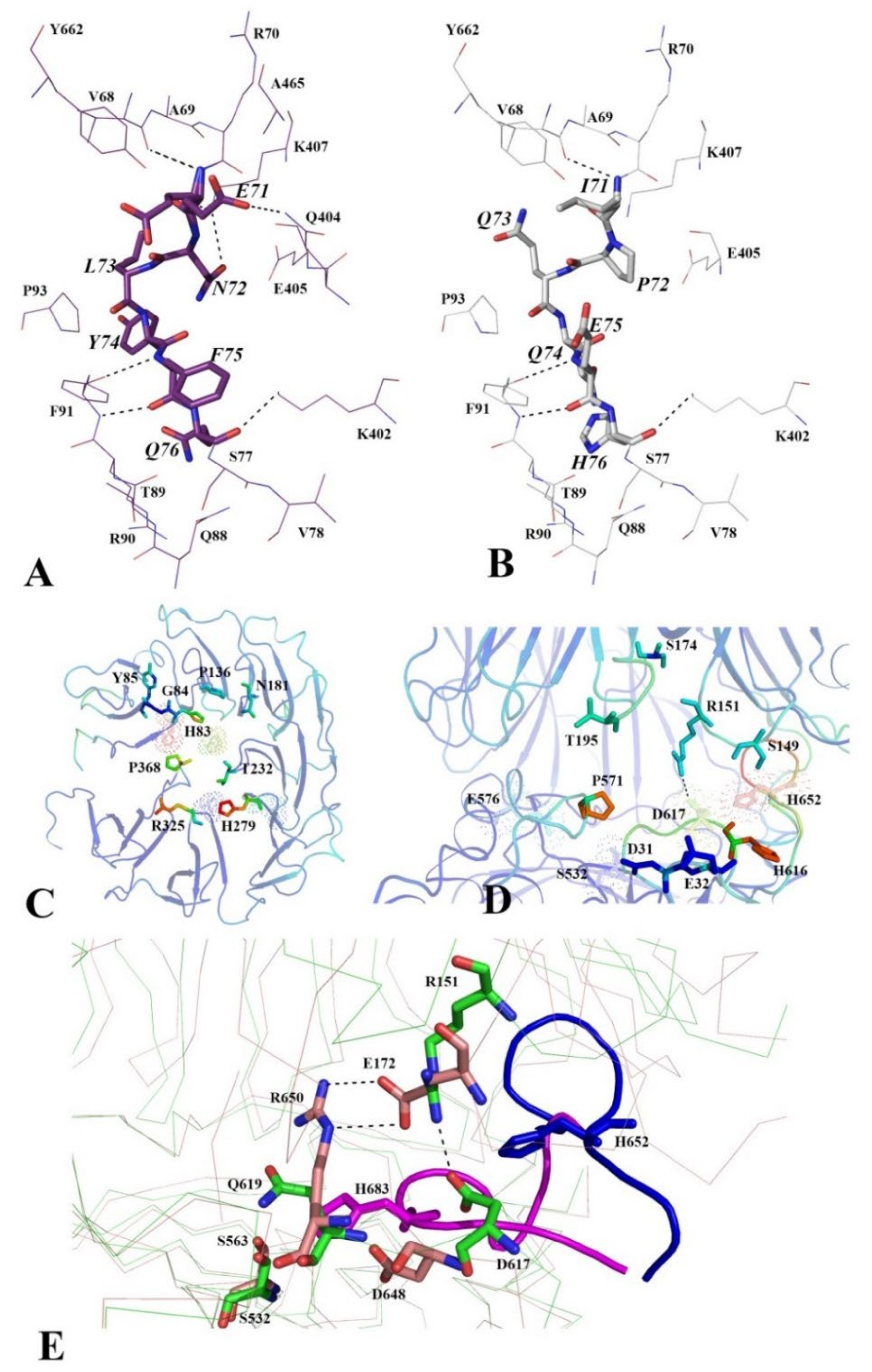
3.2.4. Functionally Important Interdomain Salt Bridge (SB1) Conserved in Protozoan OpB and Bacterial PEP Is Abscent in PSPmod
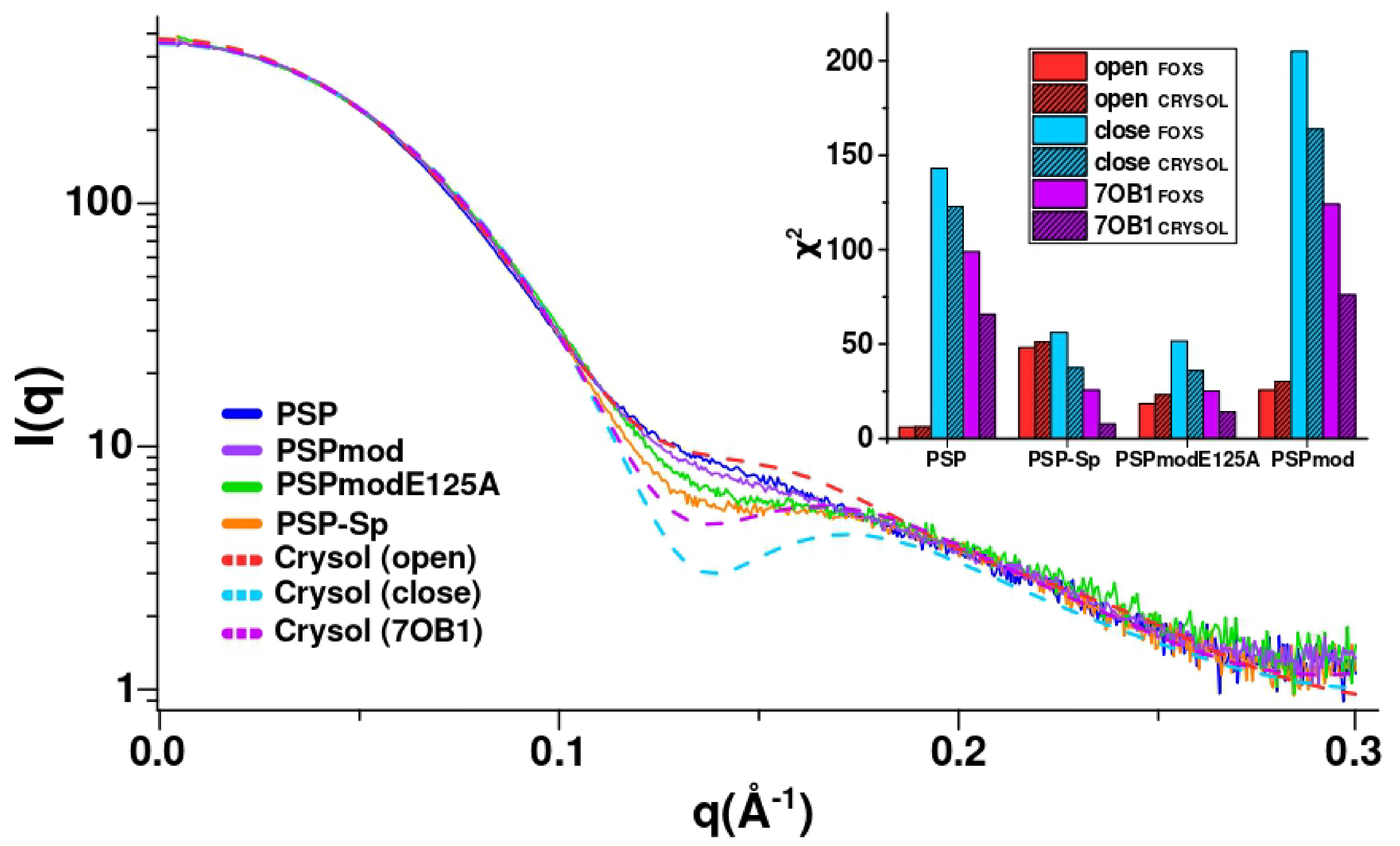
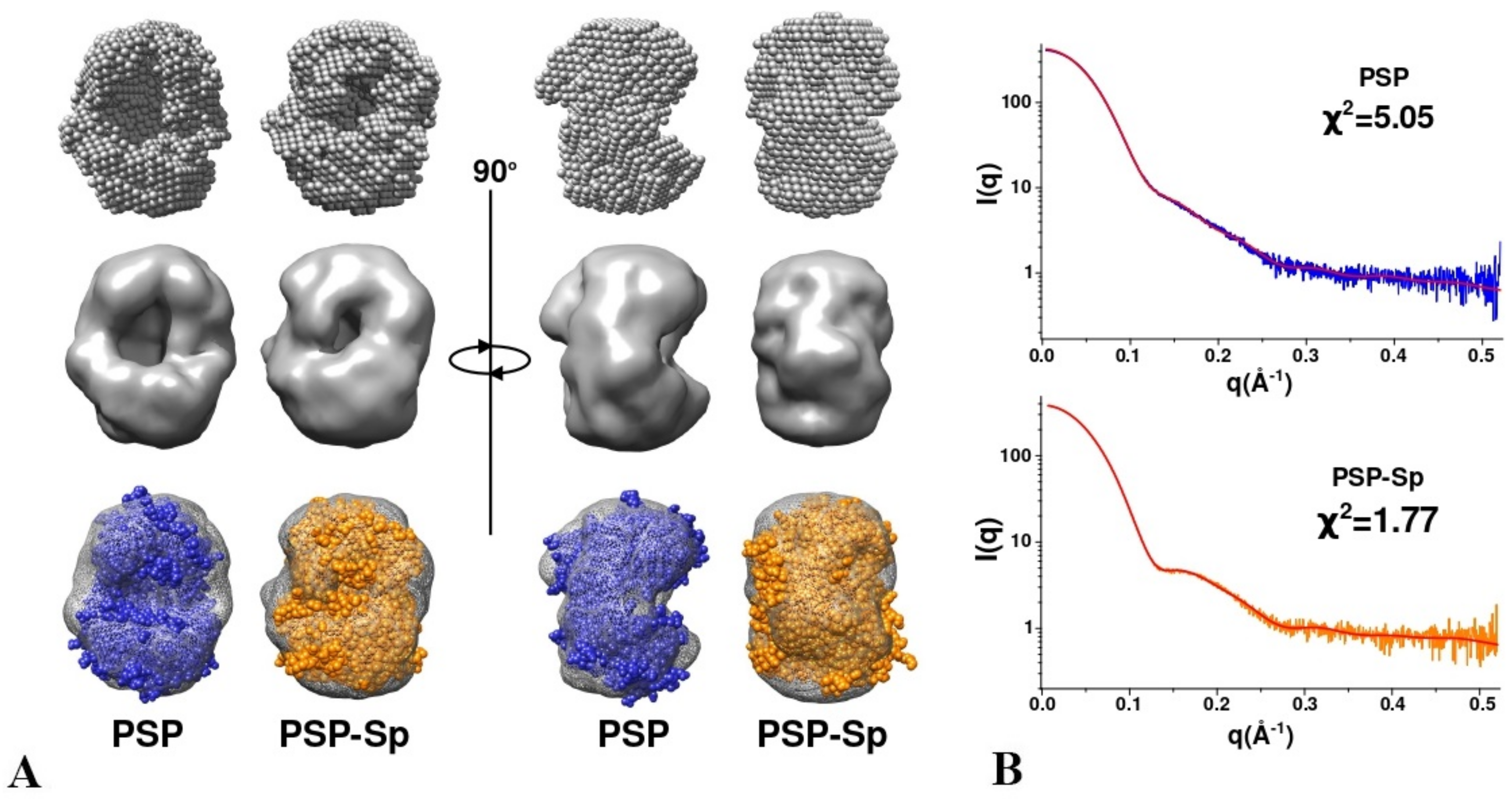
3.3. SAXS Analysis of the Conformation of PSP and Its Derivatives in Solution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Polgár, L. The prolyl oligopeptidase family. Cell. Mol. Life Sci. 2002, 59, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Motta, F.N.; Azevedo, C.D.S.; Neves, B.P.; de Araújo, C.N.; Grellier, P.; de Santana, J.M.; Bastos, I.M.D. Oligopeptidase B, a missing enzyme in mammals and a potential drug target for trypanosomatid diseases. Biochimie 2019, 167, 207–216. [Google Scholar] [CrossRef]
- Morty, R.E.; Pellé, R.; Vadász, I.; Uzcanga, G.L.; Seeger, W.; Bubis, J. Oligopeptidase B from Trypanosoma evansi. A parasite peptidase that inactivates atrial natriuretic factor in the bloodstream of infected hosts. J. Biol. Chem. 2005, 280, 10925–10937. [Google Scholar] [CrossRef] [Green Version]
- Coetzer, T.H.; Goldring, J.P.D.; Huson, L.E. Oligopeptidase B: A processing peptidase involved in pathogenesis. Biochimie 2008, 90, 336–344. [Google Scholar] [CrossRef]
- Swenerton, R.K.; Zhang, S.; Sajid, M.; Medzihradszky, K.F.; Craik, C.S.; Kelly, B.L.; McKerrow, J.H. The Oligopeptidase B of Leishmania Regulates Parasite Enolase and Immune Evasion. J. Biol. Chem. 2011, 286, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Bivona, A.E.; Alberti, A.S.; Matos, M.N.; Cerny, N.; Cardoso, A.C.; Morales, C.; González, G.; Cazorla, S.I.; Malchiodi, E.L. Trypanosoma cruzi 80 kDa prolyl oligopeptidase (Tc80) as a novel immunogen for Chagas disease vaccine. PLoS Negl. Trop. Dis. 2018, 12, e0006384. [Google Scholar] [CrossRef] [PubMed]
- Kanatani, A.; Masuda, T.; Shimoda, T.; Misoka, F.; Lin, X.S.; Yoshimoto, T.; Tsuru, D. Protease II from Escherichia coli: Sequencing and Expression of the Enzyme Gene and Characterization of the Expressed Enzyme1. J. Biochem. 1991, 110, 315–320. [Google Scholar] [CrossRef]
