
Construction and Analysis of an Enzyme-Constrained Metabolic Model of Corynebacterium glutamicum
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
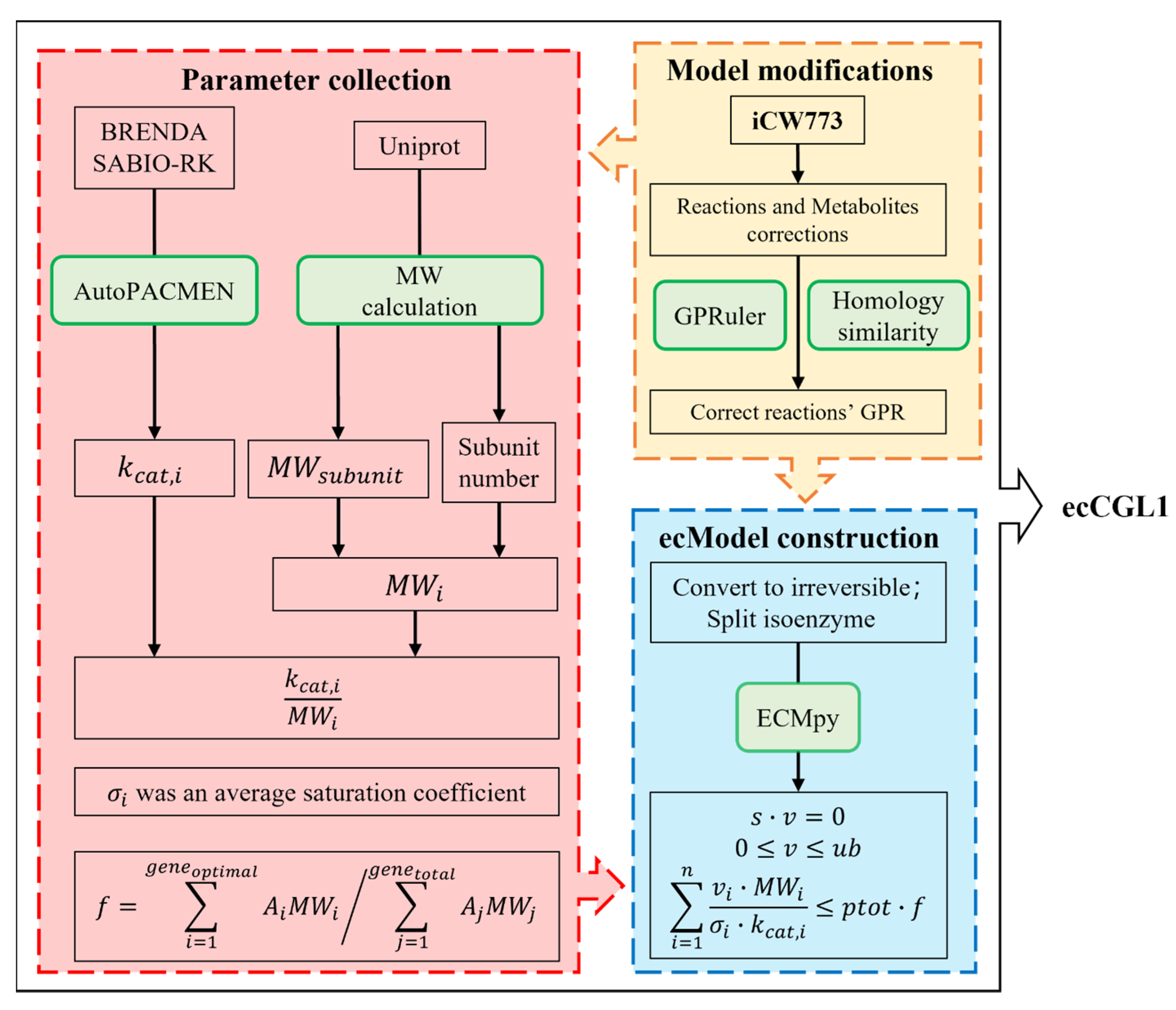
2. Materials and Methods
2.1. Model Calibration
- (1)
- Metabolite correction: ‘(e)’ to ‘_e’, ‘-D’ to ‘__D’, ‘-L’ to ‘__L’, ‘-R’ to ‘__R’ and other ‘-’ to ‘_’.
- (2)
- Reaction correction: ‘-’ to ‘__’ in reactions beginning with ‘EX’ and ‘-’ to ‘__’ in other reactions.
