Efficacy of HDAC Inhibitors Belinostat and Panobinostat against Cisplatin-Sensitive and Cisplatin-Resistant Testicular Germ Cell Tumors

 ,
,  , , , , , ,
, , , , , ,  and
and 
Abstract
:Simple Summary
Abstract
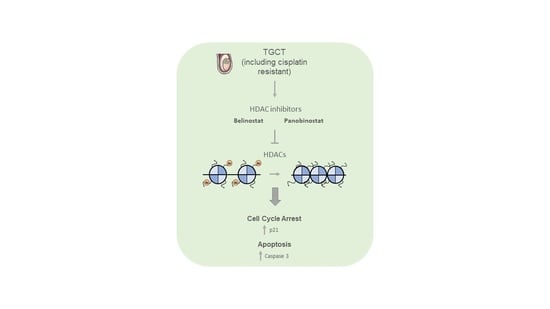
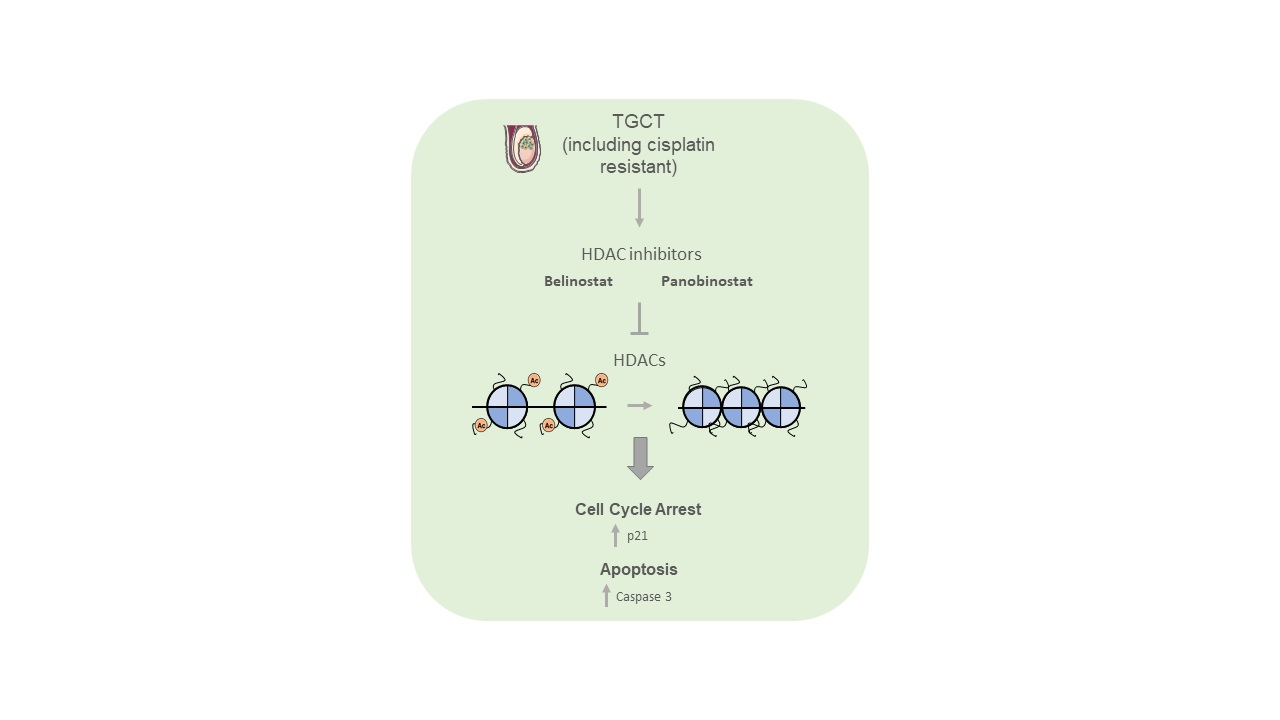{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
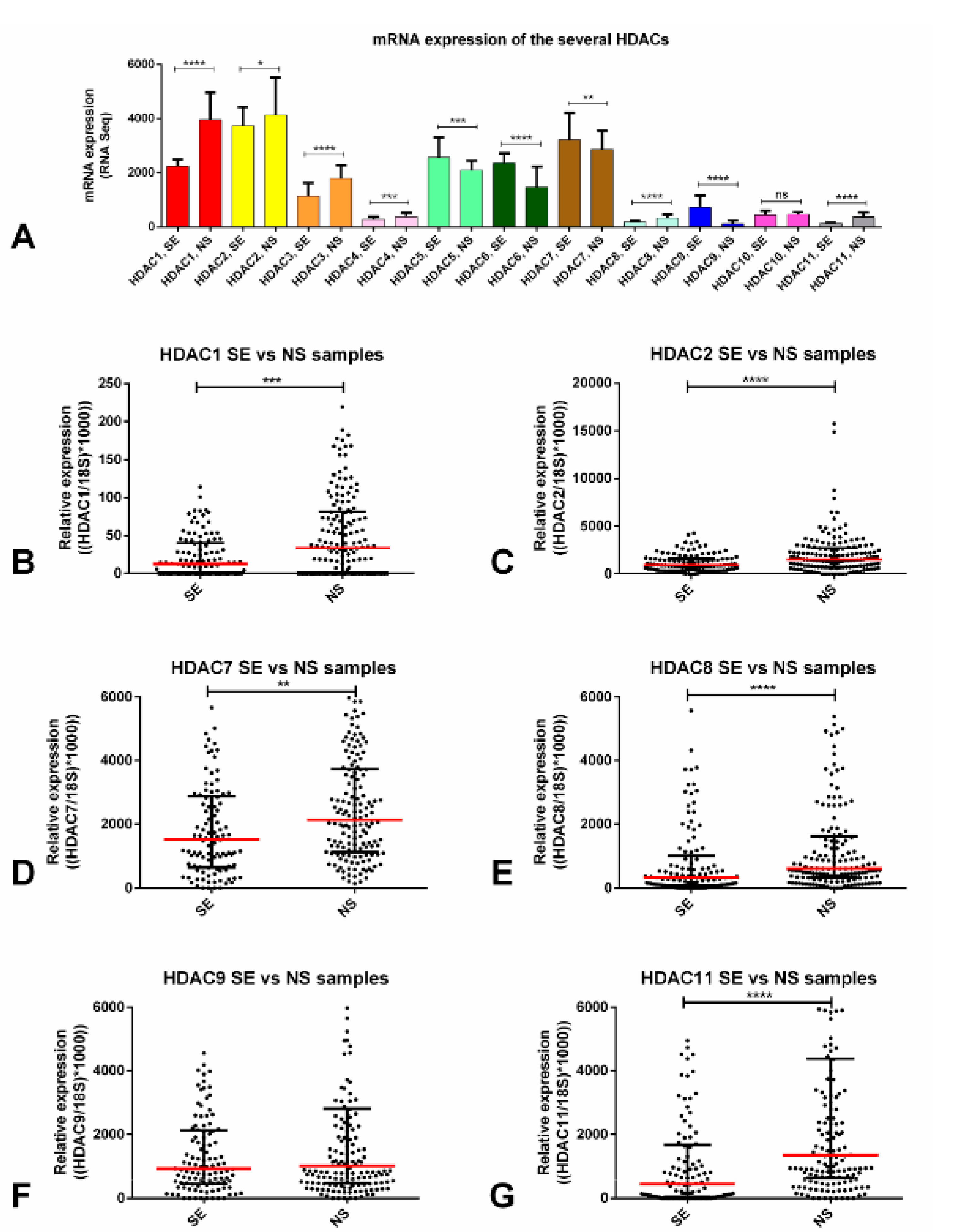
2.1. HDACs Are Differentially Expressed among TGCT Patient Samples, Including Those Exposed to Cisplatin: In Silico Analyses and Validation
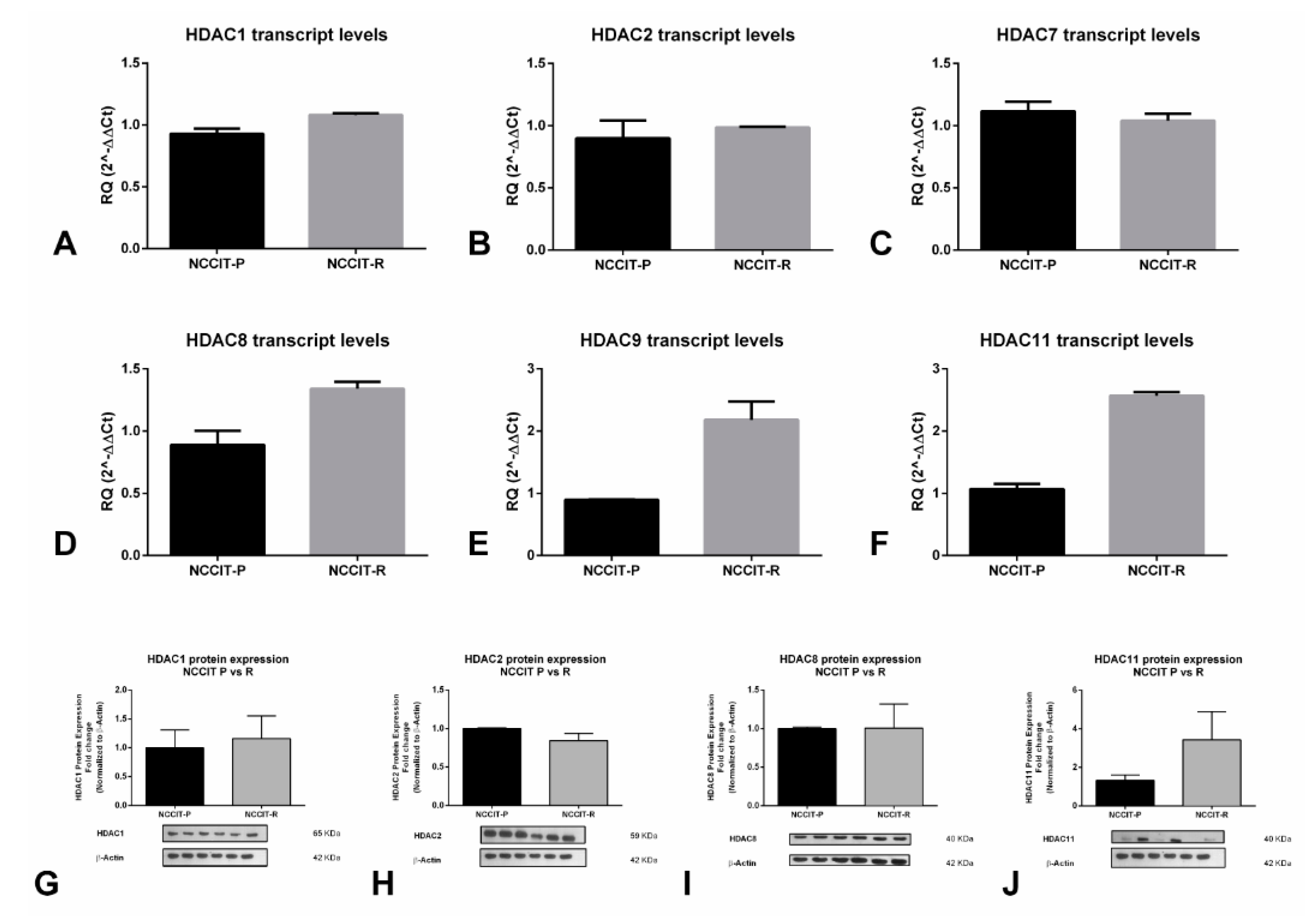
2.2. HDACs Are Differentially Expressed among (T)GCT Cell Lines, Including the Resistant and Parental Subclones
2.3. Validation of Cisplatin Resistance in the (T)GCT Cell Lines
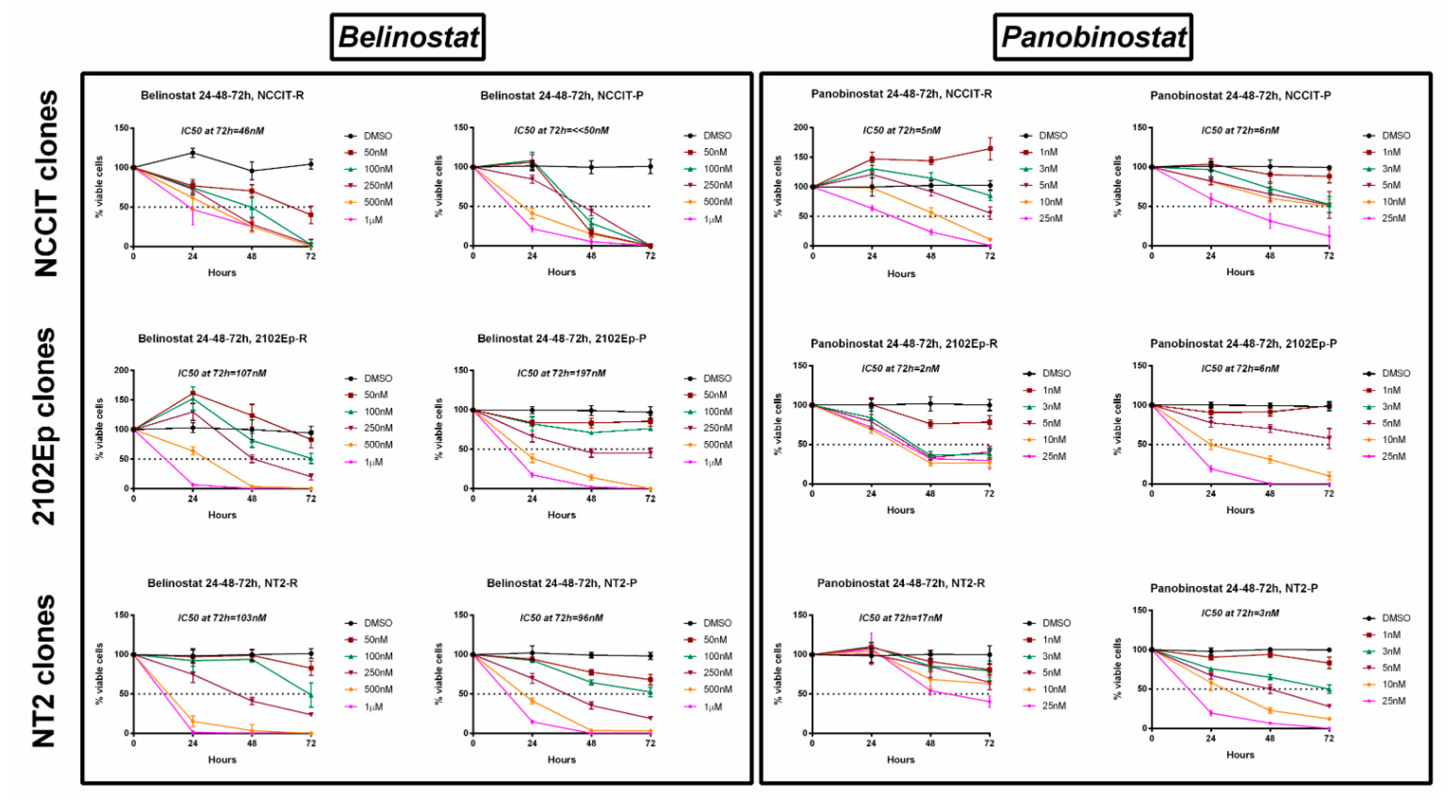
2.4. Treatment of Cisplatin-Sensitive and Cisplatin-Resistant Cell Lines with Belinostat or Panobinostat Decreases Cell Viability
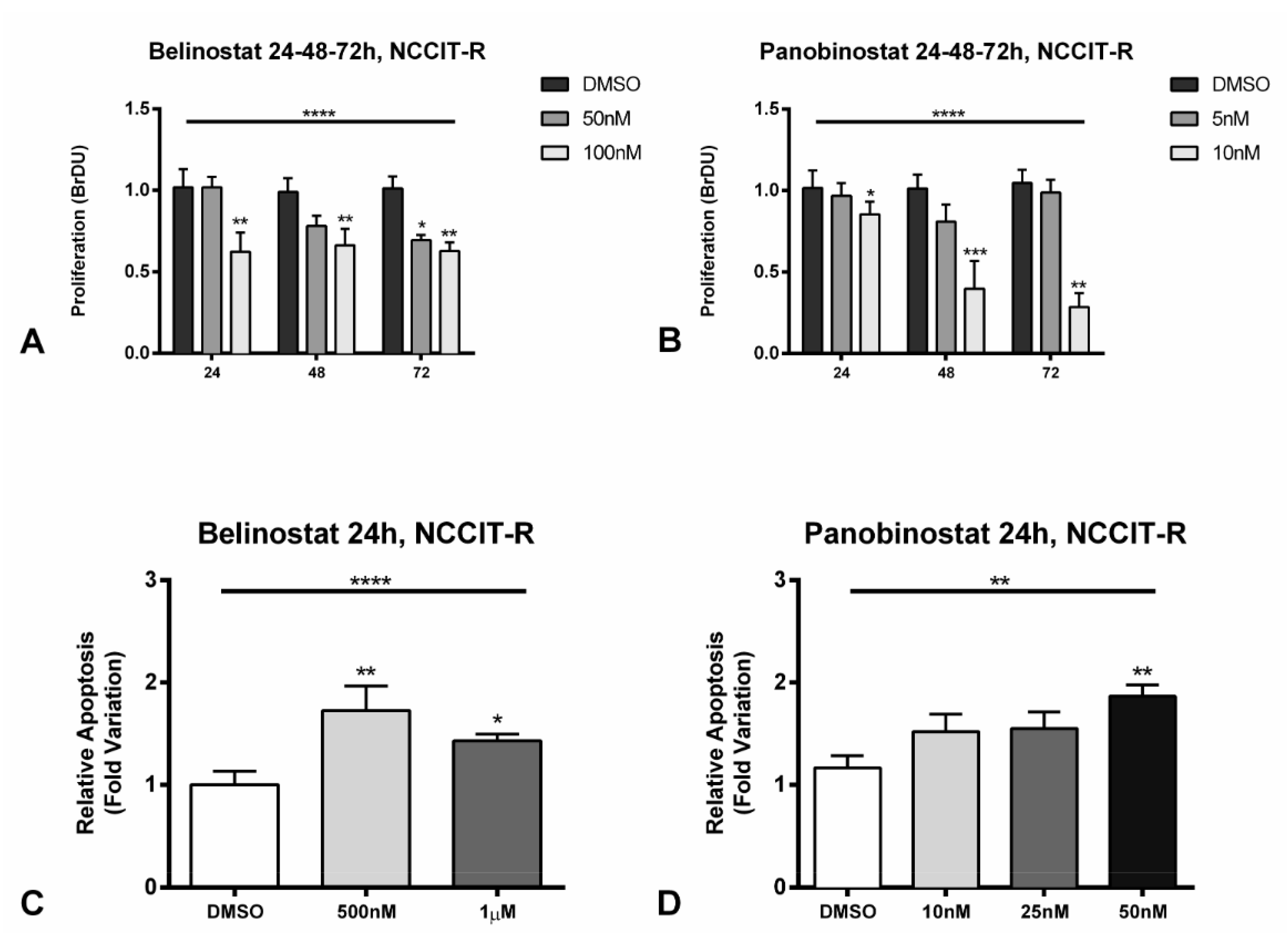
2.5. Treatment of Cisplatin-Resistant Cell Line NCCIT-R with Belinostat or Panobinostat Induces Cell Cycle Arrest and Promotes Apoptosis by Targeting Related Signaling Pathways, and Increases Acetylation Levels
2.6. Pre-Treatment of Cisplatin-Resistant Cell Line NCCIT-R with Non-Toxic Concentrations of Belinostat Increases Sensitivity to Cisplatin
3. Discussion
4. Materials and Methods
4.1. In Silico Analysis
4.2. Patient Samples
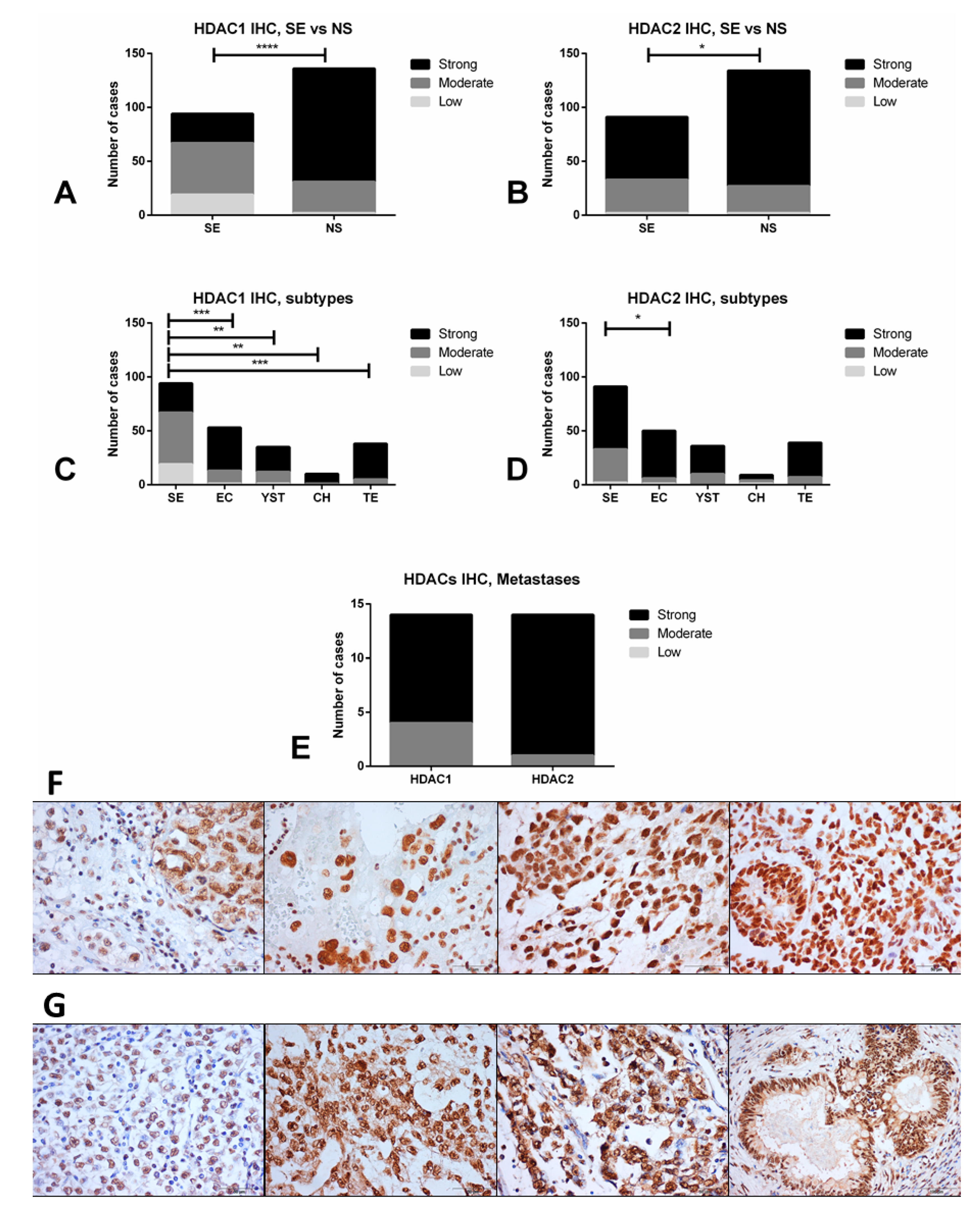
4.3. Immunohistochemistry
4.4. Cell Lines and Drugs
4.5. RNA Extraction, cDNA Synthesis, and RT-qPCR
4.6. Protein Extraction and Quantification
4.7. Western Blot
4.8. Immunofluorescence
4.9. Cell Viability Assays
4.10. Proliferation Assays
4.11. Apoptosis Assays
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lobo, J.; Gillis, A.J.M.; Jeronimo, C.; Henrique, R.; Looijenga, L.H.J. Human Germ Cell Tumors are Developmental Cancers: Impact of Epigenetics on Pathobiology and Clinic. Int. J. Mol. Sci. 2019, 20, 258. [Google Scholar] [CrossRef] [Green Version]
- Oosterhuis, J.W.; Looijenga, L.H.J. Human germ cell tumours from a developmental perspective. Nat. Rev. Cancer 2019, 19, 522–537. [Google Scholar] [CrossRef]
- Lobo, J.; Costa, A.L.; Vilela-Salgueiro, B.; Rodrigues, A.; Guimaraes, R.; Cantante, M.; Lopes, P.; Antunes, L.; Jeronimo, C.; Henrique, R. Testicular germ cell tumors: Revisiting a series in light of the new WHO classification and AJCC staging systems, focusing on challenges for pathologists. Hum. Pathol. 2018, 82, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Honecker, F.; Aparicio, J.; Berney, D.; Beyer, J.; Bokemeyer, C.; Cathomas, R.; Clarke, N.; Cohn-Cedermark, G.; Daugaard, G.; Dieckmann, K.P.; et al. ESMO Consensus Conference on testicular germ cell cancer: Diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, 1658–1686. [Google Scholar] [CrossRef] [PubMed]