- Mattiuzzo, M.; De Gobba, C.; Runti, G.; Mardirossian, M.; Bandiera, A.; Gennaro, R.; Scocchi, M. Proteolytic Activity of Escherichia coli Oligopeptidase B Against Proline-Rich Antimicrobial Peptides. J. Microbiol. Biotechnol. 2014, 24, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Fülöp, V.; Böcskei, Z.; Polgár, L. Prolyl Oligopeptidase: An Unusual β-Propeller Domain Regulates Proteolysis. Cell 1998, 94, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Rea, D.; Fülöp, V. Structure-Function Properties of Prolyl Oligopeptidase Family Enzymes. Cell Biophys. 2006, 44, 349–365. [Google Scholar] [CrossRef]
- Shan, L.; Mathews, I.I.; Khosla, C. Structural and mechanistic analysis of two prolyl endopeptidases: Role of interdomain dynamics in catalysis and specificity. Proc. Natl. Acad. Sci. USA 2005, 102, 3599–3604. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Chen, C.; Davies, D.R.; Chiu, T.K. Induced-fit Mechanism for Prolyl Endopeptidase. J. Biol. Chem. 2010, 285, 21487–21495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmat, V.; Domokos, K.; Menyhárd, D.K.; Palló, A.; Szeltner, Z.; Szamosi, I.; Beke-Somfai, T.; Náray-Szabó, G.; Polgár, L. Structure and Catalysis of Acylaminoacyl Peptidase: Closed and open subunits of a dimer oligopeptidase. J. Biol. Chem. 2011, 286, 1987–1998. [Google Scholar] [CrossRef] [Green Version]
- Menyhárd, D.K.; Orgován, Z.; Szeltner, Z.; Szamosi, I.; Harmat, V. Catalytically distinct states captured in a crystal lattice: The substrate-bound and scavenger states of acylaminoacyl peptidase and their implications for functionality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Szeltner, Z.; Rea, D.; Juhász, T.; Renner, V.; Fülöp, V.; Polgár, L. Concerted Structural Changes in the Peptidase and the Propeller Domains of Prolyl Oligopeptidase are Required for Substrate Binding. J. Mol. Biol. 2004, 340, 627–637. [Google Scholar] [CrossRef]
- Kichik, N.; Tarragó, T.; Claasen, B.; Gairí, M.; Millet, O.; Giralt, E. 15N Relaxation NMR Studies of Prolyl Oligopeptidase, an 80 kDa Enzyme, Reveal a Pre-existing Equilibrium between Different Conformational States. ChemBioChem 2011, 12, 2737–2739. [Google Scholar] [CrossRef]
