
Epigallocatechin-3-Gallate (EGCG): New Therapeutic Perspectives for Neuroprotection, Aging, and Neuroinflammation for the Modern Age

 , and
, and
Abstract
:1. Introduction
2. Neurodegeneration: Examination of Two Prevalent Neuroregressive Disorders
2.1. Parkinson’s Disease (PD)
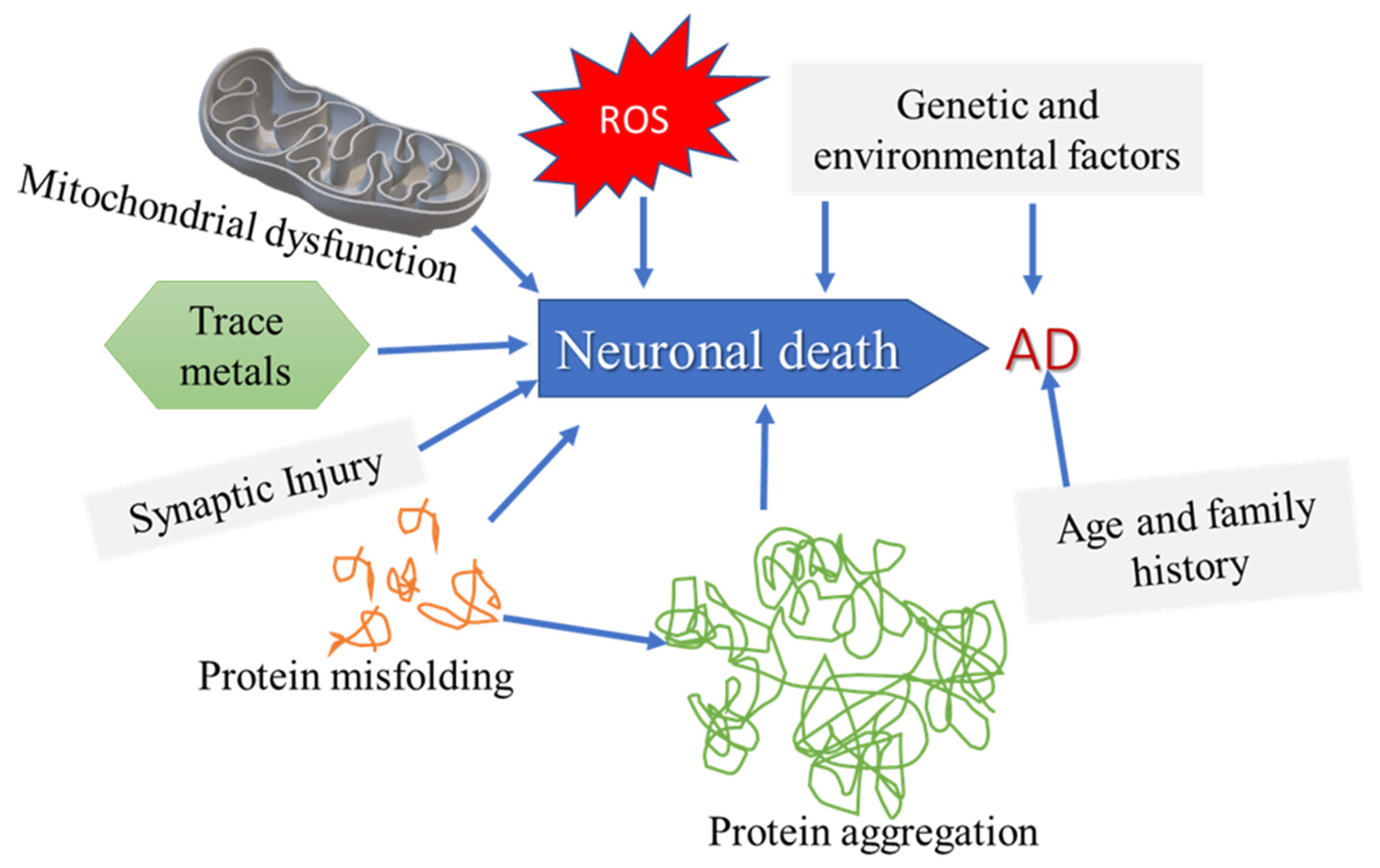
2.2. Alzheimer’s Disease (AD)
2.3. Protein Aggregation in AD: The Role of Amyloid Beta (Aβ)
2.4. Tau Protein: Neurofibrillary Tangles and Aggregation in AD
3. Role of Inflammation
3.1. Microglia and the Debilitating Effects Caused by Aging
3.2. Microglia and Tau
4. Neuroinflammatory Mediators
4.1. Oxidative Stress
4.2. Nitric Oxide
4.3. Autophagy
5. Inflammatory Signaling Pathways Involved in Neurodegeneration
6. Contributing Determinants to the Incidence of AD: Health Disparities, Sex, and Gender
7. Preventive Measures for Neurodegenerative Disease: Nutraceutical and Phytochemical Use in Neurodegenerative Disease
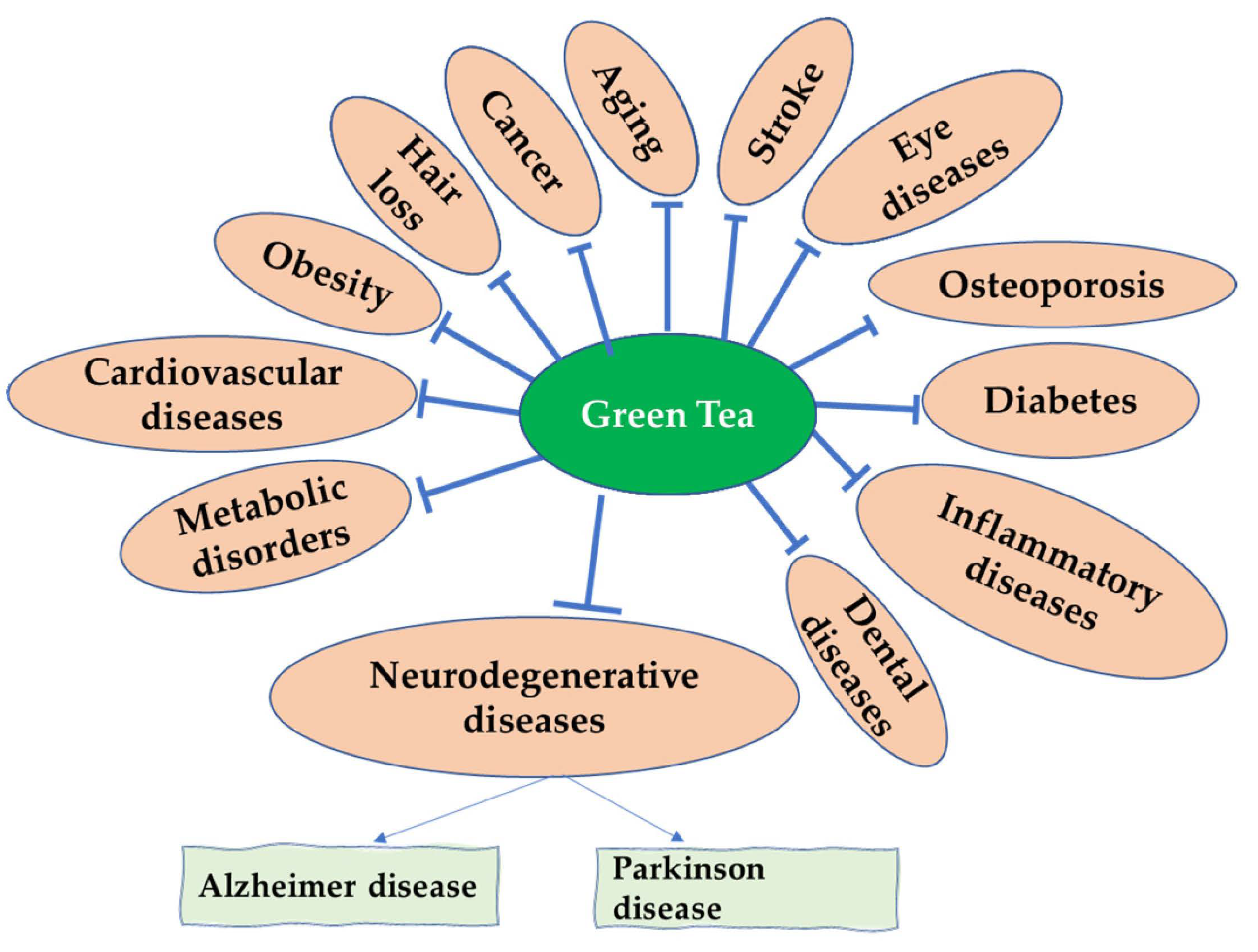
Green Tea and Its Derivatives: Therapeutic Actions Related to Inflammation and Neurodegeneration
