Negative Control of Cell Migration by Rac1b in Highly Metastatic Pancreatic Cancer Cells Is Mediated by Sequential Induction of Nonactivated Smad3 and Biglycan


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
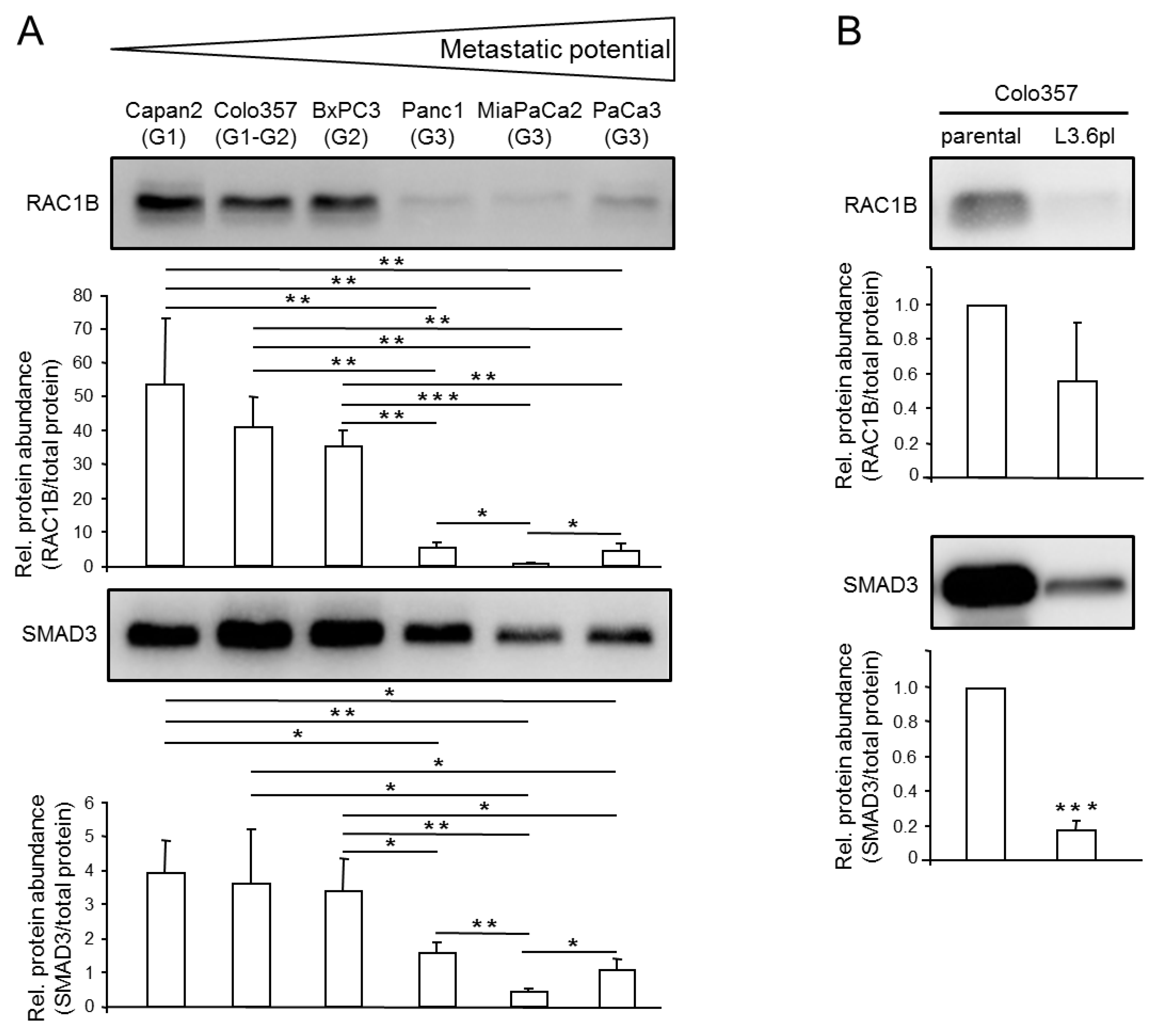
2.1. Expression Levels of RAC1B and SMAD3 are Inversely Correlated with the Metastatic Potential of PDAC-Derived Cell Lines
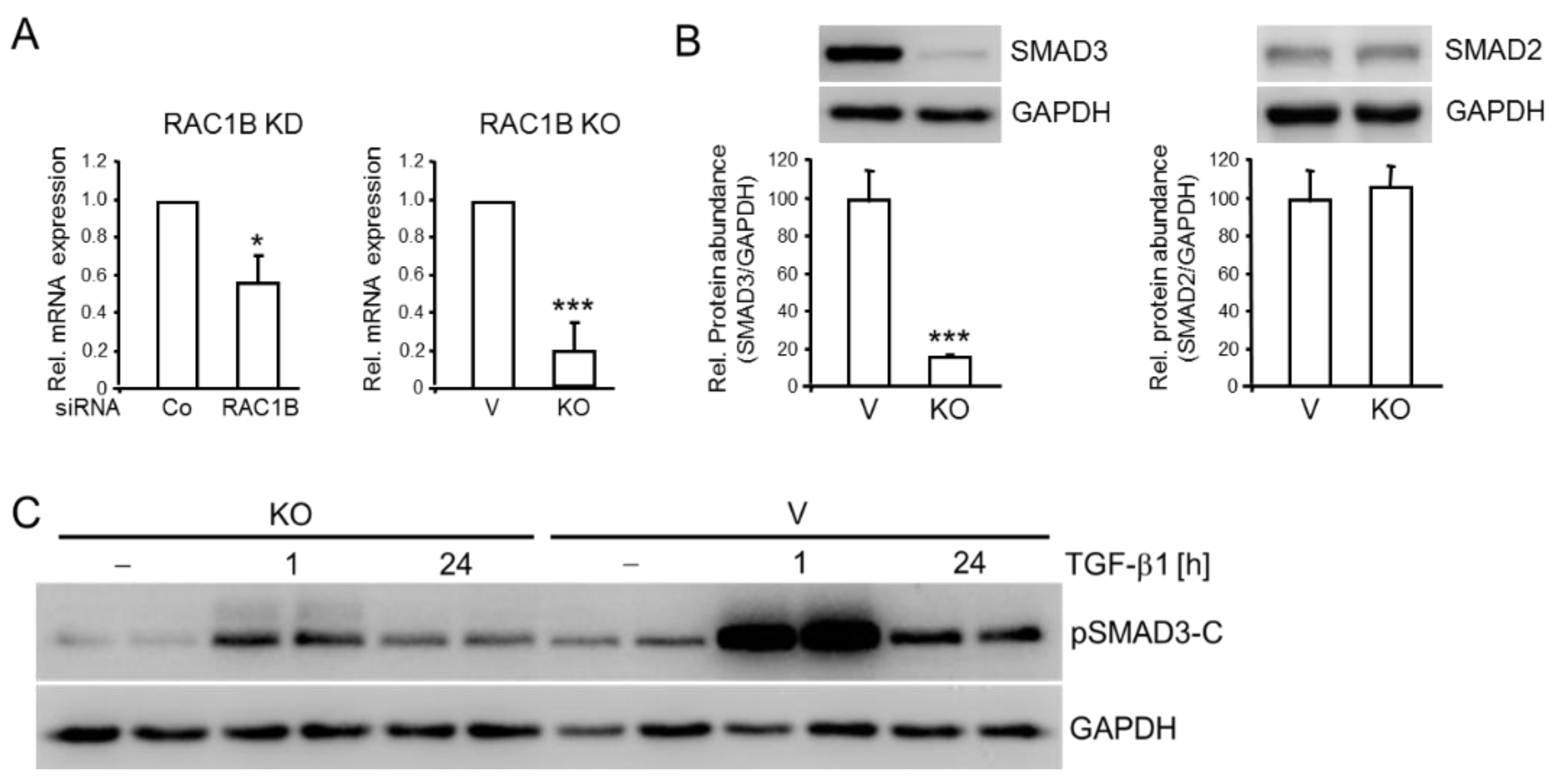
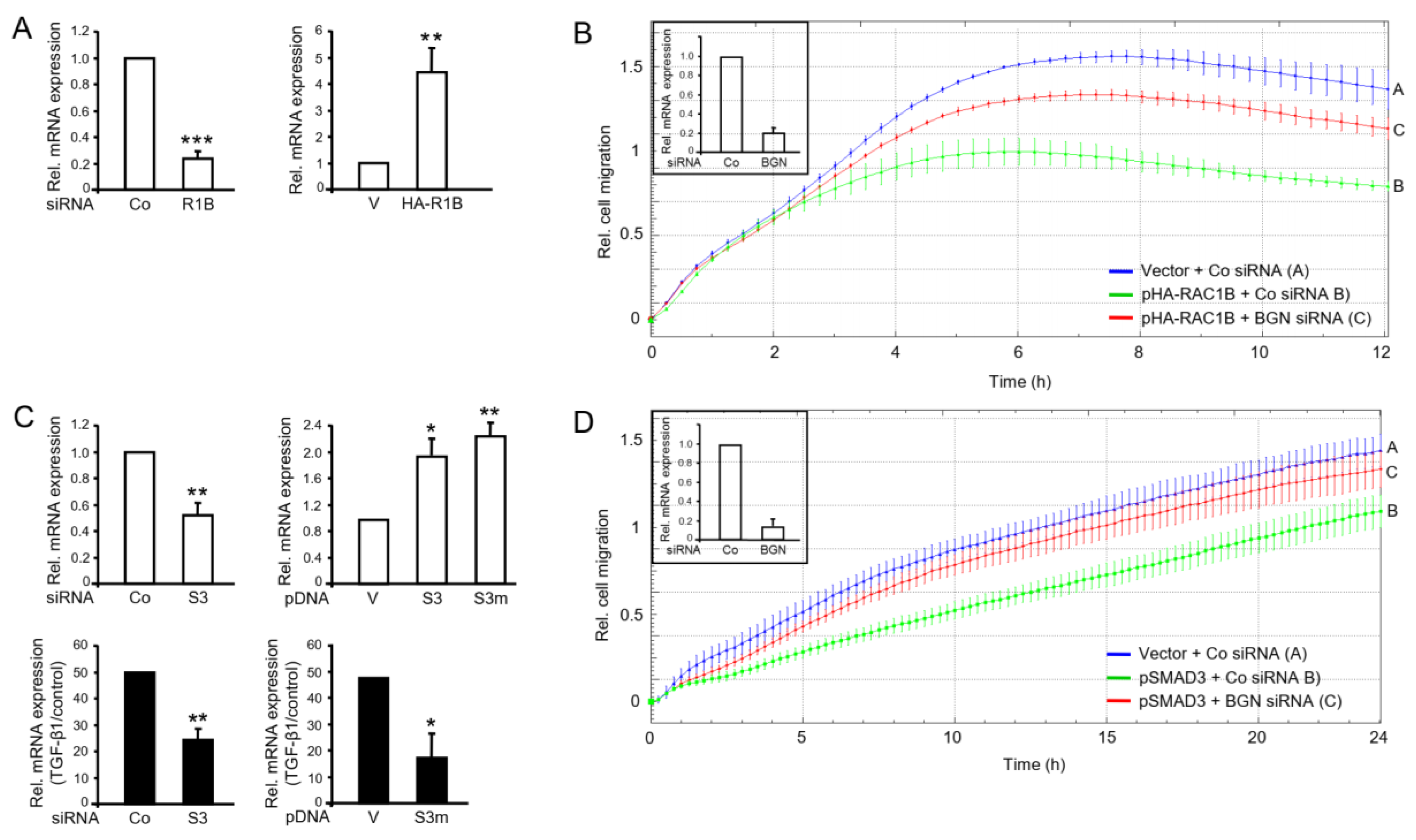
2.2. RAC1B Knockdown or Knockout Is Associated with Decreased Abundance of SMAD3
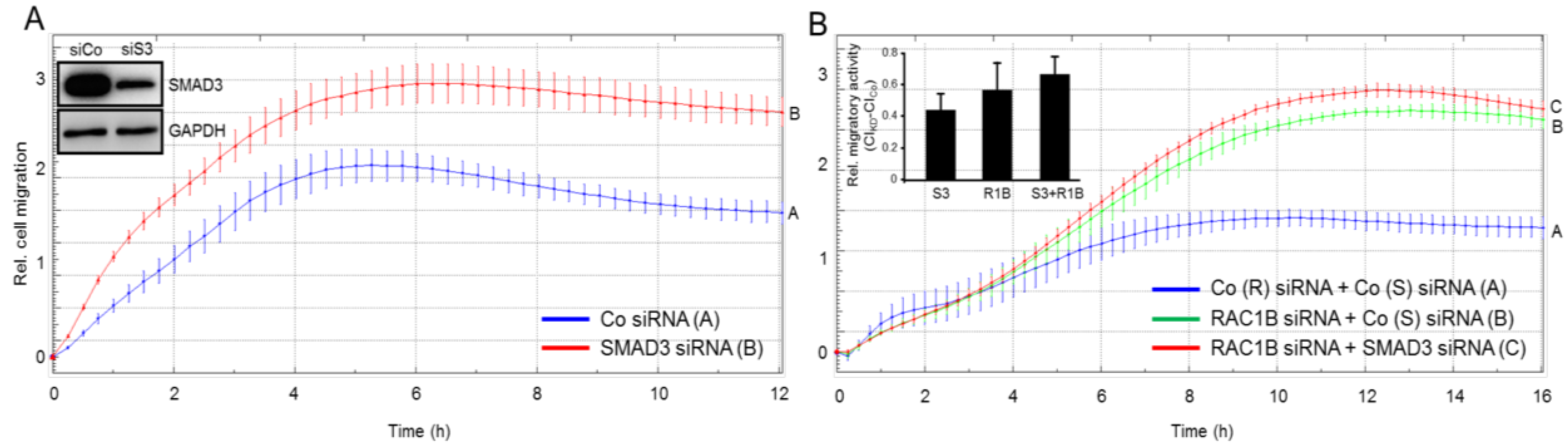
2.3. Knockdown of SMAD3 Mimics the Effects of Knockdown of RAC1B on Chemokinesis in Panc1 and PaCa3 Cells
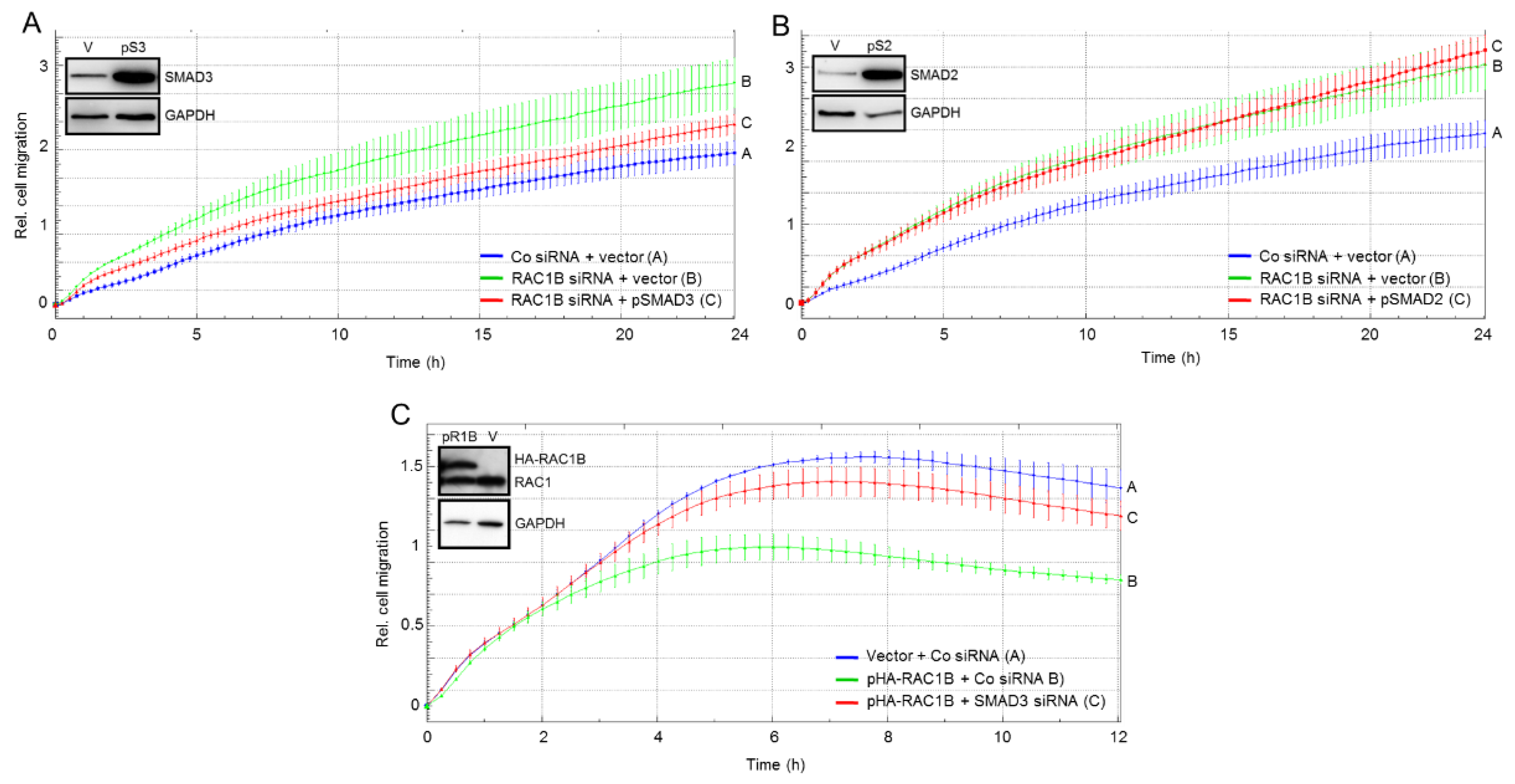
2.4. SMAD3 Mediates the Antimigratory Effect of RAC1B
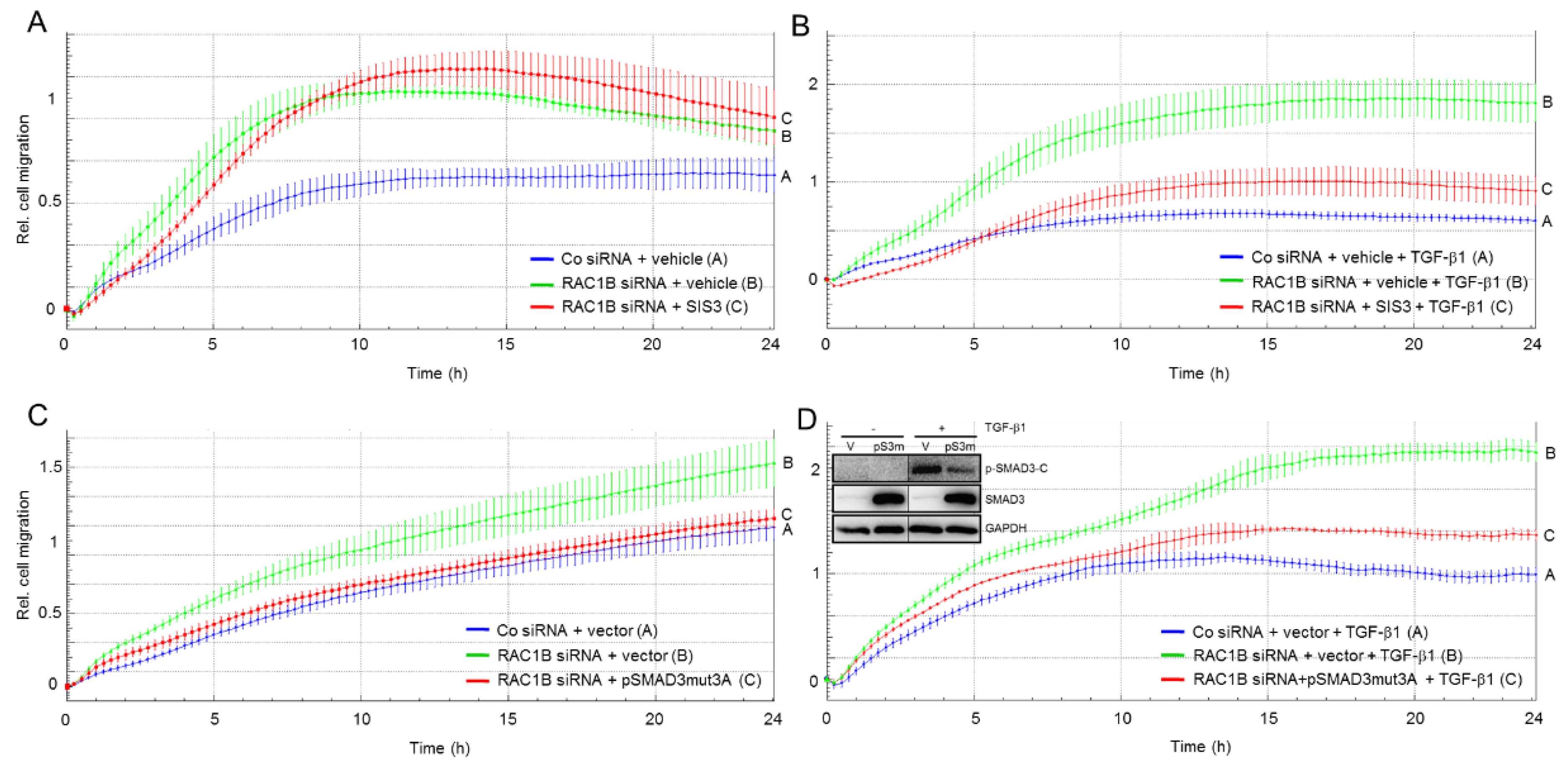
2.5. The Migration-Inhibiting Effect of SMAD3 Is Activation-Independent
2.6. Identification of Biglycan as a Common Mediator of the Antimigratory Effect of SMAD3 and RAC1B in Panc1 Cells
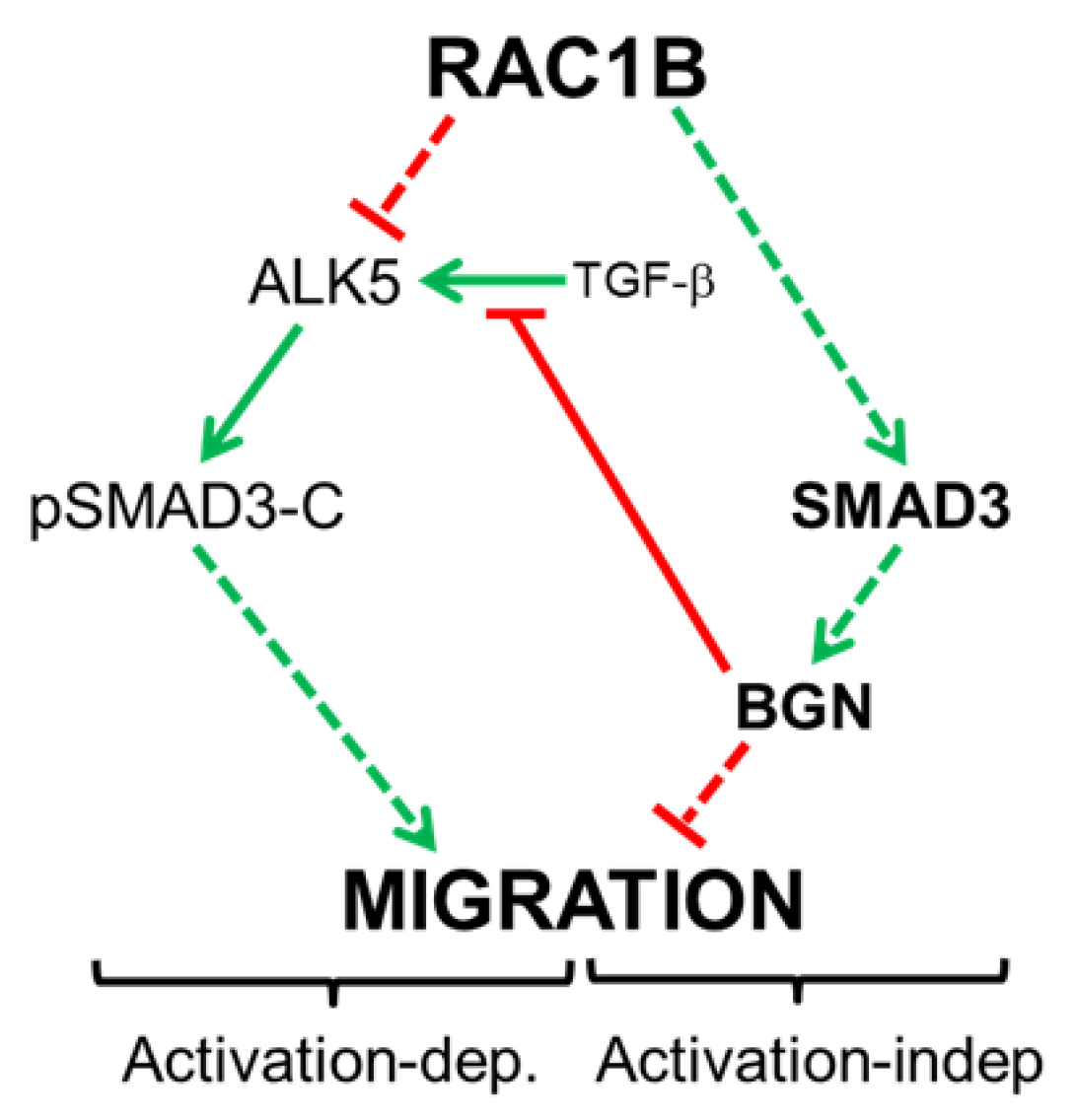
3. Discussion
4. Material and Methods
4.1. Antibodies and Reagents
4.2. Cells
4.3. Transient Transfection of siRNA and Expression Vectors
4.4. QRT-PCR Analysis
4.5. Immunoblotting
4.6. Migration Assays
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, e1338. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Siveke, J.T.; Eckel, F.; Schmid, R.M. Pancreatic cancer: Basic and clinical aspects. Gastroenterology 2005, 128, 1606–1625. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Hass, R.; Lehnert, H.; Ungefroren, H. RAC1B: A Rho GTPase with Versatile Functions in Malignant Transformation and Tumor Progression. Cells 2019, 8, e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, S.; Gosens, R.; Wieland, T.; Schmidt, M. Paving the Rho in cancer metastasis: Rho GTPases and beyond. Pharmacol. Ther. 2018, 183, 1–21. [Google Scholar] [CrossRef]
- Ungefroren, H.; Witte, D.; Lehnert, H. The role of small GTPases of the Rho/Rac family in TGF-β-induced EMT and cell motility in cancer. Dev. Dyn. 2018, 247, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H.; Sebens, S.; Giehl, K.; Helm, O.; Groth, S.; Fändrich, F.; Röcken, C.; Sipos, B.; Lehnert, H.; Gieseler, F. Rac1b negatively regulates TGF-beta1-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signalling. Oncotarget 2014, 5, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Witte, D.; Otterbein, H.; Forster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-beta1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, e17313. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; Hass, R.; von der Ohe, J.; Lehnert, H.; Ungefroren, H. The role of TGF-beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun. Signal. 2017, 15, e19. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H.; Otterbein, H.; Fiedler, C.; Mihara, K.; Hollenberg, M.D.; Gieseler, F.; Lehnert, H.; Witte, D. RAC1B Suppresses TGF-β1-Dependent Cell Migration in Pancreatic Carcinoma Cells through Inhibition of the TGF-β Type I Receptor ALK5. Cancers 2019, 11, e691. [Google Scholar] [CrossRef] [Green Version]