- Fülöp, V.; Szeltner, Z.; Polgár, L. Catalysis of serine oligopeptidases is controlled by a gating filter mechanism. EMBO Rep. 2000, 1, 277–281. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.-F.; Karttunen, M.; Mousseau, N.; Róg, T.; Bunker, A. Use of Umbrella Sampling to Calculate the Entrance/Exit Pathway for Z-Pro-Prolinal Inhibitor in Prolyl Oligopeptidase. J. Chem. Theory Comput. 2011, 7, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Sowdhamini, R. Structural Analysis of Prolyl Oligopeptidases Using Molecular Docking and Dynamics: Insights into Conformational Changes and Ligand Binding. PLoS ONE 2011, 6, e26251. [Google Scholar] [CrossRef] [Green Version]
- Szeltner, Z.; Juhász, T.; Szamosi, I.; Rea, D.; Fülöp, V.; Módos, K.; Juliano, L.; Polgár, L. The loops facing the active site of prolyl oligopeptidase are crucial components in substrate gating and specificity. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2013, 1834, 98–111. [Google Scholar] [CrossRef] [Green Version]
- Kaszuba, K.; Róg, T.; Danne, R.; Canning, P.; Fülöp, V.; Juhász, T.; Szeltner, Z.; Pierre, J.-F.S.; García-Horsman, A.; Männistö, P.T.; et al. Molecular dynamics, crystallography and mutagenesis studies on the substrate gating mechanism of prolyl oligopeptidase. Biochimie 2012, 94, 1398–1411. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Etchebest, C.; Sowdhamini, R. Decoding the structural events in substrate-gating mechanism of eukaryotic prolyl oligopeptidase using normal mode analysis and molecular dynamics simulations. Proteins 2014, 82, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Czekster, C.M.; Ludewig, H.; McMahon, S.A.; Naismith, J.H. Characterization of a dual function macrocyclase enables design and use of efficient macrocyclization substrates. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ellis-Guardiola, K.; Rui, H.; Beckner, R.L.; Srivastava, P.; Sukumar, N.; Roux, B.; Lewis, J.C. Crystal Structure and Conformational Dynamics of Pyrococcus furiosus Prolyl Oligopeptidase. Biochemistry 2019, 58, 1616–1626. [Google Scholar] [CrossRef]
- Canning, P.; Rea, D.; Morty, R.E.; Fülöp, V. Crystal Structures of Trypanosoma brucei Oligopeptidase B Broaden the Paradigm of Catalytic Regulation in Prolyl Oligopeptidase Family Enzymes. PLoS ONE 2013, 8, e79349. [Google Scholar] [CrossRef] [PubMed]