- Moul, J.W.; Dodge, R.K.; Robertson, J.E.; Paulson, D.F.; Walther, P.J. The impact of the “cisplatin era” of treatment on survival in testicular cancer. World J. Urol. 1991, 9, 45–50. [Google Scholar] [CrossRef]
- Cheng, L.; Albers, P.; Berney, D.M.; Feldman, D.R.; Daugaard, G.; Gilligan, T.; Looijenga, L.H.J. Testicular cancer. Nat. Rev. Dis. Primers 2018, 4, 29. [Google Scholar] [CrossRef]
- Chovanec, M.; Abu Zaid, M.; Hanna, N.; El-Kouri, N.; Einhorn, L.H.; Albany, C. Long-term toxicity of cisplatin in germ-cell tumor survivors. Ann. Oncol. 2017, 28, 2670–2679. [Google Scholar] [CrossRef]
- Oing, C.; Seidel, C.; Bokemeyer, C. Therapeutic approaches for refractory germ cell cancer. Expert Rev. Anticancer Ther. 2018, 18, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Alsdorf, W.H.; von Amsberg, G.; Oechsle, K.; Bokemeyer, C. Platinum-refractory germ cell tumors: An update on current treatment options and developments. World J. Urol. 2017, 35, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Schmidtova, S.; Kalavska, K.; Kucerova, L. Molecular Mechanisms of Cisplatin Chemoresistance and Its Circumventing in Testicular Germ Cell Tumors. Curr. Oncol. Rep. 2018, 20, 88. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, C.; Honecker, F. Cisplatin resistance in germ cell tumours: Models and mechanisms. Andrology 2015, 3, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaidy, K.I.; Chovanec, M.; Cheng, L. Molecular characteristics of testicular germ cell tumors: Pathogenesis and mechanisms of therapy resistance. Expert Rev. Anticancer Ther. 2020, 20, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Loveday, C.; Litchfield, K.; Proszek, P.Z.; Cornish, A.J.; Santo, F.; Levy, M.; Macintyre, G.; Holryod, A.; Broderick, P.; Dudakia, D.; et al. Genomic landscape of platinum resistant and sensitive testicular cancers. Nat. Commun. 2020, 11, 2189. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors. Cancer Drug Resist. 