
Mir-21 Suppression Promotes Mouse Hepatocarcinogenesis

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. RNA Sequencing Analysis
2.3. Histology
2.4. Immunohistochemistry
2.5. Magnetic Resonance Imaging (MRI) and CT-Scan Imaging (CT)
2.6. Real-Time PCR
2.7. Western Blot
2.8. Cell Culture
2.9. In Silico Analysis
2.9.1. List of Predicted and Validated Targets
2.9.2. Gene Ontology Enrichment Analysis
2.9.3. Identification of Genes of Potential Interest and Related Biological Functions
2.10. Statistical Analysis
3. Results
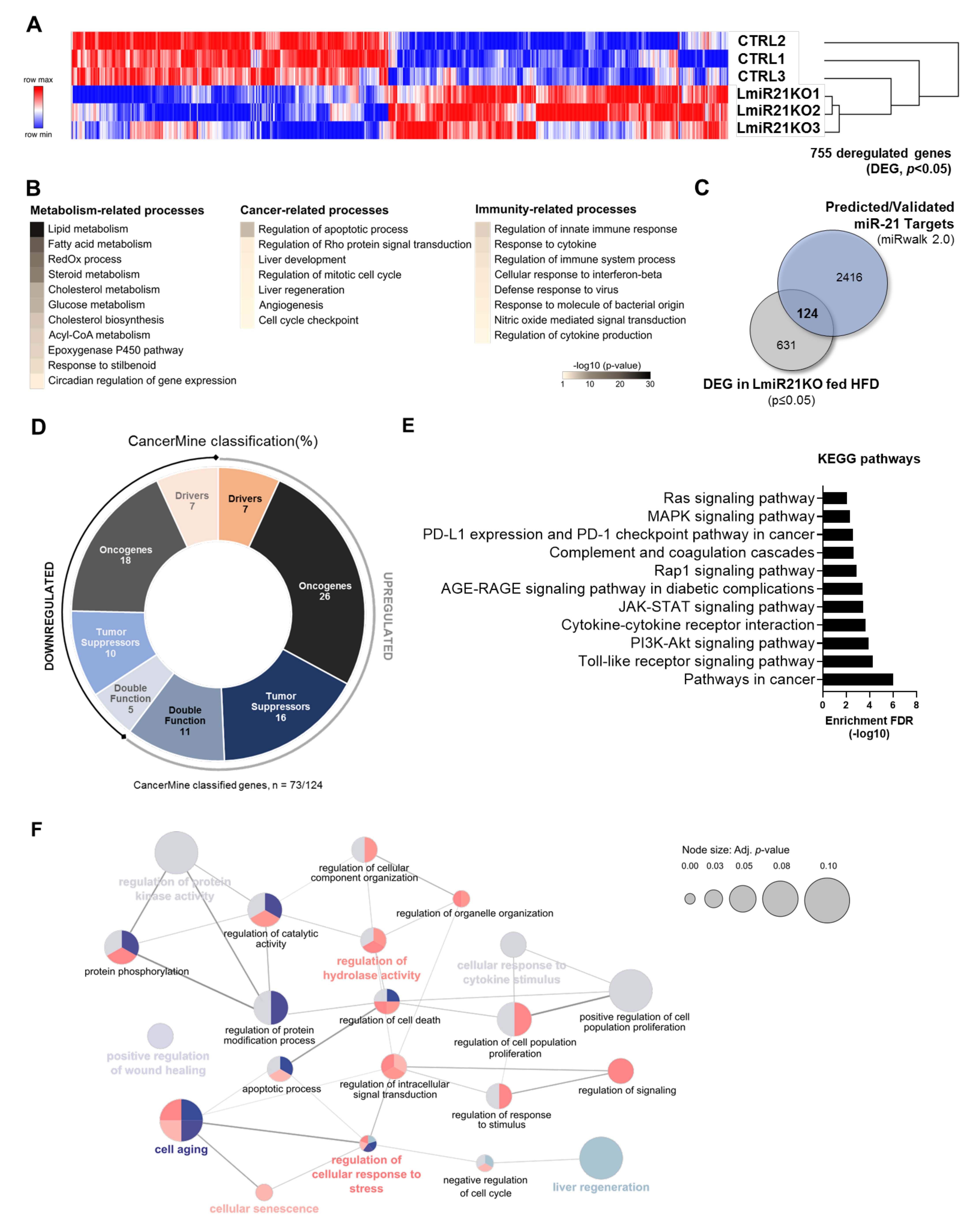
3.1. miR-21 Suppression in the Liver Deregulates Numerous Genes Relevant for Hepatocarcinogenesis and Immune Responses
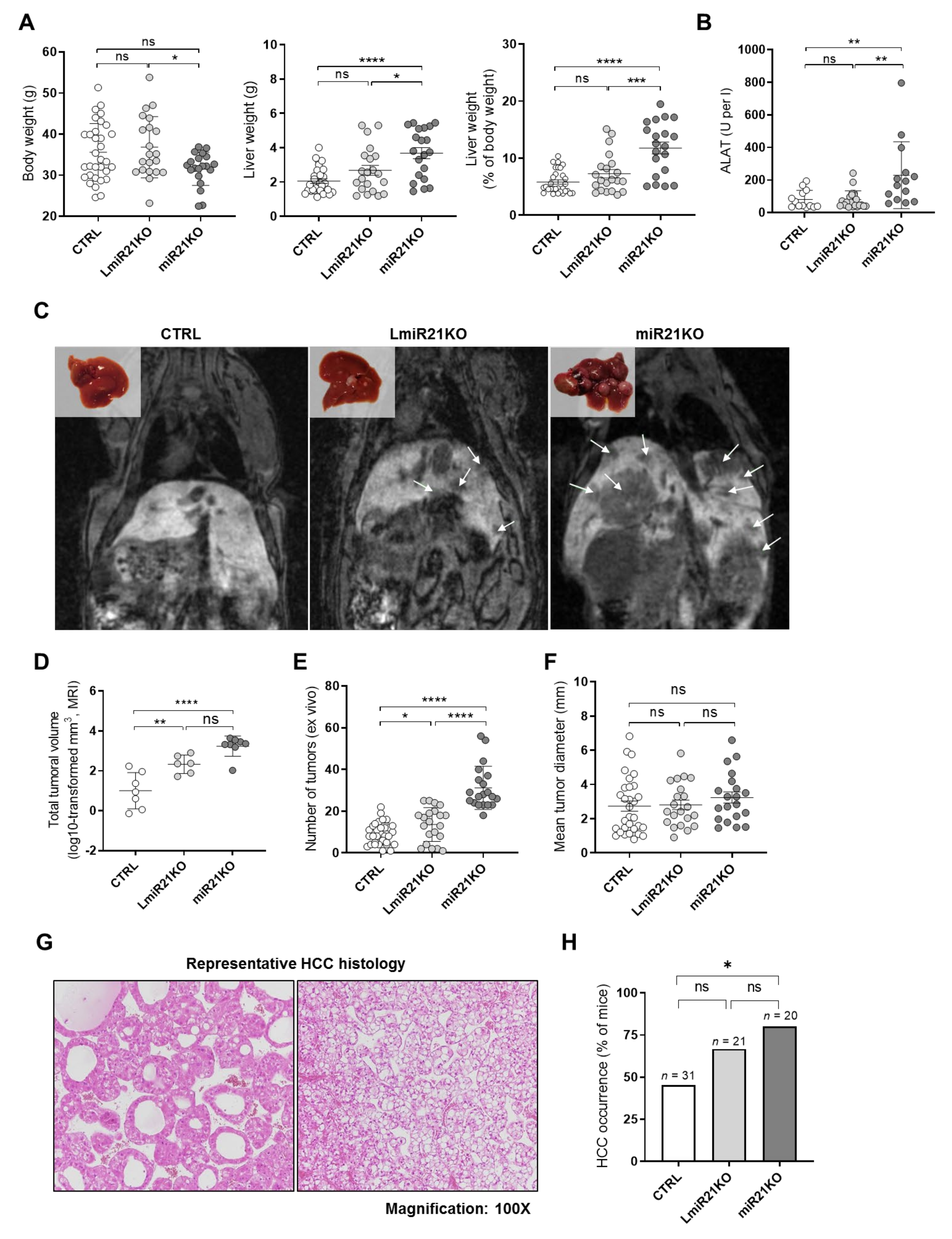
3.2. Whole-Body and Liver-Specific MiR-21 Deficiency Fosters Carcinogen (DEN)-Induced HCC Development in Mice
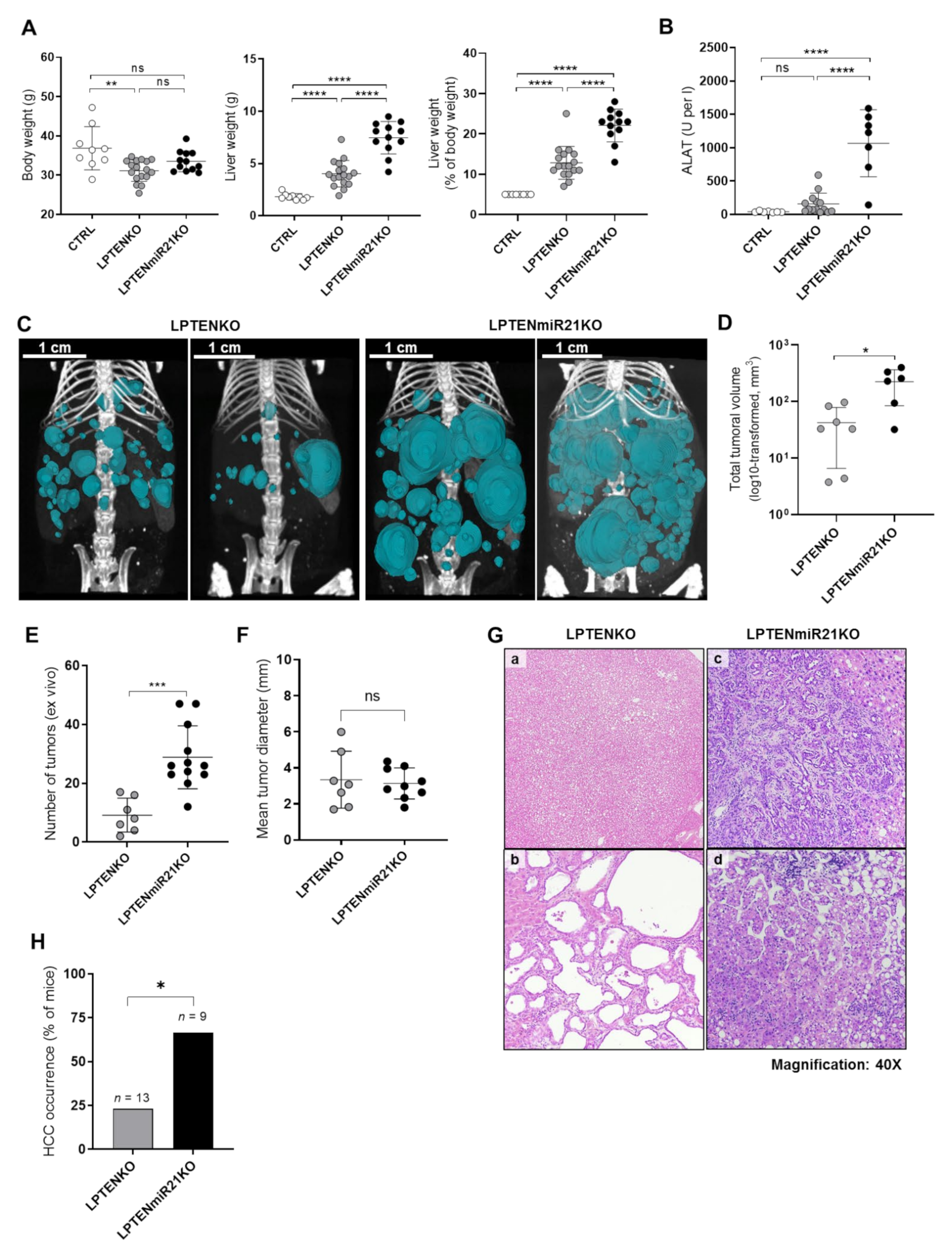
3.3. Hepatocyte-Specific miR-21 Ablation Strongly Exacerbates Inflammation, Fibrosis, and Hepatocarcinogenesis Induced by PTEN Deficiency
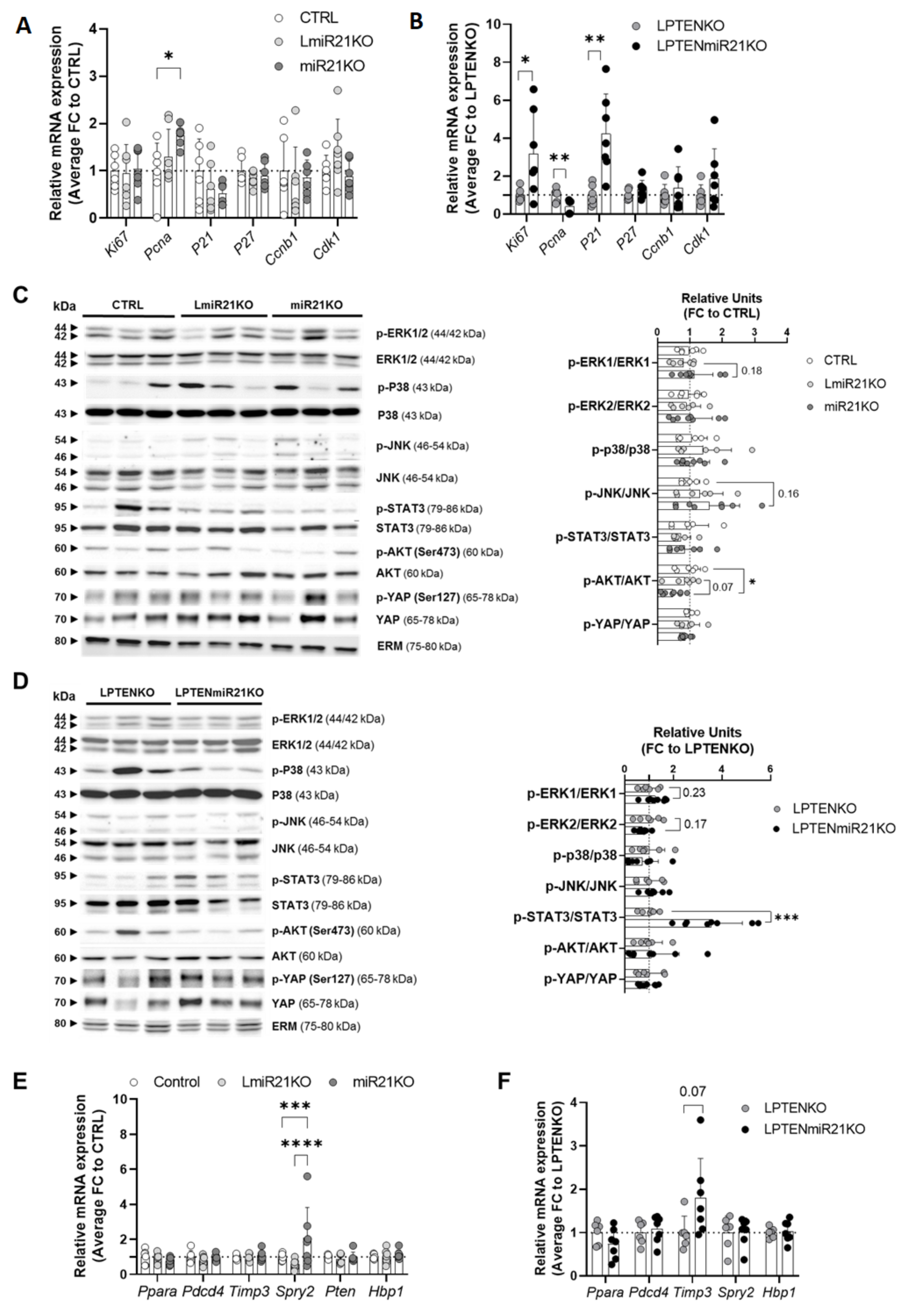
3.4. Hepatic miR-21 Suppression Is Associated with Pleiotropic and Context-Dependent Molecular Alterations driving Carcinogenesis
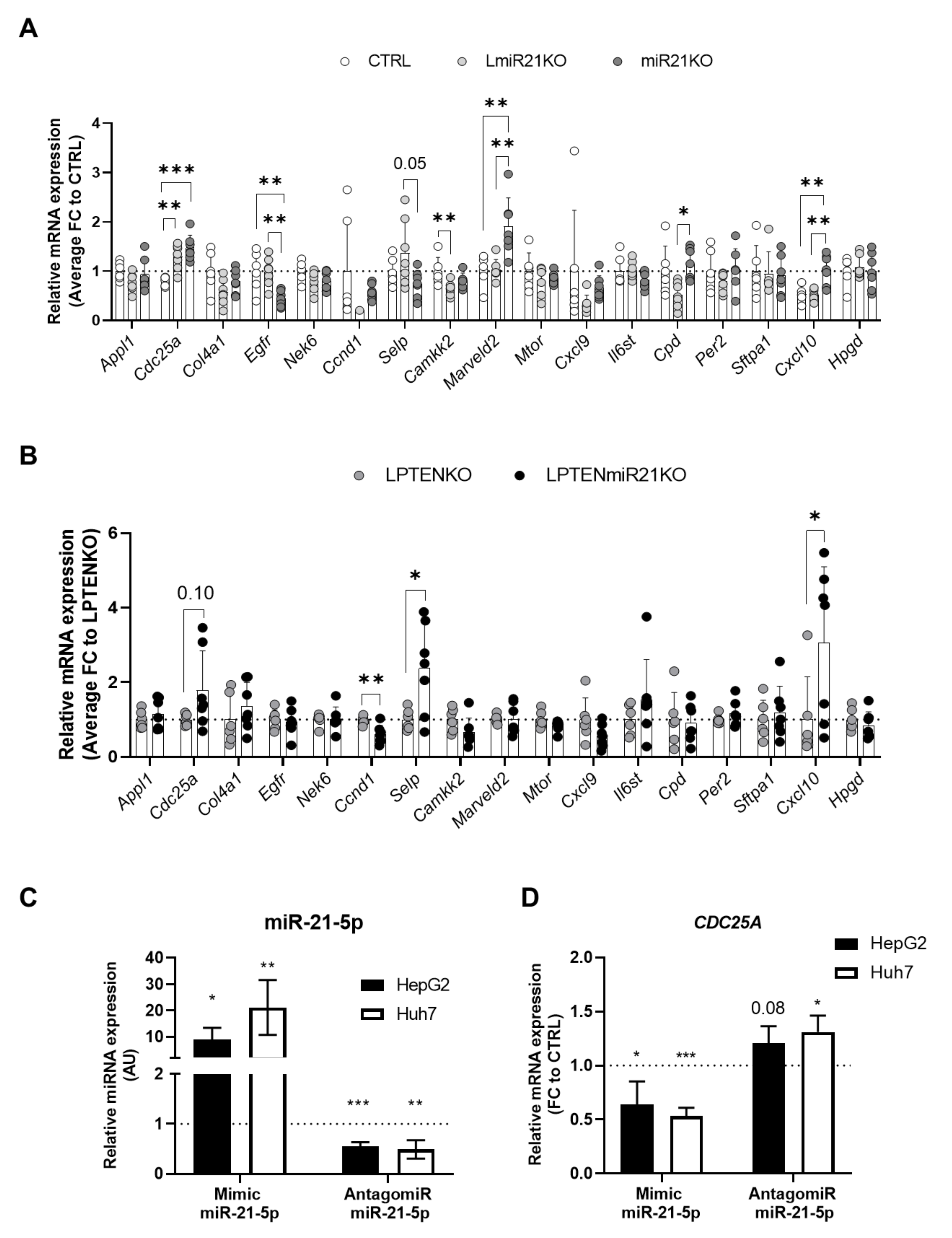
3.5. Increased Expression of Oncogenes and Deregulation of Immunity-Related Factors Are Associated with the Loss of miR-21 in the Liver
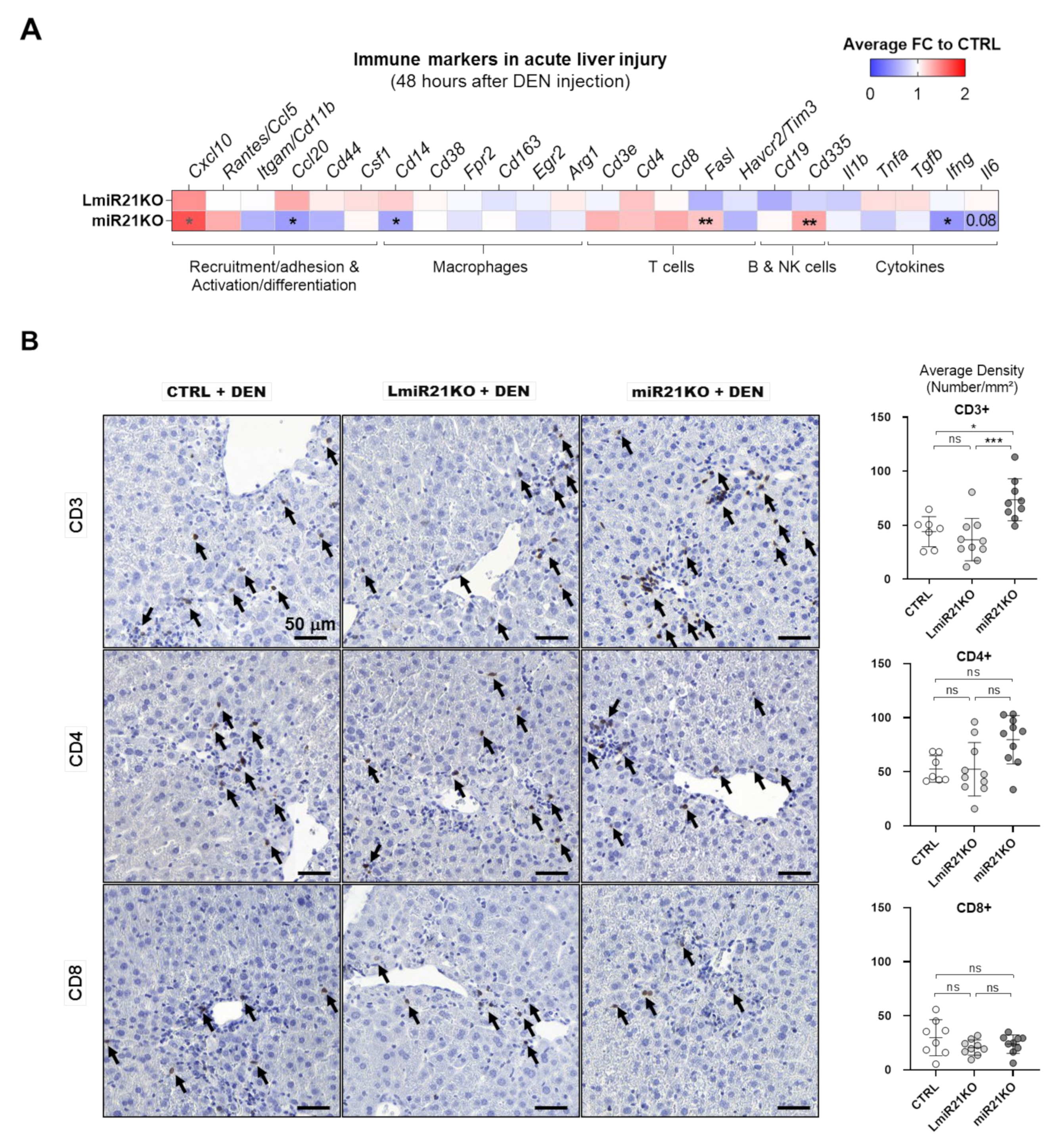
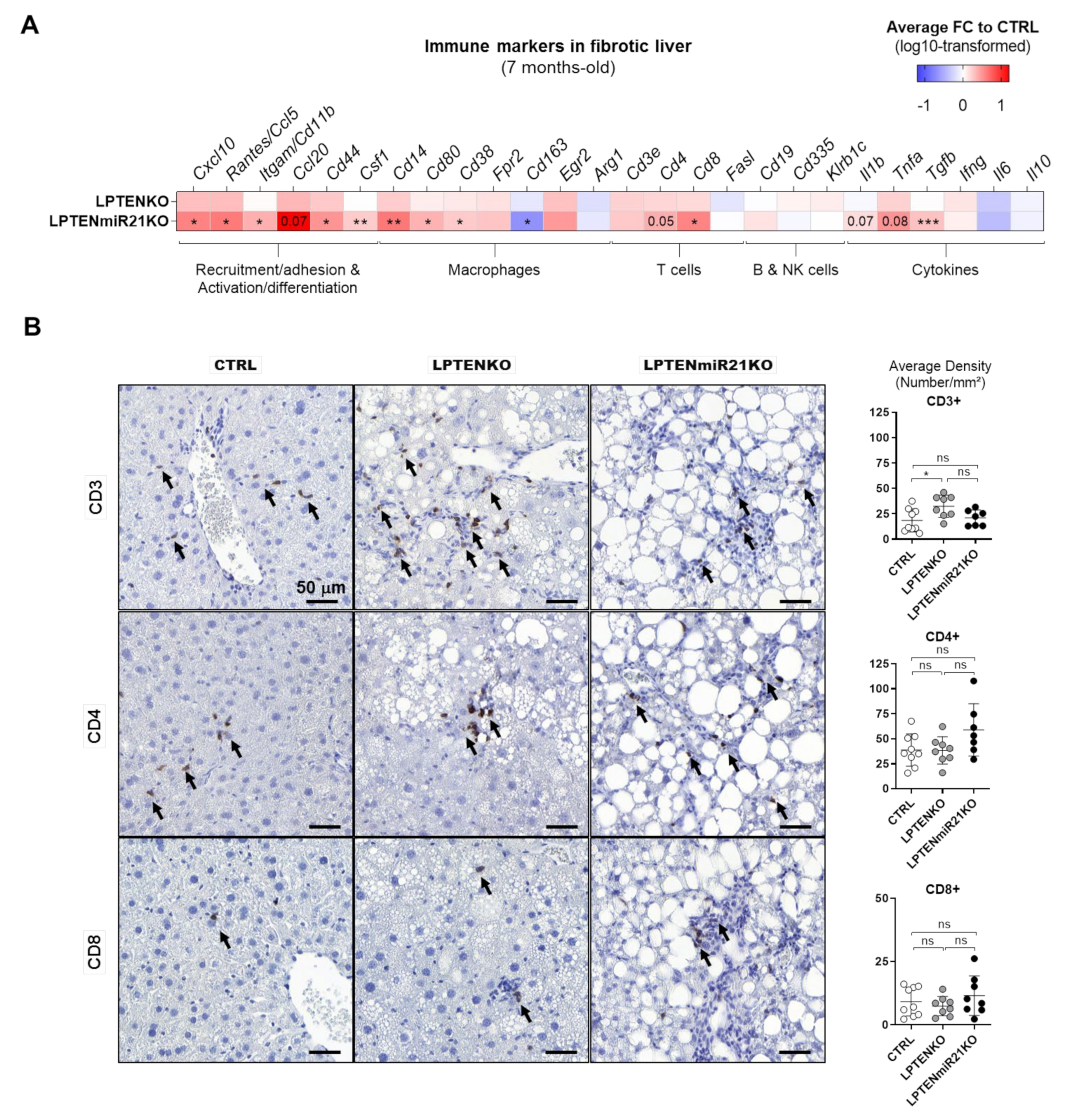
3.6. MiR-21 Deletion Differentially Affects Inflammatory and Immune Responses Induced by Acute DEN Exposure and PTEN Deficiency in the Liver
4. Discussion
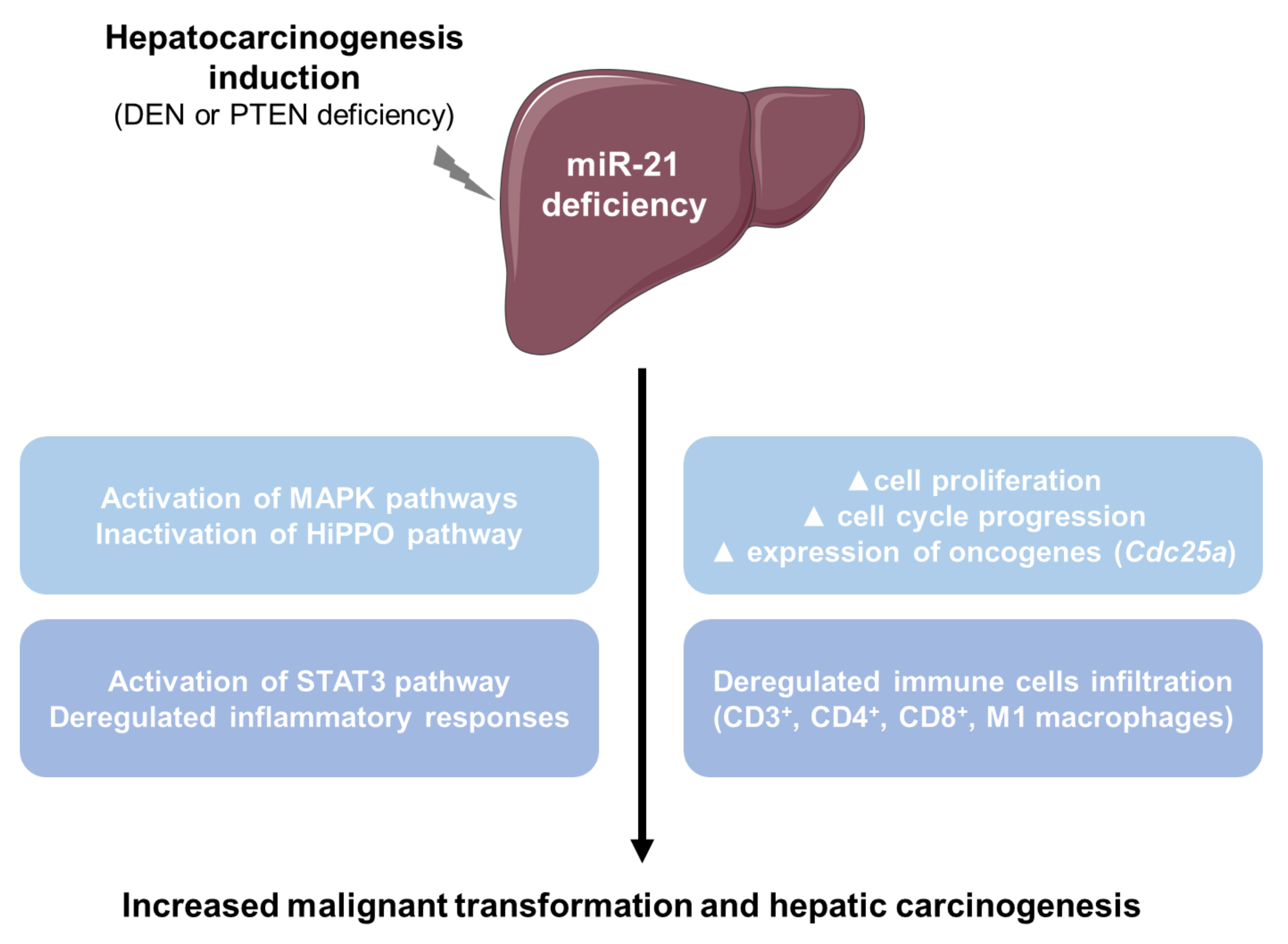
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Scwhartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2018, 17, 748–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrone, A.K.; Shpyleva, S.; Chappel, G.; Tryndyak, V.; Uehara, T.; Tsuchiya, M.; Beland, F.A.; Rusyn, I.; Pogribny, I.P. Differentially Expressed MicroRNAs Provide Mechanistic Insight into Fibrosis-Associated Liver Carcinogenesis in Mice. Mol. Carcinog. 2015, 55, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Dolicka, D.; Sobolewski, C.; Correia de Sousa, M.; Gjorgjieva, M.; Foti, M. mRNA Post-Transcriptional Regulation by AU-Rich Element-Binding Proteins in Liver Inflammation and Cancer. Int. J. Mol. Sci. 2020, 21, 6648. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; Correia de Sousa, M.; Foti, M. miRNAs and NAFLD: From Pathophysiology to Therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jiang, L.; Guan, X.-Y. The Genetic and Epigenetic Alterations in Human Hepatocellular Carcinoma: A Recent Update. Protein Cell 2014, 5, 673–691. [Google Scholar] [CrossRef] [Green Version]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouzé, E.; Blanc, J.