- Ellenrieder, V.; Hendler, S.F.; Ruhland, C.; Boeck, W.; Adler, G.; Gress, T.M. TGF-beta-induced invasiveness of pancreatic cancer cells is mediated by matrix metalloproteinase-2 and the urokinase plasminogen activator system. Int. J. Cancer 2001, 93, 204–211. [Google Scholar] [CrossRef]
- Subramanian, G.; Schwarz, R.E.; Higgins, L.; McEnroe, G.; Chakravarty, S.; Dugar, S.; Reiss, M. Targeting endogenous transforming growth factor beta receptor signaling in SMAD4-deficient human pancreatic carcinoma cells inhibits their invasive phenotype1. Cancer Res. 2004, 64, 5200–5211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, P.S.; Sipos, B.; Orlandini, S.; Sorio, C.; Real, F.X.; Lemoine, N.R.; Gress, T.; Bassi, C.; Klöppel, G.; Kalthoff, H.; et al. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 2001, 439, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drubay, V.; Skrypek, N.; Cordiez, L.; Vasseur, R.; Schulz, C.; Boukrout, N.; Duchêne, B.; Coppin, L.; Van Seuningen, I.; Jonckheere, N. TGF-βRII Knock-down in Pancreatic Cancer Cells Promotes Tumor Growth and Gemcitabine Resistance. Importance of STAT3 Phosphorylation on S727. Cancers 2018, 10, e254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, J.W.; Mattingly, C.A.; Strodel, W.E. Increased tumorigenicity in the human pancreatic cell line MIA PaCa-2 is associated with an aberrant regulation of an IGF-1 autocrine loop and lack of expression of the TGF-beta type RII receptor. J. Cell. Physiol. 1995, 165, 155–163. [Google Scholar] [CrossRef]