2019, 2, 580–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; et al. Role of promoter hypermethylation in Cisplatin treatment response of male germ cell tumors. Mol. Cancer 2004, 3, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Fazal, Z.; Corbet, A.K.; Bikorimana, E.; Rodriguez, J.C.; Khan, E.M.; Shahid, K.; Freemantle, S.J.; Spinella, M.J. Epigenetic Remodeling through Downregulation of Polycomb Repressive Complex 2 Mediates Chemotherapy Resistance in Testicular Germ Cell Tumors. Cancers 2019, 11, 796. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.L.; Lobo, J.; Jeronimo, C.; Henrique, R. The epigenetics of testicular germ cell tumors: Looking for novel disease biomarkers. Epigenomics 2017, 9, 155–169. [Google Scholar] [CrossRef]
- Liu, L.; Lian, J.; Zhang, H.; Tian, H.; Liang, M.; Yin, M.; Sun, F. MicroRNA-302a sensitizes testicular embryonal carcinoma cells to cisplatin-induced cell death. J. Cell Physiol. 2013, 228, 2294–2304. [Google Scholar] [CrossRef]
- Oing, C.; Skowron, M.A.; Bokemeyer, C.; Nettersheim, D. Epigenetic treatment combinations to effectively target cisplatin-resistant germ cell tumors: Past, present, and future considerations. Andrology 2019, 7, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, A.R.; Lobo, J.; Miranda-Goncalves, V.; Henrique, R.; Jeronimo, C. Epigenetic alterations as therapeutic targets in Testicular Germ Cell Tumours: Current and future application of ‘epidrugs’. Epigenetics 2020, 1–20. [Google Scholar] [CrossRef]
- Wongtrakoongate, P.; Li, J.; Andrews, P.W. Aza-deoxycytidine induces apoptosis or differentiation via DNMT3B and targets embryonal carcinoma cells but not their differentiated derivatives. Br. J. Cancer 2014, 110, 2131–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswal, B.K.; Beyrouthy, M.J.; Hever-Jardine, M.P.; Armstrong, D.; Tomlinson, C.R.; Christensen, B.C.; Marsit, C.J.; Spinella, M.J. Acute hypersensitivity of pluripotent testicular cancer-derived embryonal carcinoma to low-dose 5-aza deoxycytidine is associated with global DNA Damage-associated p53 activation, anti-pluripotency and DNA demethylation. PLoS ONE 2012, 7, e53003. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Verem, I.; Mansour, W.Y.; Bokemeyer, C.; Dyshlovoy, S.; Honecker, F. 5-Azacitidine Exerts Prolonged Pro-Apoptotic Effects and Overcomes Cisplatin-Resistance in Non-Seminomatous Germ Cell Tumor Cells. Int. J. Mol. Sci. 2018, 20, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albany, C.; Hever-Jardine, M.P.; von Herrmann, K.M.; Yim, C.Y.; Tam, J.; Warzecha, J.M.; Shin, L.; Bock, S.E.; Curran, B.S.; Chaudhry, A.S.; et al. Refractory testicular germ cell tumors are highly sensitive to the second generation DNA methylation inhibitor guadecitabine. Oncotarget 2017, 8, 2949–2959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostes, S.; Nettersheim, D.; Fellermeyer, M.; Schneider, S.; Hafezi, F.; Honecker, F.; Schumacher, V.; Geyer, M.; Kristiansen, G.; Schorle, H. The bromodomain inhibitor JQ1 triggers growth arrest and apoptosis in testicular germ cell tumours in vitro and in vivo. J. Cell. Mol. Med. 2017, 21, 1300–1314. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Jostes, S.; Fabry, M.; Honecker, F.; Schumacher, V.; Kirfel, J.; Kristiansen, G.; Schorle, H. A signaling cascade including ARID1A, GADD45B and DUSP1 induces apoptosis and affects the cell cycle of germ cell cancers after romidepsin treatment. Oncotarget 2016, 7, 74931–74946. [Google Scholar] [CrossRef]
- Nettersheim, D.; Gillis, A.; Biermann, K.; Looijenga, L.H.; Schorle, H. The seminoma cell line TCam-2 is sensitive to HDAC inhibitor depsipeptide but tolerates various other chemotherapeutic drugs and loss of NANOG expression. Genes Chromosom. Cancer 2011, 50, 1033–1042. [Google Scholar] [CrossRef]
- Bakardjieva-Mihaylova, V.; Skvarova Kramarzova, K.; Slamova, M.; Svaton, M.; Rejlova, K.; Zaliova, M.; Dobiasova, A.; Fiser, K.; Stuchly, J.; Grega, M.; et al. Molecular Basis of Cisplatin Resistance in Testicular Germ Cell Tumors. Cancers 2019, 11, 1316. [Google Scholar] [CrossRef] [Green Version]
- Steele, N.; Finn, P.; Brown, R.; Plumb, J.A. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef] [Green Version]
- Kalavska, K.; Conteduca, V.; De Giorgi, U.; Mego, M. Molecular Mechanisms of Resistance in Testicular Germ Cell Tumors—Clinical Implications. Curr. Cancer Drug Targets 2018, 18, 967–978. [Google Scholar] [CrossRef]
- Allen, J.C.; Kirschner, A.; Scarpato, K.R.; Morgans, A.K. Current Management of Refractory Germ Cell Tumors and Future Directions. Curr. Oncol. Rep. 2017, 19, 8. [Google Scholar] [CrossRef] [PubMed]
- Mego, M.; Svetlovska, D.; Chovanec, M.; Reckova, M.; Rejlekova, K.; Obertova, J.; Palacka, P.; Sycova-Mila, Z.; De Giorgi, U.; Mardiak, J. Phase II study of avelumab in multiple relapsed/refractory germ cell cancer. Investig. New Drugs 2019, 37, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Hentrich, M.; Lorch, A.; Glaser, D.; Rumpold, H.; Ochsenreither, S.; Richter, S.; Dieing, A.; Zschabitz, S.; Pereira, R.R.; et al. Treatment of refractory germ-cell tumours with single-agent cabazitaxel: A German Testicular Cancer Study Group case series. J. Cancer Res. Clin. Oncol. 