8. EGCG Synthesis, Structure, and Therapeutic Action
EGCG Bioavailability and Modifications
9. Epigallocatechin-3-Gallate (EGCG) Therapeutic Action in AD and PD
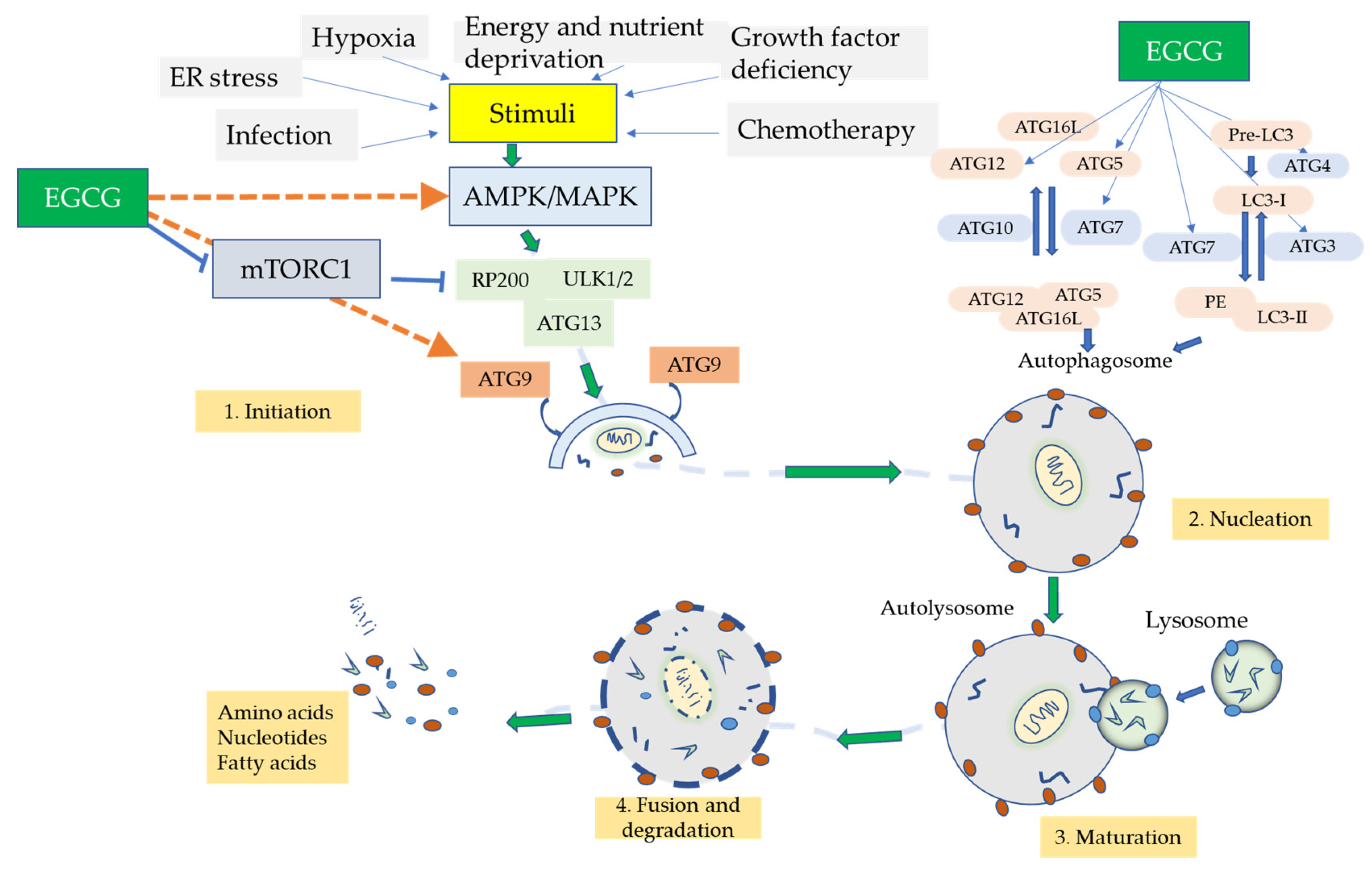
10. Autophagic Role of EGCG
11. Other EGCG Therapeutic Applications for Neuroinflammation and Neurorescue in Other Related Pathological Disorders
Role of microRNA (miRNA) in AD: Medicinal Action by EGCG
12. EGCG in Clinical Studies of AD and PD
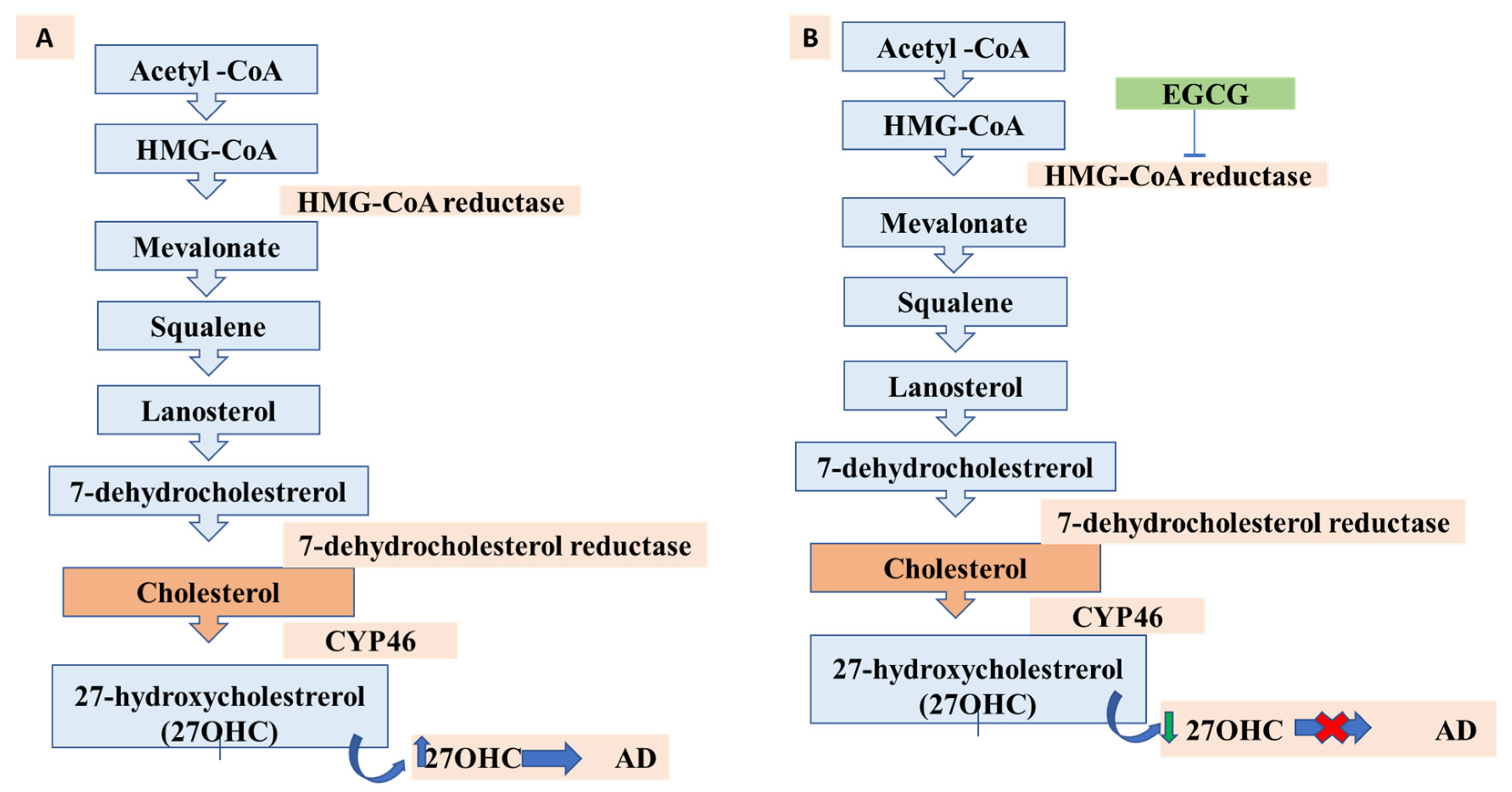
13. Lipids, Cholesterol Metabolism, and AD: A New Possibility for EGCG?
14. Future Directions
15. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Neurological Disorders: Public Health Challenges; World Health Organization: Geneva, Switzerland, 2006. [Google Scholar]
- Maresova, P.; Hruska, J.; Klimova, B.; Barakovic, S.; Krejcar, O. Activities of Daily Living and Associated Costs in the Most Widespread Neurodegenerative Diseases: A Systematic Review. Clin. Interv. Aging 2020, 15, 1841–1862. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Greenamyre, J.T. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol. Sci. 2011, 124, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J. Natural compounds may open new routes to treatment of amyloid diseases. Neurotherapeutics 2013, 10, 429–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shal, B.; Ding, W.; Ali, H.; Kim, Y.S.; Khan, S. Anti-neuroinflammatory Potential of Natural Products in Attenuation of Alzheimer’s Disease. Front. Pharmacol. 2018, 9, 548. [Google Scholar] [CrossRef]
- Sternke-Hoffmann, R.; Peduzzo, A.; Bolakhrif, N.; Haas, R.; Buell, A.K. The Aggregation Conditions Define Whether EGCG is an Inhibitor or Enhancer of α-Synuclein Amyloid Fibril Formation. Int. J. Mol. Sci. 2020, 21, 1995. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, S.; Safia; Haque, E.; Mir, S.S. Neurodegenerative Diseases: Multifactorial Conformational Diseases and Their Therapeutic Interventions. J. Neurodegener. Dis. 2013, 2013, 563481. [Google Scholar] [CrossRef] [Green Version]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Jung, U.J.; Kim, S.R. Beneficial Effects of Flavonoids Against Parkinson’s Disease. J. Med. Food 2018, 21, 421–432. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Pang, S.Y.; Ho, P.W.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef]
- Badanjak, K.; Fixemer, S.; Smajić, S.; Skupin, A.; Grünewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Chun, W.; Lee, H.J.; Kim, S.M.; Min, J.H.; Kim, D.Y.; Kim, M.O.; Ryu, H.W.; Lee, S.U. The Role of Microglia in the Development of Neurodegenerative Diseases. Biomedicines 2021, 9, 1449. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Yang, D.; Li, X.Q.; Liu, J.; Back, T.C.; Trivett, A.; Karim, B.; Barbut, D.; Zasloff, M.; Oppenheim, J.J. Alpha synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep. 2022, 38, 110090. [Google Scholar] [CrossRef]
- Booms, A.; Coetzee, G.A. Functions of Intracellular Alpha-Synuclein in Microglia: Implications for Parkinson’s Disease Risk. Front. Cell. Neurosci. 2021, 15, 759571. [Google Scholar] [CrossRef]
- Azam, S.; Haque, M.E.; Balakrishnan, R.; Kim, I.S.; Choi, D.K. The Ageing Brain: Molecular and Cellular Basis of Neurodegeneration. Front. Cell Dev. Biol. 2021, 9, 683459. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Stoker, T.B.; Barker, R.A. Recent developments in the treatment of Parkinson’s Disease. F1000Research 2020, 9. F1000 Faculty Rev-862. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Lines, L.; Sherif, N.; Wiener, J. Racial and Ethnic Disparities Among Individuals with Alzheimer’s Disease in the United States: A Literature Review; RTI Press: Durham, NC, USA, 2014. [Google Scholar] [CrossRef] [Green Version]
- Tanzi, R.E.; Bertram, L. New frontiers in Alzheimer’s disease genetics. Neuron 2001, 32, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 2011, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, C.; Toombs, J.; Lovejoy, C.; Ryan, N.S.; Paterson, R.W.; Willumsen, N.; Gkanatsiou, E.; Portelius, E.; Blennow, K.; Heslegrave, A.; et al. Familial Alzheimer’s disease patient-derived neurons reveal distinct mutation-specific effects on amyloid beta. Mol. Psychiatry 2020, 25, 2919–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, M.; Holm, M.L.; Liu, C.C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Liu, C.; Cui, G.; Zhu, M.; Kang, X.; Guo, H. Neuroinflammation in Alzheimer’s disease: Chemokines produced by astrocytes and chemokine receptors. Int. J. Clin. Exp. Pathol. 2014, 7, 8342–8355. [Google Scholar]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Cummings, J.L.; Goldman, D.P.; Simmons-Stern, N.R.; Ponton, E. The costs of developing treatments for Alzheimer’s disease: A retrospective exploration. Alzheimers Dement. 2021, in press. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Vicente, P.; Passarinha, L.A.; Silvestre, S.; Gallardo, E. Recent Developments in New Therapeutic Agents against Alzheimer and Parkinson Diseases: In-Silico Approaches. Molecules 2021, 26, 2193. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, T.; Fukutomi, R.; Shoji, Y.; Goto, S.; Isemura, M. The Beneficial Effects of Principal Polyphenols from Green Tea, Coffee, Wine, and Curry on Obesity. Molecules 2021, 26, 453. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Eroli, F.; Garcia-Ptacek, S.; Maioli, S. Brain Renin-Angiotensin System as Novel and Potential Therapeutic Target for Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 10139. [Google Scholar] [CrossRef]
- Mani, S.; Swargiary, G.; Singh, M.; Agarwal, S.; Dey, A.; Ojha, S.; Jha, N.K. Mitochondrial defects: An emerging theranostic avenue towards Alzheimer’s associated dysregulations. Life Sci. 2021, 285, 119985. [Google Scholar] [CrossRef]
- Alasmari, F.; Alshammari, M.A.; Alasmari, A.F.; Alanazi, W.A.; Alhazzani, K. Neuroinflammatory Cytokines Induce Amyloid Beta Neurotoxicity through Modulating Amyloid Precursor Protein Levels/Metabolism. BioMed Res. Int. 2018, 2018, 3087475. [Google Scholar] [CrossRef]
- Zinser, E.G.; Hartmann, T.; Grimm, M.O. Amyloid beta-protein and lipid metabolism. Biochim. Biophys. Acta 2007, 1768, 1991–2001. [Google Scholar] [CrossRef] [Green Version]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef]
- Currais, A.; Quehenberger, O.; Armando, A.M.; Daugherty, D.; Maher, P.; Schubert, D. Amyloid proteotoxicity initiates an inflammatory response blocked by cannabinoids. NPJ Aging Mech. Dis. 2016, 2, 16012. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, A.; Mela, V.; Harty, C.; Minogue, A.M.; Costello, D.A.; Kerskens, C.; Lynch, M.A. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. 2019, 29, 606–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-L.; Fan, Y.-G.; Yang, Z.-S.; Wang, Z.-Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef] [Green Version]