- Zinn, R.; Otterbein, H.; Lehnert, H.; Ungefroren, H. RAC1B: A guardian of the epithelial phenotype and protector against epithelial-mesenchymal transition. Cells 2019, 8, e1569. [Google Scholar] [CrossRef] [Green Version]
- Suemizu, H.; Monnai, M.; Ohnishi, Y.; Ito, M.; Tamaoki, N.; Nakamura, M. Identification of a key molecular regulator of liver metastasis in human pancreatic carcinoma using a novel quantitative model of metastasis in NOD/SCID/gammacnull (NOG) mice. Int. J. Oncol. 2007, 31, 741–751. [Google Scholar] [CrossRef] [Green Version]
- Vezeridis, M.P.; Tzanakakis, G.N.; Meitner, P.A.; Doremus, C.M.; Tibbetts, L.M.; Calabresi, P. In vivo selection of a highly metastatic cell line from a human pancreatic carcinoma in the nude mouse. Cancer 1992, 69, 2060–2063. [Google Scholar] [CrossRef]
- Bruns, C.J.; Harbison, M.T.; Kuniyasu, H.; Eue, I.; Fidler, I.J. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia 1999, 1, 50–62. [Google Scholar] [CrossRef] [Green Version]
- Pratesi, G.; Petrangolini, G.; Tortoreto, M.; Addis, A.; Zunino, F.; Calcaterra, C.; Merlo, A.; Tagliabue, E.; Menard, S.; Balsari, A. Antitumor efficacy of trastuzumab in nude mice orthotopically xenografted with human pancreatic tumor cells expressing low levels of HER-2/neu. J. Immunother. 2008, 31, 537–544. [Google Scholar] [CrossRef]