-F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological Subtypes of Hepatocellular Carcinoma Are Related to Gene Mutations and Molecular Tumour Classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Saborowski, A. Current Strategies for the Treatment of Intermediate and Advanced Hepatocellular Carcinoma. Cancer Treat. Rev. 2020, 82, 101946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, G.-S. Conflicting Roles of Molecules in Hepatocarcinogenesis: Paradigm or Paradox. Cancer Cell 2012, 21, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib Versus Sorafenib in First-Line Treatment of Patients with Unresectable Hepatocellular Carcinoma: A Randomised Phase 3 Non-inferiority Trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Marisi, G.; Cucchetti, A.; Ulivi, P.; Canale, M.; Canibbo, G.; Solaini, L.; Foschi, F.G.; De Matteis, S.; Ercolani, G.; Valgiusti, M.; et al. Ten Years of Sorafenib in Hepatocellular Carcinoma: Are There Any Predictive and/or Prognostic Markers? World J. Gastroenterol. 2018, 24, 4152–4163. [Google Scholar] [CrossRef]
- Chowdhury, S.M.; Wang, T.-Y.; Bachawal, S.; Devulapally, R.; Choe, J.W.; Elkacem, L.A.; Yakub, B.K.; Wang, D.S.; Tian, L.; Paulmurugan, R.; et al. Ultrasound-Guided Therapeutic Modulation of Hepatocellular Carcinoma Using Complementary microRNAs. J. Control. Release 2016, 238, 272–280. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.F. miRNA Targeting Drugs: The Next Blockbusters? In Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2017; Volume 1517, pp. 3–22. [Google Scholar]
- Correia de Sousa, M.; Gjorgjieva, M.; Dolicka, D.; Sobolewski, C.; Foti, M. Deciphering miRNAs’ Action through miRNA Editing. Int. J. Mol. Sci. 2019, 20, 6249. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Rana, T.M. Therapeutic Targeting of microRNAs: Current Status and Future Challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef]
- Otsuka, M.; Kishikawa, T.; Yoshikawa, T.; Yamagami, M.; Ohno, M.; Takata, A.; Shibata, C.; Ishibashi, R.; Koike, K. MicroRNAs and Liver Disease. J. Hum. Genet. 2016, 62, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Sánchez, D.; Arriaga-Canon, C.; Pedroza-Torres, A.; De La Rosa-Velázquez, I.A.; González-Barrios, R.; Contreras-Espinosa, L.; Montiel-Manríquez, R.; Castro-Hernández, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. The Promising Role of miR-21 as a Cancer Biomarker and Its Importance in RNA-Based Therapeutics. Mol. Ther.-Nucleic Acids 2020, 20, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Loyer, X.; Paradis, V.; Hénique, C.; Vion, A.-C.; Colnot, N.; Guerin, L.C.; Devue, C.; On, S.; Scetbun, J.; Romain, M.; et al. Liver microRNA-21 Is Overexpressed in Non-Alcoholic Steatohepatitis and Contributes to the Disease in Experimental Models by Inhibiting PPARα Expression. Gut 2015, 65, 1882–1894. [Google Scholar] [CrossRef] [Green Version]