- (3)
- Adding UniProt ID information to the annotation, which is the basis for obtaining kinetic parameters.
2.2. Correction of GPR Relationship
2.3. Acquisition of Quantitative Subunit Composition
2.4. Construction of ecCGL1
2.5. Calibration of the Original kcat Values
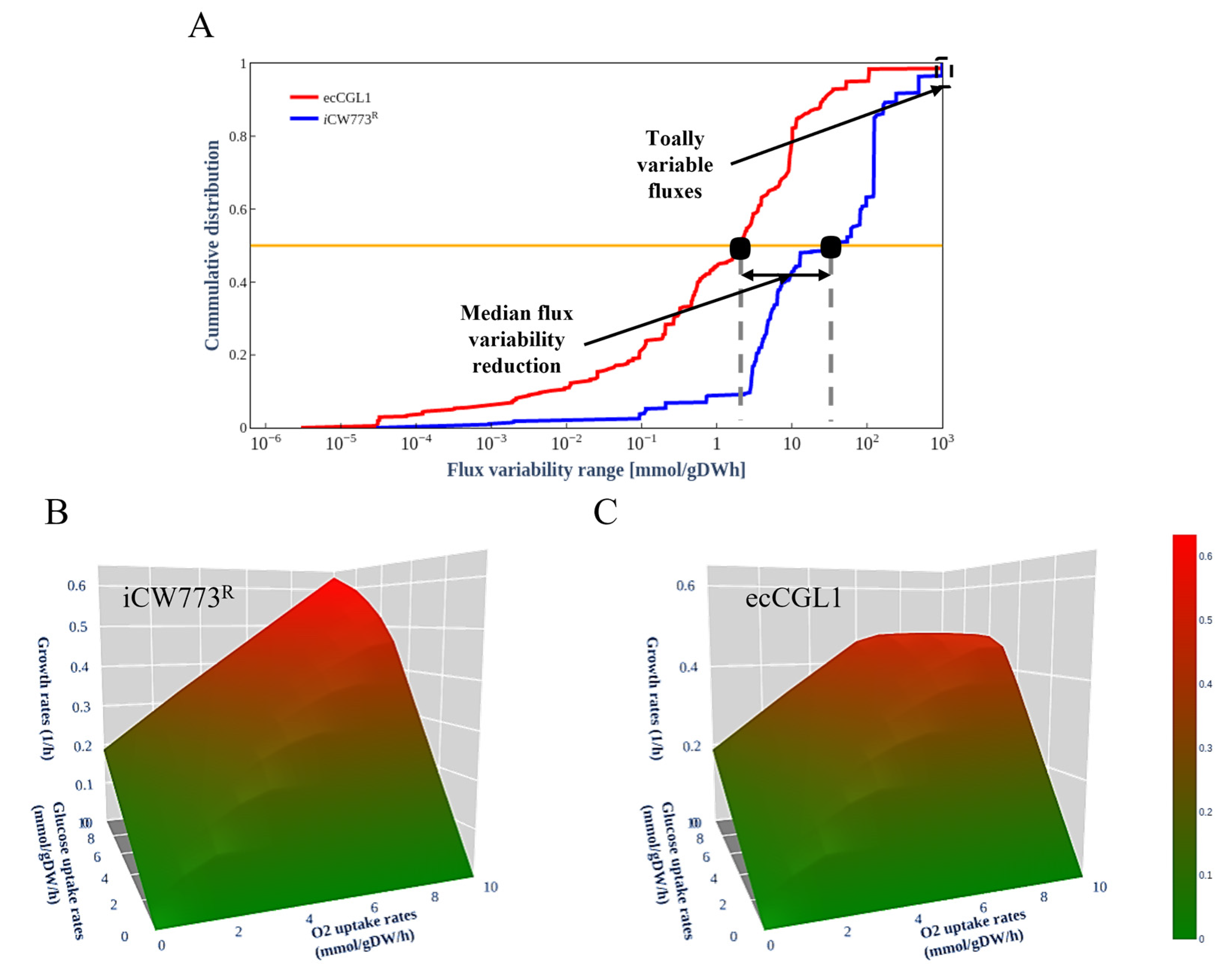
2.6. Comparative Flux Variability Analysis
2.7. Phenotype Phase Plane (PhPP) Analysis
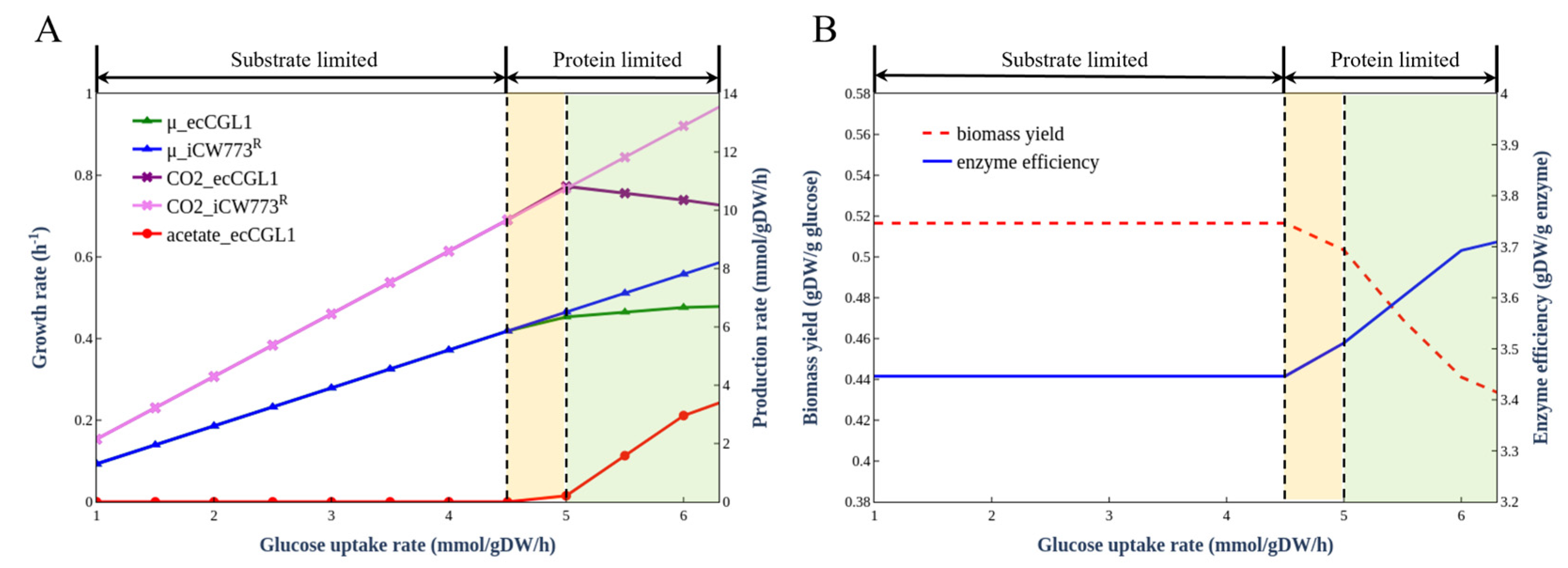
2.8. Simulation of Overflow Metabolism
2.9. Prediction of Metabolic Engineering Targets
3. Results
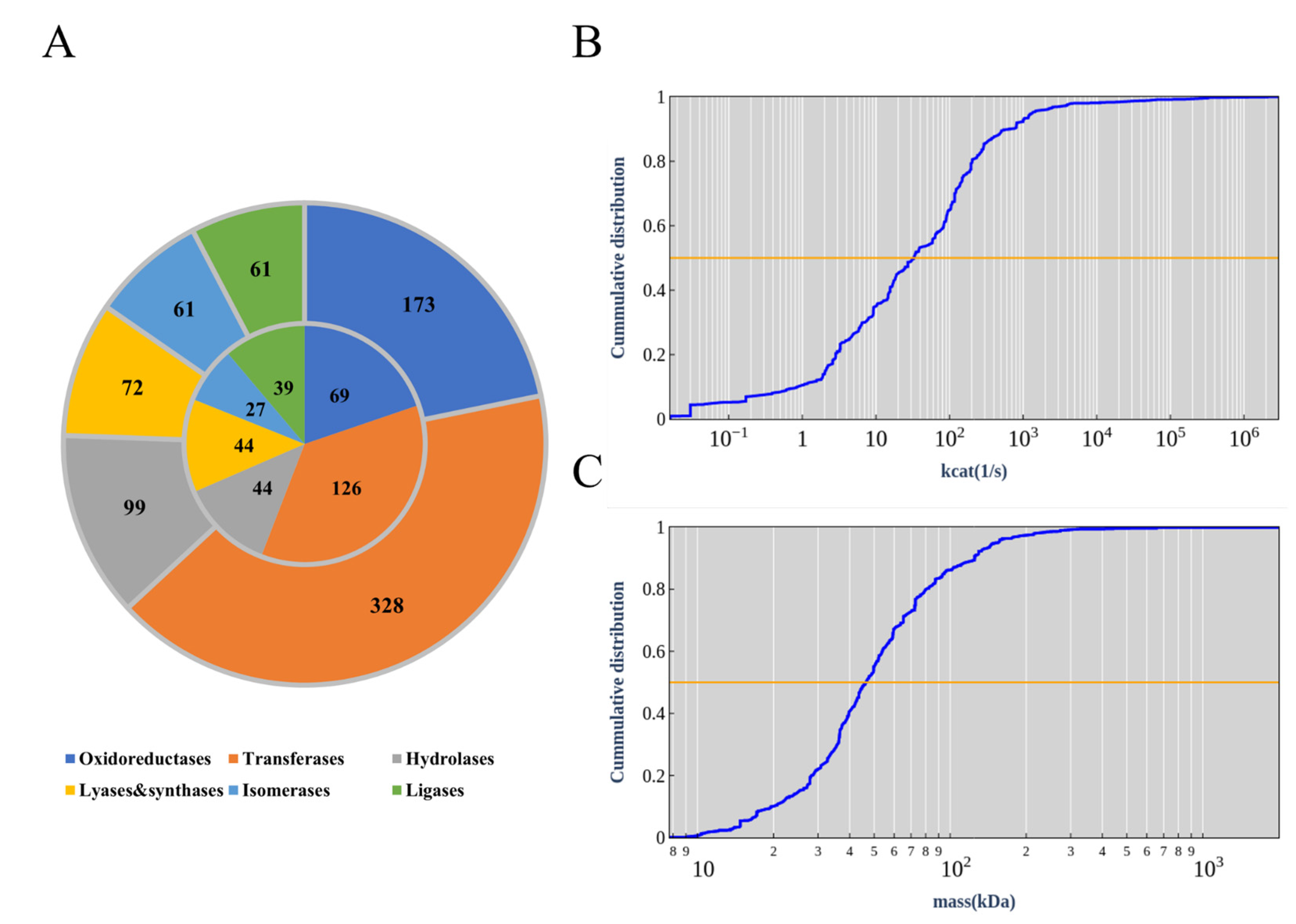
3.1. GPR Correction of iCW773
3.2. EcModel Calibration
3.3. Basic Information of ecCGL1
3.4. EcCGL1 Reduces the Solution Space
3.5. Simulation of Overflow Metabolism
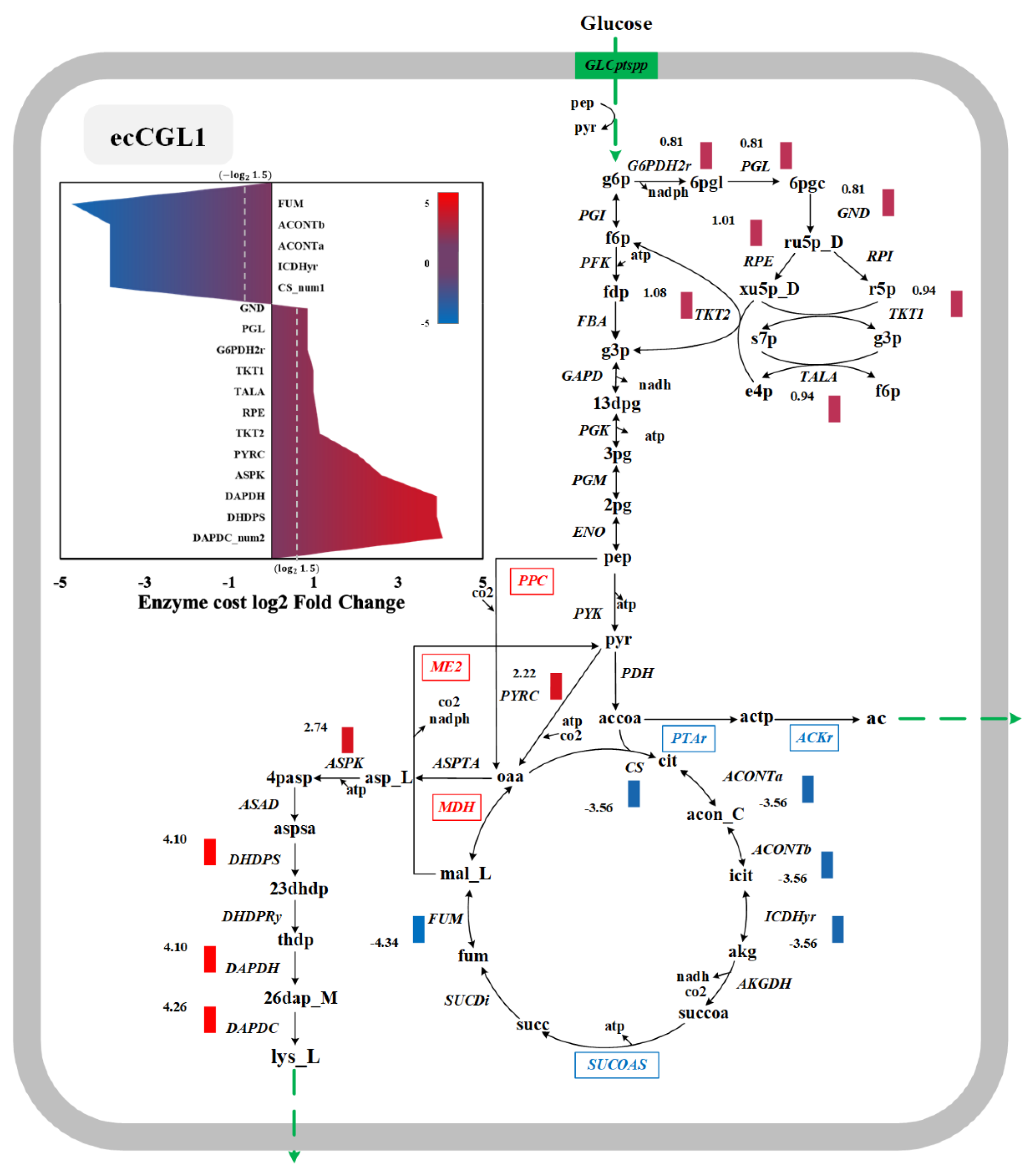
3.6. Exploration of the Targets Based on Enzyme Cost
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wolf, S.; Becker, J.; Tsuge, Y.; Kawaguchi, H.; Kondo, A.; Marienhagen, J.; Bott, M.; Wendisch, V.F.; Wittmann, C. Advances in metabolic engineering of Corynebacterium glutamicum to produce high-value active ingredients for food, feed, human health, and well-being. Essays Biochem. 2021, 65, 197–212. [Google Scholar] [PubMed]
- Becker, J.; Wittmann, C. Advanced biotechnology: Metabolically engineered cells for the bio-based production of chemicals and fuels, materials, and health-care products. Angew. Chem. Int. Ed. 2015, 54, 3328–3350. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, S.; Nogales, J. Recent advances in model-assisted metabolic engineering. Curr. Opin. Syst. Biol. 2021, 28, 100392. [Google Scholar] [CrossRef]
- Han, T.; Kim, G.B.; Lee, S.Y. Glutaric acid production by systems metabolic engineering of an L-lysine–overproducing Corynebacterium glutamicum. Proc. Natl. Acad. Sci. USA 2020, 117, 30328–30334. [Google Scholar] [CrossRef] [PubMed]