2020, 146, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Fenner, M.; Oing, C.; Dieing, A.; Gauler, T.; Oechsle, K.; Lorch, A.; Hentrich, M.; Kopp, H.G.; Bokemeyer, C.; Honecker, F. Everolimus in patients with multiply relapsed or cisplatin refractory germ cell tumors: Results of a phase II, single-arm, open-label multicenter trial (RADIT) of the German Testicular Cancer Study Group. J. Cancer Res. Clin. Oncol. 2019, 145, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Adra, N.; Einhorn, L.H.; Althouse, S.K.; Ammakkanavar, N.R.; Musapatika, D.; Albany, C.; Vaughn, D.; Hanna, N.H. Phase II trial of pembrolizumab in patients with platinum refractory germ-cell tumors: A Hoosier Cancer Research Network Study GU14-206. Ann. Oncol. 2018, 29, 209–214. [Google Scholar] [CrossRef]
- Necchi, A.; Giannatempo, P.; Raggi, D.; Mariani, L.; Colecchia, M.; Fare, E.; Monopoli, F.; Calareso, G.; Ali, S.M.; Ross, J.S.; et al. An Open-label Randomized Phase 2 study of Durvalumab Alone or in Combination with Tremelimumab in Patients with Advanced Germ Cell Tumors (APACHE): Results from the First Planned Interim Analysis. Eur. Urol. 2019, 75, 201–203. [Google Scholar] [CrossRef]
- Beyrouthy, M.J.; Garner, K.M.; Hever, M.P.; Freemantle, S.J.; Eastman, A.; Dmitrovsky, E.; Spinella, M.J. High DNA methyltransferase 3B expression mediates 5-aza-deoxycytidine hypersensitivity in testicular germ cell tumors. Cancer Res. 2009, 69, 9360–9366. [Google Scholar] [CrossRef] [Green Version]
- Matei, D.; Ghamande, S.; Roman, L.; Alvarez Secord, A.; Nemunaitis, J.; Markham, M.J.; Nephew, K.P.; Jueliger, S.; Oganesian, A.; Naim, S.; et al. A Phase I Clinical Trial of Guadecitabine and Carboplatin in Platinum-Resistant, Recurrent Ovarian Cancer: Clinical, Pharmacokinetic, and Pharmacodynamic Analyses. Clin. Cancer Res. 2018, 24, 2285–2293. [Google Scholar] [CrossRef] [Green Version]
- Roth, B.J.; Elson, P.; Sledge, G.W., Jr.; Einhorn, L.H.; Trump, D.L. 5-Azacytidine (NSC 102816) in refractory germ cell tumors. A phase II trial of the Eastern Cooperative Oncology Group. Investig. New Drugs 1993, 11, 201–202. [Google Scholar] [CrossRef]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Moreira-Silva, F.; Camilo, V.; Gaspar, V.; Mano, J.F.; Henrique, R.; Jeronimo, C. Repurposing Old Drugs into New Epigenetic Inhibitors: Promising Candidates for Cancer Treatment? Pharmaceutics 2020, 12, 410. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Omisanjo, O.A.; Biermann, K.; Hartmann, S.; Heukamp, L.C.; Sonnack, V.; Hild, A.; Brehm, R.; Bergmann, M.; Weidner, W.; Steger, K. DNMT1 and HDAC1 gene expression in impaired spermatogenesis and testicular cancer. Histochem. Cell Biol. 2007, 127, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, F.R.; Hasler, A.; Bode, P.K.; Adams, H.; Seifert, H.H.; Sulser, T.; Moch, H.; Barghorn, A.; Kristiansen, G. Expression of histone deacetylases 1, 2 and 3 in histological subtypes of testicular germ cell tumours. Histol. Histopathol. 2011, 26, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Henrique, R.; Jeronimo, C. The Role of DNA/Histone Modifying Enzymes and Chromatin Remodeling Complexes in Testicular Germ Cell Tumors. Cancers 2018, 11, 6. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Lan, R.; Zhang, X.; Zhu, L.; Chen, F.; Xu, Z.; Liu, Y.; Ye, T.; Sun, H.; Lu, F.; et al. LSD1 regulates pluripotency of embryonic stem/carcinoma cells through histone deacetylase 1-mediated deacetylation of histone H4 at lysine 16. Mol. Cell. Biol. 2014, 34, 158–179. [Google Scholar] [CrossRef] [Green Version]
- Bora-Singhal, N.; Mohankumar, D.; Saha, B.; Colin, C.M.; Lee, J.Y.; Martin, M.W.; Zheng, X.; Coppola, D.; Chellappan, S. Novel HDAC11 inhibitors suppress lung adenocarcinoma stem cell self-renewal and overcome drug resistance by suppressing Sox2. Sci. Rep. 2020, 10, 4722. [Google Scholar] [CrossRef]
- Eini, R.; Stoop, H.; Gillis, A.J.; Biermann, K.; Dorssers, L.C.; Looijenga, L.H. Role of SOX2 in the etiology of embryonal carcinoma, based on analysis of the NCCIT and NT2 cell lines. PLoS ONE 2014, 9, e83585. [Google Scholar] [CrossRef] [Green Version]
- Nettersheim, D.; Arndt, I.; Sharma, R.; Riesenberg, S.; Jostes, S.; Schneider, S.; Holzel, M.; Kristiansen, G.; Schorle, H. The cancer/testis-antigen PRAME supports the pluripotency network and represses somatic and germ cell differentiation programs in seminomas. Br. J. Cancer 2016, 115, 454–464. [Google Scholar] [CrossRef] [Green Version]
- Nettersheim, D.; Berger, D.; Jostes, S.; Skowron, M.; Schorle, H. Deciphering the molecular effects of romidepsin on germ cell tumours: DHRS2 is involved in cell cycle arrest but not apoptosis or induction of romidepsin effectors. J. Cell. Mol. Med. 2019, 23, 670–679. [Google Scholar] [CrossRef]