- Calo, N.; Ramadori, P.; Sobolewski, C.; Romero, Y.; Maeder, C.; Fournier, M.; Rantakari, P.; Zhang, F.-P.; Poutanen, M.; Dufour, J.-F.; et al. Stress-Activated miR-21/miR-21* in Hepatocytes Promotes Lipid and Glucose Metabolic Disorders Associated with High-Fat Diet Consumption. Gut 2016, 65, 1871–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Ng, R.; Chen, X.; Steer, C.J.; Song, G. MicroRNA-21 Is a Potential Link Between Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma via Modulation of the HBP1-p53-Srebp1c Pathway. Gut 2015, 65, 1850–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, P.M.; Afonso, M.B.; Simão, A.L.; Carvalho, C.C.; Trindade, A.; Duarte, A.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, M.P.C.; et al. miR-21 Ablation and Obeticholic Acid Ameliorate Nonalcoholic Steatohepatitis in Mice. Cell Death Dis. 2017, 8, 2748. [Google Scholar] [CrossRef]
- Zhang, T.; Yang, Z.; Kusumanchi, P.; Han, S.; Liangpunsakul, S. Critical Role of microRNA-21 in the Pathogenesis of Liver Diseases. Front. Med. 2020, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenaar, T.R.; Zabludoff, S.; Ahn, S.-M.; Allerson, C.; Arlt, H.; Baffa, R.; Cao, H.; Davis, S.; Garcia-Echeverria, C.; Gaur, R.; et al. Anti-miR-21 Suppresses Hepatocellular Carcinoma Growth via Broad Transcriptional Network Deregulation. Mol. Cancer Res. 2015, 13, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Sheedy, F.J. Turning 21: Induction of miR-21 as a Key Switch in the Inflammatory Response. Front. Immunol. 2015, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Henson, R.; Wehbe–Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 Regulates Expression of the PTEN Tumor Suppressor Gene in Human Hepatocellular Cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-S.; Yu, W.; Cui, H.; Wang, Y.-J.; Zhang, L.; Han, F.; Huang, T. Increased Expression of miR-21 Predicts Poor Prognosis in Patients with Hepatocellular Carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 7234–7238. [Google Scholar]
- Bao, L.; Yan, Y.; Xu, C.; Ji, W.; Shen, S.; Xu, G.; Zeng, Y.; Sun, B.; Qian, H.; Chen, L.; et al. MicroRNA-21 Suppresses PTEN and hSulf-1 Expression and Promotes Hepatocellular Carcinoma Progression through AKT/ERK Pathways. Cancer Lett. 2013, 337, 226–236. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, Z.-X.; Song, W.-J.; Li, Q.-J.; Yang, F.; Wang, D.-S.; Zhang, N.; Dou, K.-F. MicroRNA-21 Regulates the Migration and Invasion of a Stem-Like Population in Hepatocellular Carcinoma. Int. J. Oncol. 2013, 43, 661–669. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chu, Y.; Xu, M.; Zhang, X.; Zhou, Y. miR-21 promotes cell migration and invasion of hepatocellular carcinoma by targeting KLF5. Oncol. Lett. 2018, 17, 2221–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscaglia, L.E.; Li, Y. Apoptosis and the Target Genes of microRNA-21. Chin. J. Cancer 2011, 30, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Yan-Nan, B.; Zhao-Yan, Y.; Li-Xi, L.; Jiang, Y.; Qing-Jie, X.; Yong, Z. MicroRNA-21 accelerates hepatocyte proliferation in vitro via PI3K/Akt signaling by targeting PTEN. Biochem. Biophys. Res. Commun. 2014, 443, 802–807. [Google Scholar] [CrossRef]