- Zha, J.; Zang, Y.; Mattozzi, M.; Plassmeier, J.; Gupta, M.; Wu, X.; Clarkson, S.; Koffas, M.A.G. Metabolic engineering of Corynebacterium glutamicum for anthocyanin production. Microb. Cell Factories 2018, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Q.; Wu, X.Y.; Xu, X.; Tan, X.; Li, Z.; Zhang, B. Production of L-glutamate family amino acids in Corynebacterium glutamicum: Physiological mechanism, genetic modulation, and prospects. Synth. Syst. Biotechnol. 2021, 6, 302–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, J.; Shang, X.; Wang, B.; Liu, S.; Chai, X.; Tan, T.; Zhang, Y.; Wen, T. A new genome-scale metabolic model of Corynebacterium glutamicum and its application. Biotechnol. Biofuels 2017, 10, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orth, J.D.; Thiele, I.; Palsson, B.O. What is flux balance analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, H.G. Observations on the carbohydrate metabolism of tumours. Biochem. J. 1929, 23, 536–545. [Google Scholar] [CrossRef]
- Berkhout, J.; Bosdriesz, E.; Nikerel, E.; Molenaar, D.; de Ridder, D.; Teusink, B.; Bruggeman, F.J. How biochemical constraints of cellular growth shape evolutionary adaptations in metabolism. Genetics 2013, 194, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K.; O’Brien, E.J.; Lerman, J.A.; Zengler, K.; Palsson, B.O.; Feist, A.M. Reconstruction and modeling protein translocation and compartmentalization in Escherichia coli at the genome-scale. BMC Syst. Biol. 2014, 8, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, A.; Nielsen, J.; Palsson, B.O. Metabolic Models of Protein Allocation Call for the Kinetome. Cell Syst. 2017, 5, 538–541. [Google Scholar] [CrossRef] [Green Version]
- Beg, Q.K.; Vazquez, A.; Ernst, J.; De Menezes, M.A.; Bar-Joseph, Z.; Barabási, A.L.; Oltvai, Z.N. Intracellular crowding defines the mode and sequence of substrate uptake by Escherichia coli and constrains its metabolic activity. Proc. Natl. Acad. Sci. USA 2007, 104, 12663–12668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Nielsen, J. Mathematical modeling of proteome constraints within metabolism. Curr. Opin. Syst. Biol. 2021, 25, 50–56. [Google Scholar] [CrossRef]