- McLuskey, K.; Paterson, N.G.; Bland, N.D.; Isaacs, N.W.; Mottram, J. Crystal Structure of Leishmania major Oligopeptidase B Gives Insight into the Enzymatic Properties of a Trypanosomatid Virulence Factor. J. Biol. Chem. 2010, 285, 39249–39259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhailova, A.G.; Rakitina, T.V.; Timofeev, V.I.; Karlinsky, D.M.; Korzhenevskiy, D.A.; Agapova, Y.; Vlaskina, A.V.; Ovchinnikova, M.V.; Gorlenko, V.A.; Rumsh, L.D. Activity modulation of the oligopeptidase B from Serratia proteamaculans by site-directed mutagenesis of amino acid residues surrounding catalytic triad histidine. Biochimie 2017, 139, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, D.E.; Mikhailova, A.G.; Timofeev, V.I.; Agapova, Y.; Karlinsky, D.M.; Komolov, A.S.; Korzhenevskiy, D.A.; Vlaskina, A.V.; Rumsh, L.D.; Rakitina, T.V. Molecular dynamics complemented by site-directed mutagenesis reveals significant difference between the interdomain salt bridge networks stabilizing oligopeptidases B from bacteria and protozoa in their active conformations. J. Biomol. Struct. Dyn. 2020, 38, 4868–4882. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.-B.; Wang, G.-Q.; Du, P.; Zhu, D.-X.; Wang, M.-W.; Jiang, X.-Y. High-level expression and purification of Escherichia coli oligopeptidase B. Protein Expr. Purif. 2006, 47, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Morty, R.E.; Fülöp, V.; Andrews, N.W. Substrate Recognition Properties of Oligopeptidase B from Salmonella enterica Serovar Typhimurium. J. Bacteriol. 2002, 184, 3329–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhailova, A.G.; Khairullin, R.F.; Demidyuk, I.V.; Kostrov, S.V.; Grinberg, N.V.; Burova, T.V.; Grinberg, V.Y.; Rumsh, L.D. Cloning, sequencing, expression, and characterization of thermostability of oligopeptidase B from Serratia proteamaculans, a novel psychrophilic protease. Protein Expr. Purif. 2014, 93, 63–76. [Google Scholar] [CrossRef]