- Zhao, J.; Tang, N.; Wu, K.; Dai, W.; Ye, C.; Shi, J.; Zhang, J.; Ning, B.; Zeng, X.; Lin, Y. MiR-21 Simultaneously Regulates ERK1 Signaling in HSC Activation and Hepatocyte EMT in Hepatic Fibrosis. PLoS ONE 2014, 9, e108005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Jiao, J.; Cermelli, S.; Muir, K.; Jung, K.H.; Zou, R.; Rashid, A.; Gagea, M.; Zabludoff, S.; Kalluri, R.; et al. miR-21 Inhibition Reduces Liver Fibrosis and Prevents Tumor Development by Inducing Apoptosis of CD24+ Progenitor Cells. Cancer Res. 2015, 75, 1859–1867. [Google Scholar] [CrossRef] [Green Version]
- Caviglia, J.M.; Yan, J.; Jang, M.-K.; Gwak, G.-Y.; Affo, S.; Yu, L.; Olinga, P.; Friedman, R.A.; Chen, X.; Schwabe, R.F. MicroRNA-21 and Dicer are Dispensable for Hepatic Stellate Cell Activation and the Development of Liver Fibrosis. Hepatology 2017, 67, 2414–2429. [Google Scholar] [CrossRef] [Green Version]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-Specific Pten Deficiency Results in Steatohepatitis and Hepatocellular Carcinomas. J. Clin. Investig. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [Green Version]
- Peyrou, M.; Bourgoin, L.; Poher, A.-L.; Altirriba, J.; Maeder, C.; Caillon, A.; Fournier, M.; Montet, X.; Rohner-Jeanrenaud, F.; Foti, M. Hepatic PTEN Deficiency Improves Muscle Insulin Sensitivity and Decreases Adiposity in Mice. J. Hepatol. 2015, 62, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender Disparity in Liver Cancer Due to Sex Differences in MyD88-Dependent IL-6 Production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An Online Resource for Prediction of microRNA Binding Sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioin-Formatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics Enrichment Tools: Paths Toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lever, J.; Zhao, E.Y.; Grewal, J.; Jones, M.R.; Jones, S.J.M. CancerMine: A Literature-Mined Resource for Drivers, Oncogenes and Tumor Suppressors in Cancer. Nat. Methods 2019, 16, 505–507. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. Cluego: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Galon, J.; Mlecnik, B. Cluepedia Cytoscape Plugin: Pathway Insights Using Integrated Experimental and In Silico Data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-Set Enrichment Tool for Animals and Plants. Bioinformatics 2019, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Connor, F.; Rayner, T.F.; Aitken, S.; Feig, C.; Lukk, M.; Santoyo-Lopez, J.; Odom, D.T. Mutational Landscape of a Chemically-Induced Mouse Model of Liver Cancer. J. Hepatol. 2018, 69, 840–850. [Google Scholar] [CrossRef]
- Schulien, I.; Hasselblatt, P. Diethylnitrosamine-Induced Liver Tumorigenesis in Mice. Methods Cell Biol. 2021, 163, 137–152. [Google Scholar]
- McGill, M.R. The Past and Present of Serum Aminotransferases and the Future of Liver Injury Biomarkers. EXCLI J. 2016, 15, 817. [Google Scholar] [PubMed]