- Pîrşcoveanu, D.F.V.; Pirici, I.; Tudorică, V.; Bălşeanu, T.A.; Albu, V.C.; Bondari, S.; Bumbea, A.M.; Pîrşcoveanu, M. Tau protein in neurodegenerative diseases—A review. Rom. J. Morphol. Embryol. 2017, 58, 1141–1150. [Google Scholar] [PubMed]
- Jouanne, M.; Rault, S.; Voisin-Chiret, A.S. Tau protein aggregation in Alzheimer’s disease: An attractive target for the development of novel therapeutic agents. Eur. J. Med. Chem. 2017, 139, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gao, F.; Wang, D.; Li, C.; Fu, Y.; He, W.; Zhang, J. Tau Pathology in Parkinson’s Disease. Front. Neurol. 2018, 9, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef] [Green Version]
- Yanamadala, V.; Friedlander, R.M. Complement in neuroprotection and neurodegeneration. Trends Mol. Med. 2010, 16, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Dalakas, M.C.; Alexopoulos, H.; Spaeth, P.J. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat. Rev. Neurol. 2020, 16, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Schartz, N.D.; Tenner, A.J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflamm. 2020, 17, 354. [Google Scholar] [CrossRef]
- Gruver, A.L.; Hudson, L.L.; Sempowski, G.D. Immunosenescence of ageing. J. Pathol. 2007, 211, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, L.G.; Angelo, Y.S.; Iglesias, A.H.; Peron, J.P.S. Unraveling the Link Between Mitochondrial Dynamics and Neuroinflammation. Front. Immunol. 2021, 12, 624919. [Google Scholar] [CrossRef]
- Domercq, M.; Vázquez-Villoldo, N.; Matute, C. Neurotransmitter signaling in the pathophysiology of microglia. Front. Cell. Neurosci. 2013, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Doens, D.; Fernández, P.L. Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef] [Green Version]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]
- Zhao, X.; Eyo, U.B.; Murugan, M.; Wu, L.-J. Microglial interactions with the neurovascular system in physiology and pathology. Dev. Neurobiol. 2018, 78, 604–617. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Mputhia, Z.; Hone, E.; Tripathi, T.; Sargeant, T.; Martins, R.; Bharadwaj, P. Autophagy Modulation as a Treatment of Amyloid Diseases. Molecules 2019, 24, 3372. [Google Scholar] [CrossRef] [Green Version]
- Edler, M.K.; Mhatre-Winters, I.; Richardson, J.R. Microglia in Aging and Alzheimer’s Disease: A Comparative Species Review. Cells 2021, 10, 1138. [Google Scholar] [CrossRef] [PubMed]
- Leyns, C.E.G.; Holtzman, D.M. Glial contributions to neurodegeneration in tauopathies. Mol. Neurodegener. 2017, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; Bolós, M.; Avila, J. Microglia in Alzheimer’s Disease in the Context of Tau Pathology. Biomolecules 2020, 10, 1439. [Google Scholar] [CrossRef]
- Meraz-Ríos, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernández, J.; Campos-Peña, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Brasier, A.R. The nuclear factor-κB–interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 2010, 86, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Torres-Acosta, N.; O’Keefe, J.H.; O’Keefe, E.L.; Isaacson, R.; Small, G. Therapeutic Potential of TNF- α Inhibition for Alzheimer’s Disease Prevention. J. Alzheimer’s Dis. 2020, 78, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Steeland, S.; Libert, C.; Vandenbroucke, R.E. A New Venue of TNF Targeting. Int. J. Mol. Sci. 2018, 19, 1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizi, G.; Navabi, S.S.; Al-Shukaili, A.; Seyedzadeh, M.H.; Yazdani, R.; Mirshafiey, A. The Role of Inflammatory Mediators in the Pathogenesis of Alzheimer’s Disease. Sultan Qaboos Univ. Med. J. 2015, 15, e305–e316. [Google Scholar] [CrossRef] [PubMed]
- Tublin, J.M.; Adelstein, J.M.; Del Monte, F.; Combs, C.K.; Wold, L.E. Getting to the Heart of Alzheimer Disease. Circ. Res. 2019, 124, 142–149. [Google Scholar] [CrossRef]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Wallace, J.L. Nitric oxide as a regulator of inflammatory processes. Mem. Inst. Oswaldo Cruz. 2005, 100 (Suppl. S1), 5–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.-Y.; Hong, F.-F.; Yang, S.-L. The Roles of Nitric Oxide Synthase/Nitric Oxide Pathway in the Pathology of Vascular Dementia and Related Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 4540. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Tripathi, P.; Kashyap, L.; Singh, V. The role of nitric oxide in inflammatory reactions. FEMS Immunol. Med. Microbiol. 2007, 51, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C.; Bal-Price, A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol. Neurobiol. 2003, 27, 325–355. [Google Scholar] [CrossRef]
- Togo, T.; Katsuse, O.; Iseki, E. Nitric oxide pathways in Alzheimer’s disease and other neurodegenerative dementias. Neurol. Res. 2004, 26, 563–566. [Google Scholar] [CrossRef]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef]
- Stacchiotti, A.; Corsetti, G. Natural Compounds and Autophagy: Allies Against Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 555409. [Google Scholar] [CrossRef]
- Lapaquette, P.; Guzzo, J.; Bretillon, L.; Bringer, M.A. Cellular and Molecular Connections between Autophagy and Inflammation. Mediat. Inflamm. 2015, 2015, 398483. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Akkoç, Y.; L1g, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s Disease Pathogenesis: Role of Autophagy and Mitophagy Focusing in Microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ocak, U.; Gao, L.; Tu, S.; Lenahan, C.J.; Zhang, J.; Shao, A. Selective autophagy as a therapeutic target for neurological diseases. Cell. Mol. Life Sci. 2021, 78, 1369–1392. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140443. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Bhaumik, E.; Raychaudhuri, U.; Chakraborty, R. Role of nutraceuticals in human health. J. Food Sci. Technol. 2012, 49, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Tran, M.; Reddy, P.H. Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease. Front. Neurosci. 2021, 14, 1297. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Aslkhodapasandhokmabad, H.; Henninger, N.; Demillard, L.J.; Nikanfar, M.; Nourazarian, A.; Ren, J. Targeting autophagy in neurodegenerative diseases: From molecular mechanisms to clinical therapeutics. Clin. Exp. Pharmacol. Physiol. 2021, 48, 943–953. [Google Scholar] [CrossRef]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, V.; Sharma, S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 2021, 421, 117253. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Jones, S.V.; Kounatidis, I. Nuclear Factor-Kappa B and Alzheimer Disease, Unifying Genetic and Environmental Risk Factors from Cell to Humans. Front. Immunol. 2017, 8, 1805. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Lee, H.G.; Raina, A.K.; Perry, G.; Smith, M.A. The Role of Mitogen-Activated Protein Kinase Pathways in Alzheimer’s Disease. Neurosignals 2002, 11, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Kostenko, S.; Sveinbjørnsson, B. The Role of Mitogen-Activated Protein Kinase-Activated Protein Kinases (MAPKAPKs) in Inflammation. Genes 2013, 4, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef] [PubMed]