- Boyko, K.M.; Rakitina, T.V.; Korzhenevskiy, D.A.; Vlaskina, A.V.; Agapova, Y.K.; Kamashev, D.E.; Kleymenov, S.Y.; Popov, V. Structural basis of the high thermal stability of the histone-like HU protein from the mollicute Spiroplasma melliferum KC3. Sci. Rep. 2016, 6, 36366. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, D.E.; Nikolaeva, A.Y.; Lazarenko, V.A.; Dorovatovskii, P.V.; Timofeev, V.; Vlaskina, A.V.; Korzhenevskiy, D.A.; Mikhailova, A.G.; Rakitina, T.V. Screening of Conditions that Facilitate Crystallization of Oligopeptidase B from Serratia Proteamaculans by Differential Scanning Fluorimetry. Crystallogr. Rep. 2020, 65, 264–268. [Google Scholar] [CrossRef]
- Petrenko, D.E.; Nikolaeva, A.Y.; Lazarenko, V.A.; Dorovatovskiy, P.V.; Timofeev, V.I.; Vlaskina, A.V.; Korzhenevskiy, D.A.; Mikhailova, A.G.; Boyko, K.M.; Rakitina, T.V. Crystallographic Study of Mutants and Complexes of Oligopeptidase B from Serratia proteamaculans. Crystallogr. Rep. 2020, 65, 909–914. [Google Scholar] [CrossRef]
- Long, F.; Vagin, A.A.; Young, P.; Murshudov, G.N. BALBES: A molecular-replacement pipeline. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.; Cowtan, K.D. Features and development of Coot. Acta Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Collaborative Computational Project Number 4. The CCP4 suite: Programs for protein crystallography. Acta Cryst. 1994, 50, 760–763. [Google Scholar] [CrossRef]
- Holm, L. Using Dali for Protein Structure Comparison. Methods Mol. Biol. 2020, 2112, 29–42. [Google Scholar] [CrossRef]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef]
- Hopkins, J.B.; Gillilan, R.E.; Skou, S. BioXTAS RAW: Improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 2017, 50, 1545–1553. [Google Scholar] [CrossRef] [Green Version]
- Svergun, D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Rambo, R.P.; Tainer, J. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 2013, 496, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I. Restoring Low Resolution Structure of Biological Macromolecules from Solution Scattering Using Simulated Annealing. Biophys. J. 1999, 76, 2879–2886. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.; Sali, A. Accurate SAXS Profile Computation and its Assessment by Contrast Variation Experiments. Biophys. J. 2013, 105, 962–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL– a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Mikhailova, A.G.; Nekrasov, A.N.; Zinchenko, A.A.; Rakitina, T.V.; Korzhenevsky, D.A.; Lipkin, A.V.; Razguljaeva, O.A.; Ovchinnikova, M.V.; Gorlenko, V.A.; Rumsh, L.D. Truncated variants of Serratia proteamaculans oligopeptidase B having different activities. Biochemestry (Moscow) 2015, 80, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikova, M.V.; Mikhailova, A.G.; Karlinsky, D.M.; Gorlenko, V.A.; Rumsh, L.D. Reversible Cyclic Thermal Inactivation of Oligopeptidase B from Serratia proteamaculans. Acta Nat. 2018, 10, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Farhadian, S.; Shareghi, B.; Saboury, A.A. Exploring the thermal stability and activity of α-chymotrypsin in the presence of spermine. J. Biomol. Struct. Dyn. 2016, 35, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, J.; Ismail, N.I.M.; Kubo, M.; Kinoshita, K.; Inoue, M.; Yuasa, K.; Nishimoto, M.; Matsuki, H.; Tsuji, A. Possible role of inter-domain salt bridges in oligopeptidase B from Trypanosoma brucei: Critical role of Glu172 of non-catalytic -propeller domain in catalytic activity and Glu490 of catalytic domain in stability of OPB. J. Biochem. 2013, 154, 465–473. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB ID Proteins | 7OB1 PSPmod | 7NE5 PSPmodS532A | 7NE4 PSPmodE12A |
|---|---|---|---|
| Data collection | |||
| Diffraction source | K4.4 beamline, NRC “Kurchatov Institute” | K4.4 beamline, NRC “Kurchatov Institute” | K4.4 beamline, NRC “Kurchatov Institute” |
| Wavelength (Å) | 0.79272 | 0.79272 | 0.79272 |
| Temperature (K) | 100 | 100 | 100 |
| Detector | CCD | CCD | CCD |
| Space group | P212121 | P212121 | P212121 |
| a, b, c (Å) | 73.21; 101.02; 108.89 | 70.71, 100.40, 108.67 | 68.84, 98.56, 108.26 |
| α, β, γ (°) | 90.0 | 90.0 | 90.0 |
| Unique reflections | 55364 (3999) | 63282 (4622) | 20453 (1476) |
| Resolution range (Å) | 20.0–2.00 (2.10–2.00) | 47.8–1.88 (1.93–1.88) | 44.9–2.72 (2.79–2.72) |
| Completeness (%) | 99.90 (99.89) | 99.80 (99.78) | 99.92 (99.86) |