- Febbraio, M.A.; Reibe, S.; Shalapour, S.; Ooi, J.G.; Watt, M.J.; Karin, M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab. 2019, 29, 18–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Chen, S.; Zhu, Z.; Yu, L.; Ren, Y.; Jiang, M.; Weng, J.; Li, B. miR-21 Promotes EGF-Induced Pancreatic Cancer Cell Proliferation by Targeting Spry2. Cell Death Dis. 2018, 9, 1157. [Google Scholar] [CrossRef]
- Liu, F.; Zheng, S.; Liu, T.; Liu, Q.; Liang, M.; Li, X.; Sheyhidin, I.; Lu, X.; Liu, W. MicroRNA-21 Promotes the Proliferation and Inhibits Apoptosis in Eca109 via Activating ERK1/2/MAPK Pathway. Mol. Cell Biochem. 2013, 381, 115–125. [Google Scholar] [CrossRef]
- Jin, Y. 3,3′-Diindolylmethane Inhibits Breast Cancer Cell Growth via miR-21-Mediated Cdc25A Degradation. Mol. Cell. Biochem. 2011, 358, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zou, F.; Zhang, X.; Li, H.; Dulak, A.; Tomko, R.J.; Lazo, J.S.; Wang, Z.; Zhang, L.; Yu, J. microRNA-21 Negatively Regulates Cdc25A and Cell Cycle Progression in Colon Cancer Cells. Cancer Res. 2009, 69, 8157–8165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.Q.; Lin, H.Z.; Mandal, A.K.; Huang, J.; Diehl, A.M. Disrupted Signaling and Inhibited Regeneration in Obese Mice with Fatty Livers: Implications for Nonalcoholic Fatty Liver Disease Pathophysiology. Hepatology 2001, 34, 694–706. [Google Scholar] [CrossRef]
- Ezquer, F.; Bahamonde, J.; Huang, Y.-L.; Ezquer, M. Administration of Multipotent Mesenchymal Stromal Cells Restores Liver Regeneration and Improves Liver Function in Obese Mice with Hepatic Steatosis After Partial Hepatectomy. Stem Cell Res. Ther. 2017, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, A.; Kukla, M.; Adamek, B.; Tabor, K.; Banasik, G.; Horyniecki, M.; Halota, W. Steatosis Influences Hepatocytes Proliferative Potential in Chronic Hepatitis C Patients. Pol. J. Pathol. 2018, 69, 388–394. [Google Scholar] [CrossRef]
- Jiang, L.-H.; Ge, M.-H.; Hou, X.-X.; Cao, J.; Hu, S.-S.; Lu, X.-X.; Han, J.; Wu, Y.-C.; Liu, X.; Zhu, X.; et al. miR-21 Regulates Tumor Progression Through the miR-21-PDCD4-Stat3 Pathway in Human Salivary Adenoid Cystic Carcinoma. Lab. Investig. 2015, 95, 1398–1408. [Google Scholar] [CrossRef]
- Mei, Y.; Bian, C.; Li, J.; Du, Z.; Zhou, H.; Yang, Z.; Zhao, R.C.H. miR-21 Modulates the ERK-MAPK Signaling Pathway by Regulating SPRY2 Expression During Human Mesenchymal Stem Cell Differentiation. J. Cell. Biochem. 2012, 114, 1374–1384. [Google Scholar] [CrossRef]
- Hong, Y.; Ye, M.; Wang, F.; Fang, J.; Wang, C.; Luo, J.; Liu, J.; Liu, J.; Liu, L.; Zhao, Q.; et al. MiR-21-3p Promotes Hepatocellular Carcinoma Progression via SMAD7/YAP1 Regulation. Front. Oncol. 2021, 11, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Selaru, F.M.; Olaru, A.V.; Kan, T.; David, S.; Cheng, Y.; Mori, Y.; Yang, J.; Paun, B.; Jin, Z.; Agarwal, R.; et al. MicroRNA-21 Is Overexpressed in Human Cholangiocarcinoma and Regulates Programmed Cell Death 4 and Tissue Inhibitor of Metalloproteinase 3. Hepatology 2009, 49, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Hatley, M.; Patrick, D.