- Adadi, R.; Volkmer, B.; Milo, R.; Heinemann, M.; Shlomi, T. Prediction of microbial growth rate versus biomass yield by a metabolic network with kinetic parameters. PLoS Comput. Biol. 2012, 8, e1002575. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, B.J.; Zhang, C.; Nilsson, A.; Lahtvee, P.-J.; Kerkhoven, E.J.; Nielsen, J. Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Mol. Syst. Biol. 2017, 13, 935. [Google Scholar] [CrossRef] [PubMed]
- Bekiaris, P.S.; Klamt, S. Automatic construction of metabolic models with enzyme constraints. BMC Bioinform. 2020, 21, 19. [Google Scholar] [CrossRef]
- Mao, Z.; Zhao, X.; Yang, X.; Zhang, P.; Du, J.; Yuan, Q.; Ma, H. ECMpy, a Simplified Workflow for Constructing Enzymatic Constrained Metabolic Network Model. Biomolecules 2022, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Domenzain, I.; Sánchez, B.; Anton, M.; Kerkhoven, E.J.; Millán-Oropeza, A.; Henry, C.; Siewers, V.; Morrissey, J.P.; Sonnenschein, N.; Nielsen, J. Reconstruction of a catalogue of genome-scale metabolic models with enzymatic constraints using GECKO 2.0. Nat. Commun. 2022, 13, 3766. [Google Scholar] [CrossRef]
- Jeske, L.; Placzek, S.; Schomburg, I.; Chang, A.; Schomburg, D. BRENDA in 2019: A European ELIXIR core data resource. Nucleic Acids Res. 2019, 47, D542–D549. [Google Scholar] [CrossRef] [PubMed]
- Wittig, U.; Rey, M.; Weidemann, A.; Kania, R.; Müller, W. SABIO-RK: An updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Res. 2018, 46, D656–D660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.; Luo, Q.; Guo, L.; Gao, C.; Xu, N.; Zhang, L.; Liu, L.; Chen, X. Improving lysine production through construction of an Escherichia coli enzyme-constrained model. Biotechnol. Bioeng. 2020, 117, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhuang, Y.; Xia, J. Integration of enzyme constraints in a genome-scale metabolic model of Aspergillus niger improves phenotype predictions. Microb. Cell Factories 2021, 20, 125. [Google Scholar] [CrossRef]