- Jinnin, M.; Ihn, H.; Tamaki, K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol. Pharmacol. 2006, 69, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.B.; Lenschow, W.; Tiede, K.; Fischer, J.W.; Kalthoff, H.; Ungefroren, H. Smad4/DPC4-dependent regulation of biglycan gene expression by transforming growth factor-beta in pancreatic tumor cells. J. Biol. Chem. 2002, 277, 36118–36128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carl, C.; Flindt, A.; Hartmann, J.; Dahlke, M.; Rades, D.; Dunst, J.; Lehnert, H.; Gieseler, F.; Ungefroren, H. Ionizing radiation induces a motile phenotype in human carcinoma cells in vitro through hyperactivation of the TGF-beta signaling pathway. Cell. Mol. Life Sci. 2016, 73, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Matsuzaki, K.; Murata, M.; Yamaguchi, T.; Suwa, K.; Okazaki, K. Clinico-Pathological Importance of TGF-β/Phospho-Smad Signaling during Human Hepatic Fibrocarcinogenesis. Cancers 2018, 10, e183. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Canaff, L.; Hendy, G.N.; Hisa, I.; Sugimoto, T.; Chihara, K.; Kaji, H. Role of Smad3, acting independently of transforming growth factor-β, in the early induction of Wnt-β-catenin signaling by parathyroid hormone in mouse osteoblastic cells. J. Cell. Biochem. 2009, 108, 285–294. [Google Scholar] [CrossRef]