- Razani, E.; Pourbagheri-Sigaroodi, A.; Safaroghli-Azar, A.; Zoghi, A.; Shanaki-Bavarsad, M.; Bashash, D. The PI3K/Akt signaling axis in Alzheimer’s disease: A valuable target to stimulate or suppress? Cell Stress Chaperones 2021, 26, 871–887. [Google Scholar] [CrossRef]
- Emeny, R.T.; Carpenter, D.O.; Lawrence, D.A. Health disparities: Intracellular consequences of social determinants of health. Toxicol. Appl. Pharmacol. 2021, 416, 115444. [Google Scholar] [CrossRef]
- González, H.M.; Tarraf, W.; Fornage, M.; González, K.A.; Chai, A.; Youngblood, M.; Abreu, M.L.A.; Zeng, D.; Thomas, S.; Talavera, G.A.; et al. A research framework for cognitive aging and Alzheimer’s disease among diverse US Latinos: Design and implementation of the Hispanic Community Health Study/Study of Latinos-Investigation of Neurocognitive Aging (SOL-INCA). Alzheimers Dement. 2019, 15, 1624–1632. [Google Scholar] [CrossRef]
- McDonough, I.M. Beta-amyloid and Cortical Thickness Reveal Racial Disparities in Preclinical Alzheimer’s Disease. NeuroImage Clin. 2017, 16, 659–667. [Google Scholar] [CrossRef]
- Geronimus, A.T. The weathering hypothesis and the health of African-American women and infants: Evidence and speculations. Ethn. Dis. 1992, 2, 207–221. [Google Scholar]
- Powell, W.R.; Buckingham, W.R.; Larson, J.L.; Vilen, L.; Yu, M.; Salamat, M.S.; Bendlin, B.B.; Rissman, R.A.; Kind, A.J.H. Association of Neighborhood-Level Disadvantage With Alzheimer Disease Neuropathology. JAMA Netw. Open 2020, 3, e207559. [Google Scholar] [CrossRef]
- Saadi, A.; Himmelstein, D.U.; Woolhandler, S.; Mejia, N.I. Racial disparities in neurologic health care access and utilization in the United States. Neurology 2017, 88, 2268–2275. [Google Scholar] [CrossRef]
- Xiong, C.; Luo, J.; Coble, D.; Agboola, F.; Kukull, W.; Morris, J.C. Complex interactions underlie racial disparity in the risk of developing Alzheimer’s disease dementia. Alzheimers Dement. 2020, 16, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Yanguas-Casás, N. Sex Differences in Neurodegenerative Diseases. SM J. Neurol. Disord. Stroke 2017, 3, 1014. [Google Scholar]
- Hanamsagar, R.; Bilbo, S.D. Sex differences in neurodevelopmental and neurodegenerative disorders: Focus on microglial function and neuroinflammation during development. J. Steroid Biochem. Mol. Biol. 2016, 160, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrew, M.K.; Tierney, M.C. The puzzle of sex, gender and Alzheimer’s disease: Why are women more often affected than men? Women’s Health 2018, 14. [Google Scholar] [CrossRef] [Green Version]
- Laws, K.R.; Irvine, K.; Gale, T.M. Sex differences in cognitive impairment in Alzheimer’s disease. World J. Psychiatry 2016, 6, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Singh, M. Sex differences in cognitive impairment and Alzheimer’s disease. Front. Neuroendocrinol. 2014, 35, 385–403. [Google Scholar] [CrossRef] [Green Version]
- Bloomberg, M.; Dugravot, A.; Dumurgier, J.; Kivimaki, M.; Fayosse, A.; Steptoe, A.; Britton, A.; Singh-Manoux, A.; Sabia, S. Sex differences and the role of education in cognitive ageing: Analysis of two UK-based prospective cohort studies. Lancet Public Health 2021, 6, e106–e115. [Google Scholar] [CrossRef]
- Villa, A.; Vegeto, E.; Poletti, A.; Maggi, A. Estrogens, Neuroinflammation, and Neurodegeneration. Endocr. Rev. 2016, 37, 372–402. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, V.E.; Rizzi, L.; Bresciani, E.; Omeljaniuk, R.J.; Torsello, A. Androgen Therapy in Neurodegenerative Diseases. J. Endocr. Soc. 2020, 4, bvaa120. [Google Scholar] [CrossRef]
- Weber, C.; Clyne, A. Sex differences in the blood–brain barrier and neurodegenerative diseases. APL Bioeng. 2021, 5, 011509. [Google Scholar] [CrossRef]
- Lejri, I.; Grimm, A.; Eckert, A. Mitochondria, Estrogen and Female Brain Aging. Front. Aging Neurosci. 2018, 10, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zárate, S.; Stevnsner, T.; Gredilla, R. Role of Estrogen and Other Sex Hormones in Brain Aging. Neuroprotection and DNA Repair. Front. Aging Neurosci. 2017, 9, 430. [Google Scholar] [CrossRef] [Green Version]
- Horstman, A.M.; Dillon, E.L.; Urban, R.J.; Sheffield-Moore, M. The role of androgens and estrogens on healthy aging and longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 1140–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Lee, C.; Kodama, L.; Gan, L. Sex Differences in Neurodegeneration: The Role of the Immune System in Humans. Biol. Psychiatry 2022, 91, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Kodama, L.; Gan, L. Do Microglial Sex Differences Contribute to Sex Differences in Neurodegenerative Diseases? Trends Mol. Med. 2019, 25, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frölich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimers Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.-T.; Ghai, G.; Ho, C.-T. Inflammatory Process and Molecular Targets for Antiinflammatory Nutraceuticals. Compr. Rev. Food Sci. Food Saf. 2004, 3, 127–139. [Google Scholar] [CrossRef]
- Prof, D.; Gupta, C.; Sharma, P.G. Importance of Phytochemicals in Nutraceuticals. J. Chin. Med. Res. Dev. 2012, 1, 70–78. [Google Scholar]
- Costa, L.G.; Garrick, J.M.; Roquè, P.J.; Pellacani, C. Mechanisms of Neuroprotection by Quercetin: Counteracting Oxidative Stress and More. Oxid. Med. Cell. Longev. 2016, 2016, 2986796. [Google Scholar] [CrossRef] [Green Version]
- Spagnuolo, C.; Moccia, S.; Russo, G.L. Anti-inflammatory effects of flavonoids in neurodegenerative disorders. Eur. J. Med. Chem. 2018, 153, 105–115. [Google Scholar] [CrossRef]
- Sharangi, A.B. Medicinal and therapeutic potentialities of tea (Camellia sinensis L.)—A review. Food Res. Int. 2009, 42, 529–535. [Google Scholar] [CrossRef]
- Chowdhury, A.; Sarkar, J.; Chakraborti, T.; Pramanik, P.K.; Chakraborti, S. Protective role of epigallocatechin-3-gallate in health and disease: A perspective. Biomed. Pharmacother. 2016, 78, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.M.; Sousa, C. A Review on the Biological Activity of Camellia Species. Molecules 2021, 26, 2178. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Hu, J.; Hu, D.; Yang, X. A Role of Gallic Acid in Oxidative Damage Diseases: A Comprehensive Review. Nat. Prod. Commun. 2019, 14. [Google Scholar] [CrossRef] [Green Version]