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; Van Rooij, E.; Olson, E.N. Modulation of K-Ras-Dependent Lung Tumorigenesis by MicroRNA-21. Cancer Cell 2010, 18, 282–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Dong, X.; Zhai, B.; Jiang, X.; Dong, D.; Li, B.; Jiang, H.; Xu, S.; Sun, X. MiR-21 Mediates Sorafenib Resistance of Hepatocellular Carcinoma Cells by Inhibiting Autophagy via the PTEN/Akt Pathway. Oncotarget 2015, 6, 28867–28881. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Brandt, S.; Medeiros, A.; Wang, S.; Wu, H.; Dent, A.; Serezani, C.H. MicroRNA 21 Is a Homeostatic Regulator of Macrophage Polarization and Prevents Prostaglandin E2-Mediated M2 Generation. PLoS ONE 2015, 10, e0115855. [Google Scholar] [CrossRef]
- He, W.; Wang, C.; Mu, R.; Liang, P.; Huang, Z.; Zhang, J.; Dong, L. MiR-21 Is Required for Anti-Tumor Immune Response in Mice: An Implication for Its Bi-Directional Roles. Oncogene 2017, 36, 4212–4223. [Google Scholar] [CrossRef]
- Karakatsanis, A.; Papaconstantinou, I.; Gazouli, M.; Lyberopoulou, A.; Polymeneas, G.; Voros, D. Expression of microRNAs, miR-21, miR-31, miR-122, miR-145, miR-146a, miR-200c, miR-221, miR-222, and miR-223 in Patients with Hepatocellular Carcinoma or Intrahepatic Cholangiocarcinoma and Its Prognostic Significance. Mol. Carcinog. 2013, 52, 297–303. [Google Scholar] [CrossRef]
- Guo, X.; Lv, X.; Lv, X.; Ma, Y.; Chen, L.; Chen, Y. Circulating miR-21 Serves as a Serum Biomarker for Hepatocellular Carcinoma and Correlated with Distant Metastasis. Oncotarget 2017, 8, 44050–44058. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.R.; Yang, C.H.; Pfeffer, L.M. The Role of miR-21 in Cancer. Drug Dev. Res. 2015, 76, 270–277. [Google Scholar] [CrossRef]
- Jiao, W.; Leng, X.; Zhou, Q.; Wu, Y.; Sun, L.; Tan, Y.; Ni, H.; Dong, X.; Shen, T.; Liu, Y.; et al. Different miR-21-3p Isoforms and Their Different Features in Colorectal Cancer. Int. J. Cancer 2017, 141, 2103–2111. [Google Scholar] [CrossRef] [Green Version]
- Schipper, J.; Westerhuis, J.J.; Beddows, I.; Madaj, Z.; Monsma, D.; Hostetter, G.; Kiupel, M.; Conejo-Garcia, J.; Sempere, L.F. Loss of microRNA -21 Leads to Profound Stromal Remodeling and Short Survival in K-Ras -Driven Mouse Models of Pancreatic Cancer. Int. J. Cancer 2020, 147, 2265–2278. [Google Scholar] [CrossRef]
- Patrick, D.; Montgomery, R.L.; Qi, X.; Obad, S.; Kauppinen, S.; Hill, J.A.; Van Rooij, E.; Olson, E.N. Stress-Dependent Cardiac Remodeling Occurs in the Absence of microRNA-21 in Mice. J. Clin. Investig. 2010, 120, 3912–3916. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Ma, W.; Hao, B.; Hu, F.; Yan, L.; Yan, X.; Wang, Y.; Chen, Z.; Wang, Z. microRNA-21 Promotes Cardiac Fibrosis and Development of Heart Failure with Preserved Left Ventricular Ejection Fraction by Up-Regulating Bcl-2. Int. J. Clin. Exp. Pathol. 2014, 7, 565–574. [Google Scholar]