| Average redundancy | 7.84 (4.22) | 7.25 (4.31) | 6.18 (5.96) |
| 〈I/σ(I)〉 | 23.3 (5.45) | 10.15 (2.09) | 8.45 (2.11) |
| Rmrgd-F (%) | 5.2 (26) | 4.9 (31) | 6.1 (29) |
| Refinement | |||
| Rfact (%) | 20.8 | 20.9 | 25.2 |
| Rfree.(%) | 24.9 | 25.2 | 30.5 |
| Bonds (Å) | 0.01 | 0.01 | 0.004 |
| Angles (°) | 1.63 | 1.63 | 1.02 |
| Ramachandran plot | |||
| Most favoured (%) | 99.2 | 99.2 | 99.2 |
| Allowed (%) | 0.8 | 0.8 | 0.8 |
| No. atoms | |||
| Protein | 5545 | 5534 | 5531 |
| Water | 216 | 386 | 50 |
| Ligands | 70 | 28 | 42 |
| B-factor (Å2) | 28.432 | 29.393 | 28.828 |
| Enzyme | BAPNA | Z-RR-pNA | Z-KR-pNA | ||||||
|---|---|---|---|---|---|---|---|---|---|
| kcat min−1 | Km µM | kcat/Km × 10−7 M−1 min−1 | kcat min−1 | Km µM | kcat/Km × 10−7 M−1 min−1 | kcat min−1 | Km µM | kcat/Km × 10−7 M−1min−1 | |
| PSPmod | 91.7 | 188.8 | 0.049 | 157.3 | 11.18 | 1.41 | 592.8 | 124.4 | 0.477 |
| PSPmod E75 | 603.1 | 165.5 | 0.364 | 960 | 34.65 | 2.77 | 805.7 | 24.0 | 3.36 |
| PSPmod E125A | 728.7 | 265.6 | 0.274 | 424.0 | 5.92 | 8.15 | 493.3 | 11.6 | 4.25 |
| PSP | 791.2 | 78.1 | 1.01 | 2181.2 | 4.35 | 50.1 | 3045.6 | 20.8 | 14.6 |
| PDB ID | 7OB1 | 4BP8 | 4BP9 | 3IUL | 3IVM | 5N4F | 5N4C | 5T88 |
|---|---|---|---|---|---|---|---|---|
| Conformation | Interm. | Open | Closed | Open | Closed | Open | Interm. | Interm. |
| Protein | PSP | TbOpB | ApPEP | GmPEP | PfPEP | |||
| Residues # (in the crystal structure)* | 677 | 712 | 710 | 669 | 682 | 703 | 720 | 618 |
| Aligned res. #* | 677 | 668 | 665 | 605 | 650 | 517 | 659 | 600 |
| Z-score * | 61.8 | 44.0 | 46.3 | 42.5 | 41.1 | 39.6 | 41.6 | 37.8 |
| Identity, %* | 100 | 37 | 38 | 27 | 27 | 22 | 21 | 22 |
| RMSD, Å* | 0 | 3.8 | 2.2 | 4.5 | 2.8 | 4.0 | 2.6 | 3.0 |
| Catalytic Ser-His Cα-distance, Å | 18.2 | 18.5 | 8.3 | N/a ** | 8.3 | N/a ** | 15.6 | 23.6 |
| Cat. S-OG Cat. H-NE2 distance, Å | 13.9 | 18.3 | 3.5 | N/a ** | 3.3 | N/a ** | N/a *** | 17.4 |
| Catalytic Asp-His Cα-distance, Å | 10.6 | 7.6 | 4.5 | N/a ** | 4.5 | N/a ** | 8.4 | 10.9 |
| Cat. D-OD2 Cat. H-ND1, distance, Å | 9.0 | 11.8 | 3.1 | N/a ** | 2.9 | N/a ** | 10.6 | 7.0 |
| Center of mass distance, Å | 32.3 | 36.7 | 30.4 | 38.7 | 30.7 | 39.4 | 32.0 | 30.9 |