- Massaiu, I.; Pasotti, L.; Sonnenschein, N.; Rama, E.; Cavaletti, M.; Magni, P.; Calvio, C.; Herrgard, M.J. Integration of enzymatic data in Bacillus subtilis genome-scale metabolic model improves phenotype predictions and enables in silico design of poly--glutamic acid production strains. Microb. Cell Factories 2019, 18, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Yuan, L.; Lu, H.; Li, G.; Chen, Y.; Engqvist, M.K.M.; Kerkhoven, E.J.; Nielsen, J. Deep learning-based kcat prediction enables improved enzyme-constrained model reconstruction. Nat. Catal. 2022, 5, 662–672. [Google Scholar] [CrossRef]
- Di Filippo, M.; Damiani, C.; Pescini, D. GPRuler: Metabolic gene-protein-reaction rules automatic reconstruction. PLoS Comput. Biol. 2021, 17, e1009550. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar]
- Meldal, B.H.M.; Orchard, S. Searching and Extracting Data from the EMBL-EBI Complex Portal. In Protein Complex Assembly: Methods and Protocols; Marsh, J.A., Ed.; Springer: New York, NY, USA, 2018; pp. 377–390. [Google Scholar]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 2017, 20, 1085–1093. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2020, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Walter, T.; Veldmann, K.H.; Götker, S.; Busche, T.; Rückert, C.; Kashkooli, A.B.; Paulus, J.; Cankar, K.; Wendisch, V.F. Physiological response of corynebacterium glutamicum to indole. Microorganisms 2020, 8, 1945. [Google Scholar] [CrossRef]
- Keppel, M.; Hünnefeld, M.; Filipchyk, A.; Viets, U.; Davoudi, C.F.; Krüger, A.; Mack, C.; Pfeifer, E.; Polen, T.; Baumgart, M.; et al. HrrSA orchestrates a systemic response to heme and determines prioritization of terminal cytochrome oxidase expression. Nucleic Acids Res. 2020, 48, 6547–6562. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, J.; Chen, L.; Zeng, A.-P.; Solem, C.; Jensen, P.R. Alterations in the transcription factors GntR1 and RamA enhance the growth and central metabolism of Corynebacterium glutamicum. Metab. Eng. 2018, 48, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.S.; Ramakrishna, R.; Palsson, B.O. Characterizing the metabolic phenotype: A phenotype phase plane analysis. Biotechnol. Bioeng. 2002, 77, 27–36. [Google Scholar] [CrossRef]