- Morrisey, E.E. The Magic and Mystery of miR-21. J. Clin. Investig. 2010, 120, 3817–3819. [Google Scholar] [CrossRef] [Green Version]
- Lo, T.-F.; Tsai, W.-C.; Chen, S.-T. MicroRNA-21-3p, a Berberine-Induced miRNA, Directly Down-Regulates Human Methionine Adenosyltransferases 2A and 2B and Inhibits Hepatoma Cell Growth. PLoS ONE 2013, 8, e75628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Dov, I.Z.; Muthukumar, T.; Morozov, P.; Mueller, F.B.; Tuschl, T.; Suthanthiran, M. MicroRNA Sequence Profiles of Human Kidney Allografts with or without Tubulointerstitial Fibrosis. Transplantation 2012, 94, 1086–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, P.; Zhao, X.-D.; Shi, K.-H.; Ding, X.-S.; Tao, H. MiR-21–3p Triggers Cardiac Fibroblasts Pyroptosis in Diabetic Cardiac Fibrosis via Inhibiting Androgen Receptor. Exp. Cell Res. 2020, 399, 112464. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Fang, Q.; Chen, C.; Zhou, L.; Li, H.; Yin, Z.; Wang, Y.; Zhao, C.X.; Xiao, X.; Wang, D.W. Recombinant Adeno-Associated Virus-Mediated Delivery of MicroRNA-21-3p Lowers Hypertension. Mol. Ther. -Nucleic Acids 2018, 11, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Ma, Z.; Ran, Z. MiR-21-3p Modulates Lipopolysaccharide-Induced Inflammation and Apoptosis via Targeting TGS 4 in Retinal Pigment Epithelial Cells. Clin. Exp. Pharmacol. Physiol. 2019, 46, 883–889. [Google Scholar] [CrossRef]
- Ge, X.; Li, W.; Huang, S.; Yin, Z.; Yang, M.; Han, Z.; Han, Z.; Chen, F.; Wang, H.; Lei, P.; et al. Increased miR-21-3p in Injured Brain Microvascular Endothelial Cells after Traumatic Brain Injury Aggravates Blood–Brain Barrier Damage by Promoting Cellular Apoptosis and Inflammation through Targeting MAT2B. J. Neurotrauma 2019, 36, 1291–1305. [Google Scholar] [CrossRef]
- Degueurce, G.; DÉrrico, I.; Pich, C.; Ibberson, M.; Schütz, F.; Montagner, A.; Sgandurra, M.; Mury, L.; Jafari, P.; Boda, A.; et al. Identification of a Novel PPARbeta/Delta/miR-21-3p Axis in UV-Induced Skin Inflammation. EMBO Mol. Med. 2016, 8, 919–936. [Google Scholar] [CrossRef]
- Bose, D.; Nahar, S.; Rai, M.K.; Ray, A.; Chakraborty, K.; Maiti, S. Selective Inhibition of miR-21 by Phage Display Screened Peptide. Nucleic Acids Res. 2015, 43, 4342–4352. [Google Scholar] [CrossRef] [Green Version]
- Diaz, J.P.; Chirayil, R.; Chirayil, S.; Tom, M.; Head, K.J.; Luebke, K.J. Association of a Peptoid Ligand with the Apical Loop of Pri-miR-21 Inhibits Cleavage by Drosha. RNA 2014, 20, 528–539. [Google Scholar] [CrossRef] [Green Version]
- Shortridge, M.D.; Walker, M.J.; Pavelitz, T.; Chen, Y.; Yang, W.; Varani, G. A Macrocyclic Peptide Ligand Binds the Oncogenic MicroRNA-21 Precursor and Suppresses Dicer Pro-cessing. ACS Chem. Biol. 2017, 12, 1611–1620. [Google Scholar] [CrossRef]
- Green, C.J.; Pramfalk, C.; Morten, K.J.; Hodson, L. From Whole Body to Cellular Models of Hepatic Triglyceride Metabolism: Man Has Got to Know His Limitations. Am. J. Physiol. Metab. 2015, 308, 1–20. [Google Scholar] [CrossRef]