- Rivers, Z.T.; Oostra, D.R.; Westholder, J.S.; Vercellotti, G.M. Romidepsin-associated cardiac toxicity and ECG changes: A case report and review of the literature. J. Oncol. Pharm. Pract. 2018, 24, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagosklonny, M.V.; Bates, S.E. P21-dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Luchenko, V.L.; Salcido, C.D.; Zhang, Y.; Agama, K.; Komlodi-Pasztor, E.; Murphy, R.F.; Giaccone, G.; Pommier, Y.; Bates, S.E.; Varticovski, L. Schedule-dependent synergy of histone deacetylase inhibitors with DNA damaging agents in small cell lung cancer. Cell Cycle 2011, 10, 3119–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Blake, M.; Baek, J.H.; Kohlhagen, G.; Pommier, Y.; Carrier, F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003, 63, 7291–7300. [Google Scholar]
- Groh, T.; Hrabeta, J.; Khalil, M.A.; Doktorova, H.; Eckschlager, T.; Stiborova, M. The synergistic effects of DNA-damaging drugs cisplatin and etoposide with a histone deacetylase inhibitor valproate in high-risk neuroblastoma cells. Int. J. Oncol. 2015, 47, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Goncalves, V.; Lameirinhas, A.; Henrique, R.; Jeronimo, C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef] [Green Version]
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; van der Kuip, H. Cisplatin hypersensitivity of testicular germ cell tumors is determined by high constitutive Noxa levels mediated by Oct-4. Cancer Res. 2013, 73, 1460–1469. [Google Scholar] [CrossRef] [Green Version]
- Grande, L.; Bretones, G.; Rosa-Garrido, M.; Garrido-Martin, E.M.; Hernandez, T.; Fraile, S.; Botella, L.; de Alava, E.; Vidal, A.; Garcia del Muro, X.; et al. Transcription factors Sp1 and p73 control the expression of the proapoptotic protein NOXA in the response of testicular embryonal carcinoma cells to cisplatin. J. Biol. Chem. 2012, 287, 26495–26505. [Google Scholar] [CrossRef] [Green Version]
- Beyer, U.; Moll-Rocek, J.; Moll, U.M.; Dobbelstein, M. Endogenous retrovirus drives hitherto unknown proapoptotic p63 isoforms in the male germ line of humans and great apes. Proc. Natl. Acad. Sci. USA 2011, 108, 3624–3629. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, P.M.; Read, G. International Germ Cell Consensus Classification: A prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J. Clin. Oncol. 1997, 15, 594–603. [Google Scholar] [CrossRef]
- Costa, A.L.; Moreira-Barbosa, C.; Lobo, J.; Vilela-Salgueiro, B.; Cantante, M.; Guimaraes, R.; Lopes, P.; Braga, I.; Oliveira, J.; Antunes, L.; et al. DNA methylation profiling as a tool for testicular germ cell tumors subtyping. Epigenomics 2018, 10, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Costa, A.L.; Cantante, M.; Guimaraes, R.; Lopes, P.; Antunes, L.; Braga, I.; Oliveira, J.; Pelizzola, M.; Henrique, R.; et al. m(6)A RNA modification and its writer/reader VIRMA/YTHDF3 in testicular germ cell tumors: A role in seminoma phenotype maintenance. J. Transl. Med. 2019, 17, 79. [Google Scholar] [CrossRef] [Green Version]