- Kosuru, R.Y.; Roy, A.; Das, S.K.; Bera, S. Gallic Acid and Gallates in Human Health and Disease: Do Mitochondria Hold the Key to Success? Mol. Nutr. Food Res. 2018, 62, 1700699. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Koyama, N.; Yokoo, T.; Segawa, T.; Maeda, M.; Sawmiller, D.; Tan, J.; Town, T. Gallic acid is a dual α/β-secretase modulator that reverses cognitive impairment and remediates pathology in Alzheimer mice. J. Biol. Chem. 2020, 295, 16251–16266. [Google Scholar] [CrossRef]
- Andrade, S.; Loureiro, J.A.; Pereira, M.D.C. Influence of in vitro neuronal membranes on the anti-amyloidogenic activity of gallic acid: Implication for the therapy of Alzheimer’s disease. Arch. Biochem. Biophys. 2021, 711, 109022. [Google Scholar] [CrossRef]
- Chen, L.; Huang, G.L.; Lü, M.H.; Zhang, Y.X.; Xu, J.; Bai, S.P. Amide derivatives of Gallic acid: Design, synthesis and evaluation of inhibitory activities against in vitro α-synuclein aggregation. Bioorg. Med. Chem. 2020, 28, 115596. [Google Scholar] [CrossRef]
- Liu, Y.; Carver, J.A.; Calabrese, A.N.; Pukala, T.L. Gallic acid interacts with α-synuclein to prevent the structural collapse necessary for its aggregation. Biochim. Biophys. Acta 2014, 1844, 1481–1485. [Google Scholar] [CrossRef]
- Xu, Y.; Tang, G.; Zhang, C.; Wang, N.; Feng, Y. Gallic Acid and Diabetes Mellitus: Its Association with Oxidative Stress. Molecules 2021, 26, 7115. [Google Scholar] [CrossRef]
- Liu, Y.L.; Hsu, C.C.; Huang, H.J.; Chang, C.J.; Sun, S.H.; Lin, A.M. Gallic Acid Attenuated LPS-Induced Neuroinflammation: Protein Aggregation and Necroptosis. Mol. Neurobiol. 2020, 57, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ye, J.; Shi, R.; Zhao, B.; Liu, Z.; Lin, W.; Liu, X. Dietary protein and amino acid restriction: Roles in metabolic health and aging-related diseases. Free Radic. Biol. Med. 2022, 178, 226–242. [Google Scholar] [CrossRef]
- Hid, E.J.; Mosele, J.I.; Prince, P.D.; Fraga, C.G.; Galleano, M. (−)-Epicatechin and cardiometabolic risk factors: A focus on potential mechanisms of action. Pflug. Arch. 2022, 474, 99–115. [Google Scholar] [CrossRef]
- Cremonini, E.; Iglesias, D.E.; Kang, J.; Lombardo, G.E.; Mostofinejad, Z.; Wang, Z.; Zhu, W.; Oteiza, P.I. (−)-Epicatechin and the comorbidities of obesity. Arch. Biochem. Biophys. 2020, 690, 108505. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Liu, S.; Xu, J.; Li, W.; Duan, G.; Wang, H.; Yang, H.; Yang, Z.; Zhou, R. A new molecular mechanism underlying the EGCG-mediated autophagic modulation of AFP in HepG2 cells. Cell Death Dis. 2017, 8, e3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karas, D.; Ulrichová, J.; Valentová, K. Galloylation of polyphenols alters their biological activity. Food Chem. Toxicol. 2017, 105, 223–240. [Google Scholar] [CrossRef]
- Zwolak, I. Epigallocatechin Gallate for Management of Heavy Metal-Induced Oxidative Stress: Mechanisms of Action, Efficacy, and Concerns. Int. J. Mol. Sci. 2021, 22, 4027. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.A.; Genové, G.; Li, T.; Lütjohann, D.; Olin, M.; Mast, N.; Pikuleva, I.A.; Crick, P.; Wang, Y.; Griffiths, W.; et al. Effects of a disrupted blood-brain barrier on cholesterol homeostasis in the brain. J. Biol. Chem. 2014, 289, 23712–23722. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.Y.; Li, X.M.; Liang, J.P.; Xiang, L.P.; Wang, K.R.; Shi, Y.L.; Yang, R.; Shi, M.; Ye, J.H.; Lu, J.L.; et al. Bioavailability of Tea Catechins and Its Improvement. Molecules 2018, 23, 2346. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Ruan, C.; Zhang, Y.; Wang, J.; Han, J.; Shao, Z.; Sun, Y.; Liang, J. Bioavailability enhancement of EGCG by structural modification and nano-delivery: A review. J. Funct. Foods 2020, 65, 103732. [Google Scholar] [CrossRef]
- Pervin, M.; Unno, K.; Takagaki, A.; Isemura, M.; Nakamura, Y. Function of Green Tea Catechins in the Brain: Epigallocatechin Gallate and its Metabolites. Int. J. Mol. Sci. 2019, 20, 3630. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Kang, Z.; Yan, W. Synthesis, Stability, and Antidiabetic Activity Evaluation of (−)-Epigallocatechin Gallate (EGCG) Palmitate Derived from Natural Tea Polyphenols. Molecules 2021, 26, 393. [Google Scholar] [CrossRef] [PubMed]
- Granja, A.; Neves, A.R.; Sousa, C.T.; Pinheiro, M.; Reis, S. EGCG intestinal absorption and oral bioavailability enhancement using folic acid-functionalized nanostructured lipid carriers. Heliyon 2019, 5, e02020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaiserman, A.; Koliada, A.; Zayachkivska, A.; Lushchak, O. Nanodelivery of Natural Antioxidants: An Anti-aging Perspective. Front. Bioeng. Biotechnol. 2019, 7, 447. [Google Scholar] [CrossRef] [Green Version]
- Sneideris, T.; Sakalauskas, A.; Sternke-Hoffmann, R.; Peduzzo, A.; Ziaunys, M.; Buell, A.K.; Smirnovas, V. The Environment Is a Key Factor in Determining the Anti-Amyloid Efficacy of EGCG. Biomolecules 2019, 9, 855. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, L.; Cardim-Pires, T.R.; Foguel, D.; Palhano, F.L. Green Tea Polyphenol Epigallocatechin-Gallate in Amyloid Aggregation and Neurodegenerative Diseases. Front. Neurosci. 2021, 15, 718188. [Google Scholar] [CrossRef]
- Gonçalves, P.B.; Sodero, A.C.R.; Cordeiro, Y. Green Tea Epigallocatechin-3-gallate (EGCG) Targeting Protein Misfolding in Drug Discovery for Neurodegenerative Diseases. Biomolecules 2021, 11, 767. [Google Scholar] [CrossRef]
- Namita, P.; Mukesh, R.; Vijay, K. Camellia Sinensis (Green Tea): A Review. Glob. J. Pharmacol. 2012, 6, 52–59. [Google Scholar]
- Prasanth, M.I.; Sivamaruthi, B.S.; Chaiyasut, C.; Tencomnao, T. A Review of the Role of Green Tea (Camellia sinensis) in Antiphotoaging, Stress Resistance, Neuroprotection, and Autophagy. Nutrients 2019, 11, 474. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.C.; Wang, M.H.; Chang, K.C.; Soung, H.S.; Fang, C.H.; Lin, Y.W.; Li, K.Y.; Yang, C.C.; Tsai, C.C. Protective Effect of (−)Epigallocatechin-3-gallate on Rotenone-Induced Parkinsonism-like Symptoms in Rats. Neurotox. Res. 2020, 37, 669–682. [Google Scholar] [CrossRef]
- Guéroux, M.; Fleau, C.; Slozeck, M.; Laguerre, M.; Pianet, I. Epigallocatechin 3-Gallate as an Inhibitor of Tau Phosphorylation and Aggregation: A Molecular and Structural Insight. J. Prev. Alzheimers Dis. 2017, 4, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Ziaunys, M.; Mikalauskaite, K.; Sakalauskas, A.; Smirnovas, V. Interplay between epigallocatechin-3-gallate and ionic strength during amyloid aggregation. PeerJ 2021, 9, e12381. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, S.K.; Chidambaram, H.; Boral, D.; Gorantla, N.V.; Balmik, A.A.; Dangi, A.; Ramasamy, S.; Marelli, U.K.; Chinnathambi, S. EGCG impedes human Tau aggregation and interacts with Tau. Sci. Rep. 2020, 10, 12579. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.N.; Kwon, H.J.; Akindehin, S.; Jeong, H.W.; Lee, Y.H. Effects of Epigallocatechin-3-Gallate on Autophagic Lipolysis in Adipocytes. Nutrients 2017, 9, 680. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Chen, Y.; Wang, J.; Qiu, J.; Chang, C.; Bi, F.; Wu, X.; Liu, W. EGCG protects vascular endothelial cells from oxidative stress-induced damage by targeting the autophagy-dependent PI3K-AKT-mTOR pathway. Ann. Transl. Med. 2020, 8, 200. [Google Scholar] [CrossRef]