- Yang, F.; Chung, A.; Huang, X.R.; Lan, H.Y. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-β-dependent and -independent Smad pathways: The role of Smad3. Hypertension 2009, 54, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Chung, A.; Zhang, H.; Kong, Y.Z.; Tan, J.J.; Huang, X.R.; Kopp, J.B.; Lan, H.Y. Advanced glycation end-products induce tubular CTGF via TGF-β-independent Smad3 signaling. J. Am. Soc. Nephrol. 2010, 21, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Saika, S.; Kono-Saika, S.; Ohnishi, Y.; Sato, M.; Muragaki, Y.; Ooshima, A.; Flanders, K.C.; Yoo, J.; Anzano, M.; Liu, C.Y.; et al. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am. J. Pathol. 2004, 164, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.B.; Tian, F.; Byfield, S.D.; Stuelten, C.; Ooshima, A.; Saika, S.; Flanders, K.C. Smad3 is key to TGF-beta-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006, 17, 19–27. [Google Scholar] [CrossRef]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.M.; Cha, J.Y.; Kim, J.; Kim, D.; Trang, H.T.; Kim, Y.M.; Cho, Y.H.; Park, D.; Hong, S. Smad3 regulates E-cadherin via miRNA-200 pathway. Oncogene 2012, 31, 3051–3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Zhang, X.; Li, J.; Zhao, D.; Gao, B.; Zhou, H.; Gao, S.; Zhang, L. Silencing TRIP13 inhibits cell growth and metastasis of hepatocellular carcinoma by activating of TGF-β1/smad3. Cancer Cell Int. 2018, 18, e208. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Otsuka, T.; Kohsaka, A.; Le, H.T.; Bhawal, U.K.; Muragaki, Y. Smad3 Suppresses Epithelial Cell Migration and Proliferation via the Clock Gene Dec1, Which Negatively Regulates the Expression of Clock Genes Dec2 and Per1. Am. J. Pathol. 2019, 189, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.; Radacz, Y.; Pawlik, N.; Schoeneck, A.; Baldus, S.E.; Munding, J.; Schmiegel, W.; Schwarte-Waldhoff, I.; Reinacher-Schick, A. SMAD4 mediates mesenchymal-epithelial reversion in SW480 colon carcinoma cells. Anticancer Res. 2010, 30, 2603–2613. [Google Scholar]
- Thakur, A.K.; Nigri, J.; Lac, S.; Leca, J.; Bressy, C.; Berthezene, P.; Bartholin, L.; Chan, P.; Calvo, E.; Iovanna, J.L.; et al. TAp73 loss favors Smad-independent TGF-β signaling that drives EMT in pancreatic ductal adenocarcinoma. Cell Death Differ. 2016, 23, 1358–1370. [Google Scholar] [CrossRef] [Green Version]
- Droguett, R.; Cabello-Verrugio, C.; Riquelme, C.; Brandan, E. Extracellular proteoglycans modify TGF-beta bio-availability attenuating its signaling during skeletal muscle differentiation. Matrix Biol. 2006, 25, 332–341. [Google Scholar] [CrossRef]
- Weber, C.K.; Sommer, G.; Michl, P.; Fensterer, H.; Weimer, M.; Gansauge, F.; Leder, G.; Adler, G.; Gress, T.M. Biglycan is overexpressed in pancreatic cancer and induces G1-arrest in pancreatic cancer cell lines. Gastroenterology 2001, 121, 657–667. [Google Scholar] [CrossRef]
- Cho, S.Y.; Ha, S.Y.; Huang, S.M.; Kim, J.H.; Kang, M.S.; Yoo, H.Y.; Kim, H.H.; Park, C.K.; Um, S.H.; Kim, K.H.; et al. The prognostic significance of Smad3, Smad4, Smad3 phosphoisoform expression in esophageal squamous cell carcinoma. Med. Oncol. 2014, 31, e236. [Google Scholar] [CrossRef]