- Lobo, J.; Rodrigues, A.; Guimaraes, R.; Cantante, M.; Lopes, P.; Mauricio, J.; Oliveira, J.; Jeronimo, C.; Henrique, R. Detailed Characterization of Immune Cell Infiltrate and Expression of Immune Checkpoint Molecules PD-L1/CTLA-4 and MMR Proteins in Testicular Germ Cell Tumors Disclose Novel Disease Biomarkers. Cancers 2019, 11, 1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, J.; Gillis, A.J.M.; van den Berg, A.; Dorssers, L.C.J.; Belge, G.; Dieckmann, K.P.; Roest, H.P.; van der Laan, L.J.W.; Gietema, J.; Hamilton, R.J.; et al. Identification and Validation Model for Informative Liquid Biopsy-Based microRNA Biomarkers: Insights from Germ Cell Tumor In Vitro, In Vivo and Patient-Derived Data. Cells 2019, 8, 1637. [Google Scholar] [CrossRef] [Green Version]
- Skowron, M.A.; Vermeulen, M.; Winkelhausen, A.; Becker, T.K.; Bremmer, F.; Petzsch, P.; Schonberger, S.; Calaminus, G.; Kohrer, K.; Albers, P.; et al. CDK4/6 inhibition presents as a therapeutic option for paediatric and adult germ cell tumours and induces cell cycle arrest and apoptosis via canonical and non-canonical mechanisms. Br. J. Cancer 2020, 123, 378–391. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobo, J.; Guimarães-Teixeira, C.; Barros-Silva, D.; Miranda-Gonçalves, V.; Camilo, V.; Guimarães, R.; Cantante, M.; Braga, I.; Maurício, J.; Oing, C.; et al. Efficacy of HDAC Inhibitors Belinostat and Panobinostat against Cisplatin-Sensitive and Cisplatin-Resistant Testicular Germ Cell Tumors. Cancers 2020, 12, 2903. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12102903
Lobo J, Guimarães-Teixeira C, Barros-Silva D, Miranda-Gonçalves V, Camilo V, Guimarães R, Cantante M, Braga I, Maurício J, Oing C, et al. Efficacy of HDAC Inhibitors Belinostat and Panobinostat against Cisplatin-Sensitive and Cisplatin-Resistant Testicular Germ Cell Tumors. Cancers. 2020; 12(10):2903. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12102903
Chicago/Turabian StyleLobo, João, Catarina Guimarães-Teixeira, Daniela Barros-Silva, Vera Miranda-Gonçalves, Vânia Camilo, Rita Guimarães, Mariana Cantante, Isaac Braga, Joaquina Maurício, Christoph Oing, and et al. 2020. "Efficacy of HDAC Inhibitors Belinostat and Panobinostat against Cisplatin-Sensitive and Cisplatin-Resistant Testicular Germ Cell Tumors" Cancers 12, no. 10: 2903. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12102903