- Holczer, M.; Besze, B.; Zámbó, V.; Csala, M.; Bánhegyi, G.; Kapuy, O. Epigallocatechin-3-Gallate (EGCG) Promotes Autophagy-Dependent Survival via Influencing the Balance of mTOR-AMPK Pathways upon Endoplasmic Reticulum Stress. Oxid. Med. Cell. Longev. 2018, 2018, 6721530. [Google Scholar] [CrossRef] [Green Version]
- Khalil, H.; Tazi, M.; Caution, K.; Ahmed, A.; Kanneganti, A.; Assani, K.; Kopp, B.; Marsh, C.; Dakhlallah, D.; Amer, A.O. Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics 2016, 11, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Tasneem, S.; Liu, B.; Li, B.; Choudhary, M.I.; Wang, W. Molecular pharmacology of inflammation: Medicinal plants as anti-inflammatory agents. Pharmacol. Res. 2019, 139, 126–140. [Google Scholar] [CrossRef]
- Liang, Y.; Ip, M.S.M.; Mak, J.C.W. (−)-Epigallocatechin-3-gallate suppresses cigarette smoke-induced inflammation in human cardiomyocytes via ROS-mediated MAPK and NF-κB pathways. Phytomedicine 2019, 58, 152768. [Google Scholar] [CrossRef]
- Lakshmi, S.P.; Reddy, A.T.; Kodidhela, L.D.; Varadacharyulu, N.C. Epigallocatechin gallate diminishes cigarette smoke-induced oxidative stress, lipid peroxidation, and inflammation in human bronchial epithelial cells. Life Sci. 2020, 259, 118260. [Google Scholar] [CrossRef]
- Huang, S.C.; Kao, Y.H.; Shih, S.F.; Tsai, M.C.; Lin, C.S.; Chen, L.W.; Chuang, Y.P.; Tsui, P.F.; Ho, L.J.; Lai, J.H.; et al. Epigallocatechin-3-gallate exhibits immunomodulatory effects in human primary T cells. Biochem. Biophys. Res. Commun. 2021, 550, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kang, H.G.; Nam, S.Y.; Kim, H.M.; Jeong, H.J. Blockade of RANKL/RANK signaling pathway by epigallocatechin gallate alleviates mast cell-mediated inflammatory reactions. Int. Immunopharmacol. 2020, 88, 106872. [Google Scholar] [CrossRef] [PubMed]
- Karatas, A.; Dagli, A.F.; Orhan, C.; Gencoglu, H.; Ozgen, M.; Sahin, N.; Sahin, K.; Koca, S.S. Epigallocatechin 3-gallate attenuates arthritis by regulating Nrf2, HO-1, and cytokine levels in an experimental arthritis model. Biotechnol. Appl. Biochem. 2020, 67, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Abib, R.T.; Peres, K.C.; Barbosa, A.M.; Peres, T.V.; Bernardes, A.; Zimmermann, L.M.; Quincozes-Santos, A.; Fiedler, H.D.; Leal, R.B.; Farina, M.; et al. Epigallocatechin-3-gallate protects rat brain mitochondria against cadmium-induced damage. Food Chem. Toxicol. 2011, 49, 2618–2623. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Meng, Z. Protective effects of epigallocatechin-3-gallate against lead-induced oxidative damage. Hum. Exp. Toxicol. 2011, 30, 1521–1528. [Google Scholar] [CrossRef]
- Wu, F.; Sun, H.; Kluz, T.; Clancy, H.A.; Kiok, K.; Costa, M. Epigallocatechin-3-gallate (EGCG) protects against chromate-induced toxicity in vitro. Toxicol. Appl. Pharmacol. 2012, 258, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Du, L.; Li, J.; Song, H. Epigallocatechin-3-gallate attenuates cadmium-induced chronic renal injury and fibrosis. Food Chem. Toxicol. 2016, 96, 70–78. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Tormos, A.M.; Pérez, S.; Taléns-Visconti, R. Vascular pathology: Cause or effect in Alzheimer disease? Neurologia 2018, 33, 112–120. [Google Scholar] [CrossRef]
- Dal Lin, C.; Tona, F.; Osto, E. The crosstalk between the cardiovascular and the immune system. Vasc. Biol. 2019, 1, H83–H88. [Google Scholar] [CrossRef] [Green Version]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Jimenez, M.; Vujacic-Mirski, K.; Helmstaedter, J.; Kröller-Schön, S.; Munzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Strickland, S. Blood will out: Vascular contributions to Alzheimer’s disease. J. Clin. Investig. 2018, 128, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klohs, J. An Integrated View on Vascular Dysfunction in Alzheimer’s Disease. Neurodegener. Dis. 2019, 19, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Riegsecker, S.; Wiczynski, D.; Kaplan, M.J.; Ahmed, S. Potential benefits of green tea polyphenol EGCG in the prevention and treatment of vascular inflammation in rheumatoid arthritis. Life Sci. 2013, 93, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Adinew, G.M.; Taka, E.; Mendonca, P.; Messeha, S.S.; Soliman, K.F.A. The Anticancer Effects of Flavonoids through miRNAs Modulations in Triple-Negative Breast Cancer. Nutrients 2021, 13, 1212. [Google Scholar] [CrossRef]
- Lee, C.Y.; Ryu, I.S.; Ryu, J.H.; Cho, H.J. miRNAs as Therapeutic Tools in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 13012. [Google Scholar] [CrossRef]
- Kou, X.; Chen, N. Resveratrol as a Natural Autophagy Regulator for Prevention and Treatment of Alzheimer’s Disease. Nutrients 2017, 9, 927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Wang, Z.Y.; Ma, L.N.; Zhang, T.T.; Cao, Y.; Li, H. MicroRNAs in Alzheimer’s Disease: Function and Potential Applications as Diagnostic Biomarkers. Front. Mol. Neurosci. 2020, 13, 160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Bian, Z. Alzheimer’s Disease and microRNA-132: A Widespread Pathological Factor and Potential Therapeutic Target. Front. Neurosci. 2021, 15, 687973. [Google Scholar] [CrossRef]
- Majidinia, M.; Karimian, A.; Alemi, F.; Yousefi, B.; Safa, A. Targeting miRNAs by polyphenols: Novel therapeutic strategy for aging. Biochem. Pharmacol. 2020, 173, 113688. [Google Scholar] [CrossRef]
- Cione, E.; La Torre, C.; Cannataro, R.; Caroleo, M.C.; Plastina, P.; Gallelli, L. Quercetin, Epigallocatechin Gallate, Curcumin, and Resveratrol: From Dietary Sources to Human MicroRNA Modulation. Molecules 2019, 25, 63. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Li, Y.; Su, B. Identification of Circulating miR-125b as a Potential Biomarker of Alzheimer’s Disease in APP/PS1 Transgenic Mouse. J. Alzheimers Dis. 2017, 59, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Gruendler, R.; Hippe, B.; Sendula Jengic, V.; Peterlin, B.; Haslberger, A.G. Nutraceutical Approaches of Autophagy and Neuroinflammation in Alzheimer’s Disease: A Systematic Review. Molecules 2020, 25, 6018. [Google Scholar] [CrossRef] [PubMed]
- Tzou, F.Y.; Wen, J.K.; Yeh, J.Y.; Huang, S.Y.; Chen, G.C.; Chan, C.C. Drosophila as a model to study autophagy in neurodegenerative diseases and digestive tract. IUBMB Life 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.X.; Li, Y.B.; Zhao, R.P. Epigallocatechin Gallate Attenuates β-Amyloid Generation and Oxidative Stress Involvement of PPARγ in N2a/APP695 Cells. Neurochem. Res. 2017, 42, 468–480. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Sureda, A.; Dehpour, A.R.; Shirooie, S.; Silva, A.S.; Devi, K.P.; Ahmed, T.; Ishaq, N.; Hashim, R.; Sobarzo-Sánchez, E.; et al. Regulation of autophagy by polyphenols: Paving the road for treatment of neurodegeneration. Biotechnol. Adv. 2018, 36, 1768–1778. [Google Scholar] [CrossRef]