- Serova, M.; Tijeras-Raballand, A.; Dos Santos, C.; Albuquerque, M.; Paradis, V.; Neuzillet, C.; Benhadji, K.A.; Raymond, E.; Faivre, S.; de Gramont, A. Effects of TGF-beta signalling inhibition with galunisertib (LY2157299) in hepatocellular carcinoma models and in ex vivo whole tumor tissue samples from patients. Oncotarget 2015, 6, 21614–21627. [Google Scholar] [CrossRef] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otterbein, H.; Lehnert, H.; Ungefroren, H. Negative Control of Cell Migration by Rac1b in Highly Metastatic Pancreatic Cancer Cells Is Mediated by Sequential Induction of Nonactivated Smad3 and Biglycan. Cancers 2019, 11, 1959. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121959
Otterbein H, Lehnert H, Ungefroren H. Negative Control of Cell Migration by Rac1b in Highly Metastatic Pancreatic Cancer Cells Is Mediated by Sequential Induction of Nonactivated Smad3 and Biglycan. Cancers. 2019; 11(12):1959. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121959
Chicago/Turabian StyleOtterbein, Hannah, Hendrik Lehnert, and Hendrik Ungefroren. 2019. "Negative Control of Cell Migration by Rac1b in Highly Metastatic Pancreatic Cancer Cells Is Mediated by Sequential Induction of Nonactivated Smad3 and Biglycan" Cancers 11, no. 12: 1959. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121959