- Duarte, N.C.; Palsson, B.Ø.; Fu, P. Integrated analysis of metabolic phenotypes in Saccharomyces cerevisiae. BMC Genom. 2004, 5, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, N.E.; Hixson, K.K.; Conrad, T.M.; Lerman, J.A.; Charusanti, P.; Polpitiya, A.D.; Adkins, J.N.; Schramm, G.; Purvine, S.O.; Lopez-Ferrer, D.; et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 2010, 6, 390. [Google Scholar] [CrossRef]
- Wang, M.; Herrmann, C.J.; Simonovic, M.; Szklarczyk, D.; von Mering, C. Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 2015, 15, 3163–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadevan, R.; Schilling, C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Péquignot, C.; Dussap, C.G.; Pons, A.; Gros, J.B. Intra- and extracellular concentrations of glutamate, lactate and acetate during growth of Corynebacterium glutamicum on different media. J. Ind. Microbiol. Biotechnol. 1997, 18, 312–318. [Google Scholar] [CrossRef]
- Koch, A.L. Microbial Physiology and Ecology of Slow Growth. Microbiol. Mol. Biol. Rev. 1997, 61, 305–318. [Google Scholar] [PubMed]
- Becker, J.; Zelder, O.; Häfner, S.; Schröder, H.; Wittmann, C. From zero to hero-Design-based systems metabolic engineering of Corynebacterium glutamicum for l-lysine production. Metab. Eng. 2011, 13, 159–168. [Google Scholar] [CrossRef]
- O’Regan, M.; Thierbach, G.; Bachmann, B.; Villeval, D.; Lepage, P.; Viret, J.-F.; Lemoine, Y. Cloning and nucleotide sequence of the phosphoenolpyruvate carboxylase-coding gene of Corynebacterium glutamicum ATCC13032. Gene 1989, 77, 237–251. [Google Scholar] [CrossRef]
- Cremer, J.; Eggeling, L.; Sahm, H. Control of the Lysine Biosynthesis Sequence in Corynebacterium glutamicum as Analyzed by Overexpression of the Individual Corresponding Genes. Appl. Environ. Microbiol. 1991, 57, 1746–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Bommareddy, R.R.; Frank, D.; Rappert, S.; Zeng, A.P. Deregulation of feedback inhibition of phosphoenolpyruvate carboxylase for improved lysine production in Corynebacterium glutamicum. Appl. Environ. Microbiol. 