- Dong, X.; Tang, Y.; Zhan, C.; Wei, G. Green tea extract EGCG plays a dual role in Aβ(42) protofibril disruption and membrane protection: A molecular dynamic study. Chem. Phys. Lipids 2021, 234, 105024. [Google Scholar] [CrossRef]
- Acharya, A.; Stockmann, J.; Beyer, L.; Rudack, T.; Nabers, A.; Gumbart, J.C.; Gerwert, K.; Batista, V.S. The Effect of (−)-Epigallocatechin-3-Gallate on the Amyloid-β Secondary Structure. Biophys. J. 2020, 119, 349–359. [Google Scholar] [CrossRef]
- Mao, L.; Hochstetter, D.; Yao, L.; Zhao, Y.; Zhou, J.; Wang, Y.; Xu, P. Green Tea Polyphenol (−)-Epigallocatechin Gallate (EGCG) Attenuates Neuroinflammation in Palmitic Acid-Stimulated BV-2 Microglia and High-Fat Diet-Induced Obese Mice. Int. J. Mol. Sci. 2019, 20, 5081. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Li, X.; Hu, X.; Xu, Q.; Zhang, Y.; Liu, H.; Diao, Y.; Zhang, X.; Li, L.; Yu, J.; et al. Epigallocatechin-3-gallate prevents inflammation and diabetes-induced glucose tolerance through inhibition of NLRP3 inflammasome activation. Int. Immunopharmacol. 2021, 93, 107412. [Google Scholar] [CrossRef]
- Huang, J.B.; Zhang, Y.; Zhou, Y.B.; Wan, X.C.; Zhang, J.S. Effects of epigallocatechin gallate on lipid metabolism and its underlying molecular mechanism in broiler chickens. J. Anim. Physiol. Anim. Nutr. 2015, 99, 719–727. [Google Scholar] [CrossRef]
- Unno, K.; Nakamura, Y. Green Tea Suppresses Brain Aging. Molecules 2021, 26, 4897. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.H.; Cui, Z.Y.; Yang, A.L.; Nhu, N.T.; Ting, S.Y.; Yu, S.H.; Cheng, Y.J.; Lin, Y.Y.; Wu, X.B.; Lee, S.D. Cerebral Cortex Apoptosis in Early Aged Hypertension: Effects of Epigallocatechin-3-Gallate. Front. Aging Neurosci. 2021, 13, 705304. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sharma, A.; Kumari, A.; Gulati, A.; Padwad, Y.; Sharma, R. Epigallocatechin gallate suppresses premature senescence of preadipocytes by inhibition of PI3K/Akt/mTOR pathway and induces senescent cell death by regulation of Bax/Bcl-2 pathway. Biogerontology 2019, 20, 171–189. [Google Scholar] [CrossRef]
- Aon, M.A.; Bhatt, N.; Cortassa, S.C. Mitochondrial and cellular mechanisms for managing lipid excess. Front. Physiol. 2014, 5, 282. [Google Scholar] [CrossRef] [Green Version]
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (per) oxidation in mitochondria: An emerging target in the ageing process? Biogerontology 2017, 18, 859–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, M.J.; Tchernof, A. Lipid metabolism in the elderly. Eur. J. Clin. Nutr. 2000, 54, S121–S125. [Google Scholar] [CrossRef] [PubMed]
- Shamim, A.; Mahmood, T.; Ahsan, F.; Kumar, A.; Bagga, P. Lipids: An insight into the neurodegenerative disorders. Clin. Nutr. Exp. 2018, 20, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Gamba, P.; Staurenghi, E.; Testa, G.; Giannelli, S.; Sottero, B.; Leonarduzzi, G. A Crosstalk Between Brain Cholesterol Oxidation and Glucose Metabolism in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 556. [Google Scholar] [CrossRef] [Green Version]
- Orth, M.; Bellosta, S. Cholesterol: Its Regulation and Role in Central Nervous System Disorders. Cholesterol 2012, 2012, 292598. [Google Scholar] [CrossRef] [Green Version]
- Kopecka, J.; Godel, M.; Riganti, C. Cholesterol metabolism: At the cross road between cancer cells and immune environment. Int. J. Biochem. Cell Biol. 2020, 129, 105876. [Google Scholar] [CrossRef]
- Björkhem, I.; Meaney, S. Brain cholesterol: Long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.G.; Pfrieger, F.; Dotti, C.G. Cholesterol in brain disease: Sometimes determinant and frequently implicated. EMBO Rep. 2014, 15, 1036–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, A.E.; Mooney, K.M.; Wilkinson, S.J.; Pickles, N.A.; Mc Auley, M.T. Cholesterol metabolism: A review of how ageing disrupts the biological mechanisms responsible for its regulation. Ageing Res. Rev. 2016, 27, 108–124. [Google Scholar] [CrossRef]
- Saeedi Saravi, S.S.; Saeedi Saravi, S.S.; Arefidoust, A.; Dehpour, A.R. The beneficial effects of HMG-CoA reductase inhibitors in the processes of neurodegeneration. Metab. Brain Dis. 2017, 32, 949–965. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, H.; Ghasemi, F.; Barreto, G.E.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. The effects of statins on microglial cells to protect against neurodegenerative disorders: A mechanistic review. Biofactors 2020, 46, 309–325. [Google Scholar] [CrossRef]
- Sethi, J.K.; Hotamisligil, G.S. The role of TNF alpha in adipocyte metabolism. Semin. Cell Dev. Biol. 1999, 10, 19–29. [Google Scholar] [CrossRef]
- Chen, X.; Xun, K.; Chen, L.; Wang, Y. TNF-alpha, a potent lipid metabolism regulator. Cell Biochem. Funct. 2009, 27, 407–416. [Google Scholar] [CrossRef]
- Loving, B.A.; Bruce, K.D. Lipid and Lipoprotein Metabolism in Microglia. Front. Physiol. 2020, 11, 393. [Google Scholar] [CrossRef]
- Estrada, L.D.; Ahumada, P.; Cabrera, D.; Arab, J.P. Liver Dysfunction as a Novel Player in Alzheimer’s Progression: Looking Outside the Brain. Front. Aging Neurosci. 2019, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Farah, B.L.; Sinha, R.A.; Wu, Y.; Singh, B.K.; Bay, B.H.; Yang, C.S.; Yen, P.M. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, stimulates hepatic autophagy and lipid clearance. PLoS ONE 2014, 9, e87161. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhu, C.; Liu, T.; Zhang, W.; Liu, X.; Li, P.; Zhu, T. Epigallocatechin-3-gallate ameliorates glucolipid metabolism and oxidative stress in type 2 diabetic rats. Diabetes Vasc. Dis. Res. 2020, 17, 1479164120966998. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, D.O.; Zhou, C.; Zhang, L. A Review on the Weight-Loss Effects of Oxidized Tea Polyphenols. Molecules 2018, 23, 1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, T.R.; Marsh, S.E.; Stevens, B. Immune Signaling in Neurodegeneration. Immunity 2019, 50, 955–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
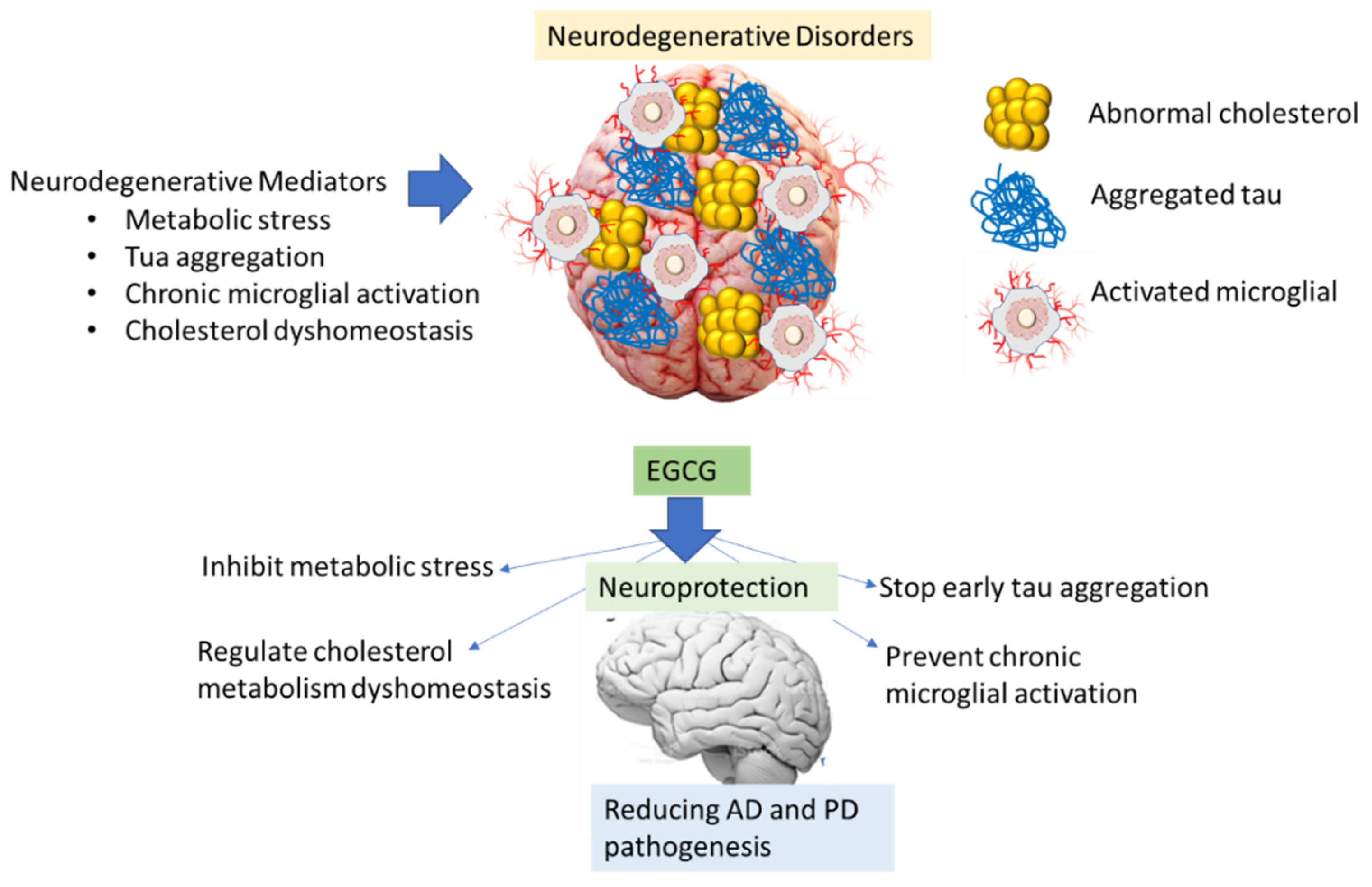{kind=link}
| Pharmacological Action | Model (In Vivo/In Vitro) | Therapeutic Focus | Reference |
|---|---|---|---|
| Anti-inflammatory | In vivo transgenic mice, human volunteers, Elegans, hamsters, Drosophila | A review of the role of green tea (Camellia sinensis) in antiphotoaging, stress resistance, neuroprotection, and autophagy | [161] |
| In vitro and in vivo airway epithelial cells, experimental autoimmune encephalomyelitis (EAE), rat aortas, RA human patients, human umbilical vein endothelial cells, and synovial fibroblasts | The prevention and treatment of vascular inflammation in rheumatoid arthritis | [185] | |