| Buried surface area, cat./prop. domain, %1 | 11.3/9.4 | 8.4/7.5 | 14.0/12.3 | 8.1/7.7 | 16.9/14.6 | 7.5/7.0 | 13.5/11.4 | 12.2/11.5 |
| Interfaceresidues, cat./prop. domain, %2 | 16.3/15.9 | 10.3/10.5 | 17.4/16.9 | 12.1/7.7 | 22.4/19.5 | 11.0/6.3 | 19.0/15.2 | 18.5/15.1 |
| ΔiG, kcal/M | −12.9 | −8.9 | −17.8 | −20.8 | −23.6 | −16.3 | −24.3 | −23 |
| Hydrogen bonds | 11 | 14 | 28 | 7 | 22 | 9 | 25 | 16 |
| Salt Bridges | 4 | 4 | 4 | - | 2 | - | 1 | 1 |
| Proteins | Rg (Å) (Guinier Approximation) | Dmax (Å) (P(r) Function) | Volume-of-Correlation V(c) |
|---|---|---|---|
| PSP | 27.4 | 80 | 433 |
| PSP-Sp | 27.2 | 76 | 434 |
| PSPmodE125A | 26.5 | 80 | 397 |
| PSPmod | 25.9 | 79 | 422 |
| Proteins | χ2 7OB1 (FOXS/CRYSOL) | χ2 Open | χ2 Close |
|---|---|---|---|
| PSP | 98.8/65.8 | 6.1 (6.2) | 143.0 (122.9) |
| PSP-Sp | 25.6/7.8 | 48.1 (51.1) | 56.3 (37.6) |
| PSPmodE125A | 25.1/14.3 | 18.5 (23.2) | 51.6 (36.0) |
| PSPmod | 124.1/76.1 | 25.6 (30.3) | 205.0 (163.8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrenko, D.E.; Timofeev, V.I.; Britikov, V.V.; Britikova, E.V.; Kleymenov, S.Y.; Vlaskina, A.V.; Kuranova, I.P.; Mikhailova, A.G.; Rakitina, T.V. First Crystal Structure of Bacterial Oligopeptidase B in an Intermediate State: The Roles of the Hinge Region Modification and Spermine. Biology 2021, 10, 1021. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10101021
Petrenko DE, Timofeev VI, Britikov VV, Britikova EV, Kleymenov SY, Vlaskina AV, Kuranova IP, Mikhailova AG, Rakitina TV. First Crystal Structure of Bacterial Oligopeptidase B in an Intermediate State: The Roles of the Hinge Region Modification and Spermine. Biology. 2021; 10(10):1021. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10101021
Chicago/Turabian StylePetrenko, Dmitry E., Vladimir I. Timofeev, Vladimir V. Britikov, Elena V. Britikova, Sergey Y. Kleymenov, Anna V. Vlaskina, Inna P. Kuranova, Anna G. Mikhailova, and Tatiana V. Rakitina. 2021. "First Crystal Structure of Bacterial Oligopeptidase B in an Intermediate State: The Roles of the Hinge Region Modification and Spermine" Biology 10, no. 10: 1021. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10101021