2014, 80, 1388–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, X.; Chai, X.; Lu, X.; Li, Y.; Zhang, Y.; Wang, G.; Zhang, C.; Liu, S.; Zhang, Y.; Ma, J.; et al. Native promoters of Corynebacterium glutamicum and its application in l-lysine production. Biotechnol. Lett. 2018, 40, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.L.; Kohlstedt, M.; Jungmann, L.; Hutter, M.; Poblete-Castro, I.; Becker, J.; Wittmann, C. Cascaded valorization of brown seaweed to produce l-lysine and value-added products using Corynebacterium glutamicum streamlined by systems metabolic engineering. Metab. Eng. 2021, 67, 293–307. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, J.; Guo, Y.; Zhang, W. Genetically modifying aspartate aminotransferase and aspartate ammonia-lyase affects metabolite accumulation in l-lysine producing strain derived from Corynebacterium glutamicum ATCC13032. J. Mol. Catal. B Enzym. 2015, 113, 82–89. [Google Scholar] [CrossRef]
- Zhou, L.-B.; Zeng, A.-P. Exploring Lysine Riboswitch for Metabolic Flux Control and Improvement of l-Lysine Synthesis in Corynebacterium glutamicum. ACS Synth. Biol. 2015, 4, 729–734. [Google Scholar] [CrossRef]
- Xu, J.; Han, M.; Zhang, J.; Guo, Y.; Qian, H.; Zhang, W. Improvement of L-lysine production combines with minimization of by-products synthesis in Corynebacterium glutamicum. J. Chem. Technol. Biotechnol. 2014, 89, 1924–1933. [Google Scholar] [CrossRef]
- van Ooyen, J.; Noack, S.; Bott, M.; Reth, A.; Eggeling, L. Improved L-lysine production with Corynebacterium glutamicum and systemic insight into citrate synthase flux and activity. Biotechnol. Bioeng. 2012, 109, 2070–2081. [Google Scholar] [CrossRef]
- Nilsson, A.; Nielsen, J. Metabolic Trade-offs in Yeast are Caused by F1F0-ATP synthase. Sci. Rep. 2016, 6, 22264. [Google Scholar] [CrossRef] [Green Version]
- Feierabend, M.; Renz, A.; Zelle, E.; Nöh, K.; Wiechert, W.; Dräger, A. High-Quality Genome-Scale Reconstruction of Corynebacterium glutamicum ATCC 13032. Front. Microbiol. 2021, 12, 3432. [Google Scholar] [CrossRef] [PubMed]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; et al. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.Y.; Kim, H.U.; Lee, S.Y. Deep learning enables high-quality and high-throughput prediction of enzyme commission numbers. Proc. Natl. Acad. Sci. USA 2019, 116, 13996–14001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Yuan, Q.; Wang, R.; Li, H.; Liao, X.; Ma, H. ECRECer: Enzyme Commission Number Recommendation and Benchmarking Based on Multiagent Dual-Core Learning. Available online: https://paperswithcode.com/paper/ecrecer-enzyme-commission-number (accessed on 30 August 2022).
- Heckmann, D.; Lloyd, C.J.; Mih, N.; Ha, Y.; Zielinski, D.C.; Haiman, Z.B.; Desouki, A.A.; Lercher, M.J.; Palsson, B.O. Machine learning applied to enzyme turnover numbers reveals protein structural correlates and improves metabolic models. Nat. Commun. 2018, 9, 5252. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Mao, Z.; Zhao, X.; Wang, R.; Zhang, P.; Cai, J.; Xue, C.; Ma, H. Integrating thermodynamic and enzymatic constraints into genome-scale metabolic models. Metab. Eng. 2021, 67, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Salvy, P.; Hatzimanikatis, V. The ETFL formulation allows multi-omics integration in thermodynamics-compliant metabolism and expression models. Nat. Commun. 2020, 11, 30. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, J.; Mao, Z.; Mao, Y.; Wu, K.; Shi, Z.; Yuan, Q.; Cai, J.; Ma, H. Construction and Analysis of an Enzyme-Constrained Metabolic Model of Corynebacterium glutamicum. Biomolecules 2022, 12, 1499. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12101499
Niu J, Mao Z, Mao Y, Wu K, Shi Z, Yuan Q, Cai J, Ma H. Construction and Analysis of an Enzyme-Constrained Metabolic Model of Corynebacterium glutamicum. Biomolecules. 2022; 12(10):1499. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12101499
Chicago/Turabian StyleNiu, Jinhui, Zhitao Mao, Yufeng Mao, Ke Wu, Zhenkun Shi, Qianqian Yuan, Jingyi Cai, and Hongwu Ma. 2022. "Construction and Analysis of an Enzyme-Constrained Metabolic Model of Corynebacterium glutamicum" Biomolecules 12, no. 10: 1499. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12101499