| In vivo: human | Molecular pharmacology of inflammation: medicinal plants as anti-inflammatory agents | [170] | |
| In vitro: adult human ventricular cardiomyocyte cell line AC16 | Suppresses cigarette smoking-induced inflammation in human cardiomyocytes, utilizing ROS-mediated MAPK and NF-κB pathways | [171] | |
| In vivo: Wistar albino female rats | Attenuates arthritis by regulating Nrf2, HO-1, and cytokine levels in an experimental arthritis model | [175] | |
| In vivo: Human mast cell line | Blockade of RANKL/RANK signaling pathway by EGCG alleviates mast cell-mediated inflammatory reactions | [174] | |
| Antioxidant/oxidative stress | In vitro: human bronchial epithelial cells | Diminishes cigarette smoke-induced oxidative stress, lipid peroxidation, and inflammation in human bronchial epithelial cells | [172] |
| In vitro: Single ventricular myocytes | Protective effects against lead-induced oxidative damage | [177] | |
| In vivo: Adult male Wistar albino rats | Attenuates cadmium-induced chronic renal injury and fibrosis | [179] | |
| multiple human and cell model systems | EGCG Management of Heavy Metal-Induced Oxidative Stress: Mechanisms of Action, Efficacy, and Concerns | [149] | |
| In vitro BEAS-2B cells and EBV-BL cells | Protects against chromate-induced toxicity in vitro | [178] | |
| In vivo Male 14-day-old Wistar rats | Protects rat brain mitochondria against cadmium-induced damage | [176] | |
| Amyloid-beta formation inhibition | In vivo human adult brain | Microglial dysfunction in brain aging and Alzheimer’s disease | [65] |
| In vitro N2a-APP695 cells | EGCG attenuates β-amyloid generation and oxidative stress involvement of PPARγ | [196] | |
| In vitro and in vivo Human embryonic kidney cells human CNS-derived neuroblastoma cells, APPHEK293 and SwAPP-N2a cells. Biomarkers of CSF, plasma, blood-derived brain exosomes, monocytes, and peripheral blood mononuclear cells | Autophagy modulation as a treatment of amyloid diseases | [197] | |
| In vitro hippocampal neurons, mouse, rat brain cells, and yeast model overexpressing htt | EGCG Amyloid Aggregation and Neurodegenerative Diseases | [158] | |
| In vivo mixed POPC/POPG (7:3) lipid bilayer | Plays a dual role in Aβ 42 protofibril disruption and membrane protection: A dynamic molecular study | [198] | |
| In vitro Synthetic Aβ42, Aβ fibril formulation | The effect of (−)-epigallocatechin-3-gallate on the amyloid-β secondary structure | [199] | |
| Autophagy | In vitro C3H10T1/2 cells and 3T3-L1 preadipocytes | Effects of EGCG on autophagic lipolysis in adipocytes | [166] |
| In vitro HEPG2 cells | A new molecular mechanism underlying the EGCG mediated autophagic modulation of AFP | [147] | |
| Human endothelial cells | Protects vascular endothelial cells from oxidative stress induced damage by targeting the autophagy dependent PI3K-Akt-mTOR pathway | [167] | |
| HEK293T cells | Promotes autophagy dependent survival via influencing the balance of mTOR -AMPK pathways upon endoplasmic reticulum stress | [168] | |
| Cholesterol/Lipid Metabolism | high-fat diet induced mouse obesity model, human volunteers, rats, | The beneficial effects of principal polyphenols from green tea, coffee, wine, and curry on obesity | [37] |
| BV2 cells and Twenty-four-week-old male C57BL/6J mice | Attenuates neuroinflammation in palmitic acid-stimulated bv-2 microglia and high-fat diet-induced obese mice | [200] | |
| C57BL/6 mice | prevents inflammation and diabetes -induced glucose tolerance through inhibition of NLRP3 inflammasome activation | [201] | |
| broiler chickens | Effects of EGCG on lipid metabolism and its underlying molecular mechanism | [202] | |
| Anti-Aging | SH-SY5Y cells, SAMP10, and ddy mice | A review of the role of green tea (Camellia sinensis) in antiphotoaging, stress resistance, neuroprotection, and autophagy | [161] |
| 36 weeks old, spontaneously hypertensive rats and male normotensive Wistar-Kyoto rats | Green tea suppresses brain aging | [203] | |
| 3T3-L1 preadipocytes | Cerebral cortex apoptosis in early aged hypertension: effects of epigallocatechin-3-gallate | [204] | |
| Wistar albino rats | Suppresses premature senescence of preadipocytes by inhibition of PI3K/Akt/mTOR pathway and induces senescent cell death by regulation of Bax/Bcl-2 pathway | [205] | |
| MicroRNA | APP/PS1 transgenic mouse model | Identification of circulating mir-125b as a potential biomarker of Alzheimer’s disease | [193] |
| chondrocytes, human THP-1 monocytic cells, and primary human fibroblasts | Quercetin, epigallocatechin gallate, curcumin, and resveratrol: from dietary sources to human microrna modulation | [192] | |
| Sprague Dawley Rats, chondrocytes, monocytes, and mice | Targeting miRNAs by polyphenols: a novel therapeutic strategy for aging | [191] |
| Study Identifier | Study Type | Study Population | Study Purpose | Study Participants | Number of Patients Recruited | Intervention | Status |
|---|---|---|---|---|---|---|---|
| NCT03978052 | Interventional | Spain | Prevention of cognitive decline in ApoE4 carriers with subjective cognitive decline after EGCG and a multimodal intervention | Alzheimer’s Disease cognitive function nutritional intervention | 200 | Dietary Supplement: EGCG, Placebo EGCG, personalized intervention, and lifestyle recommendations | Recruiting |
| NCT00951834 | Interventional | Germany | Sunphenon EGCg (Epigallocatechin-Gallate) in the early stage of Alzheimer’s disease | Alzheimer’s Disease | 21 | Drug: Epigallocatechin-Gallate Drug Placebo | Completed |
| NCT00461942 | Interventional | China | Efficacy and safety of green tea polyphenol in de novo Parkinson’s disease patients | Parkinson’s Disease | 480 | Drug: Green Tea Polyphenols (EGCG/ECG) | Completed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Payne, A.; Nahashon, S.; Taka, E.; Adinew, G.M.; Soliman, K.F.A. Epigallocatechin-3-Gallate (EGCG): New Therapeutic Perspectives for Neuroprotection, Aging, and Neuroinflammation for the Modern Age. Biomolecules 2022, 12, 371. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12030371
Payne A, Nahashon S, Taka E, Adinew GM, Soliman KFA. Epigallocatechin-3-Gallate (EGCG): New Therapeutic Perspectives for Neuroprotection, Aging, and Neuroinflammation for the Modern Age. Biomolecules. 2022; 12(3):371. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12030371
Chicago/Turabian StylePayne, Ashley, Samuel Nahashon, Equar Taka, Getinet M. Adinew, and Karam F. A. Soliman. 2022. "Epigallocatechin-3-Gallate (EGCG): New Therapeutic Perspectives for Neuroprotection, Aging, and Neuroinflammation for the Modern Age" Biomolecules 12, no. 3: 371. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12030371