- Hirschfield, H.; Bian, C.B.; Higashi, T.; Nakagawa, S.; Zeleke, T.Z.; Nair, V.; Fuchs, B.C.; Hoshida, Y. In Vitro Modeling of Hepatocellular Carcinoma Molecular Subtypes for Anti-Cancer Drug Assessment. Exp. Mol. Med. 2018, 50, e419. [Google Scholar] [CrossRef]
- Collins, S.D.; Yuen, G.; Tu, T.; Budzinska, A.; Spring, K.; Bryant, K.; Shackel, N.A. In Vitro Models of the Liver: Disease Modeling, Drug Discovery and Clinical Applications. In Hepatocellular Carcinoma; Tirnitz-Parker, J.E.E., Ed.; Codon Publications: Brisbane, Australia, 2019. [Google Scholar]
- Dolicka, D.; Sobolewski, C.; Gjorgjieva, M.; Correia de Sousa, M.; Berthou, F.; de Vito, C.; Colin, D.J.; Bejuy, O.; Fournier, M.; Maeder, C.; et al. Tristetraprolin Promotes Hepatic Inflammation and Tumor Initiation but Restrains Cancer Progression to Ma-lignancy. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 597–621. [Google Scholar] [CrossRef] [PubMed]
- Juskeviciute, E.; Dippold, R.P.; Antony, A.N.; Swarup, A.; Vadigepalli, R.; Hoek, J.B. Inhibition of miR-21 Rescues Liver Regeneration after Partial Hepatectomy in Ethanol-Fed Rats. Am. J. Physiol. Liver Physiol. 2016, 311, 794–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krichevsky, M.A.; Gabriely, G. miR-21: A Small Multi-Faceted RNA. J. Cell. Mol. Med. 2009, 13, 39–53. [Google Scholar]
- Poria, D.K.; Guha, A.; Nandi, I.; Ray, P.S. RNA-Binding Protein HuR Sequesters microRNA-21 to Prevent Translation Repression of Proinflammatory Tumor Suppressor Gene Programmed Cell Death 4. Oncogene 2015, 35, 1703–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liwak-Muir, U.; Dobson, C.C.; Naing, T.; Wylie, Q.; Chehade, L.; Baird, S.D.; Chakraborty, P.K.; Holcik, M. ERK8 Is a Novel HuR Kinase That Regulates Tumour Suppressor PDCD4 through a miR-21 Dependent Mechanism. Oncotarget 2015, 7, 1439–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essers, J.; Theil, A.F.; Baldeyron, C.; van Cappellen, A.W.; Houtsmuller, A.B.; Kanaar, R.; Vermeulen, W. Nuclear Dynamics of PCNA in DNA Replication and Repair. Mol. Cell. Biol. 2005, 25, 9350–9359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picco, V.; Pages, G. Linking JNK Activity to the DNA Damage Response. Genes Cancer 2013, 4, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.S.; Kim, G.; Lee, Y.R.; Park, Y.S.; Tak, W.Y.; Kweon, Y.-O.; Park, G.J.; Lee, W.H.; Han, Y.S.; Ha, H.T.; et al. Clinical Significance of microRNA-21 Expression in Disease Progression of Patients with Hepatocellular Carcinoma. Biomark. Med. 2018, 12, 1105–1114. [Google Scholar] [CrossRef]
- Ge, W.; Yu, D.-C.; Li, Q.-G.; Chen, X.; Zhang, C.-Y.; Ding, Y.-T. Expression of Serum miR-16, Let-7f, and miR-21 in Patients with Hepatocellular Carcinoma and Their Clinical Significances. Clin. Lab. 2014, 60, 427–434. [Google Scholar] [CrossRef]
- Jenike, A.E.; Halushka, M.K. miR-21: A Non-Specific Biomarker of All Maladies. Biomark. Res. 2021, 9, 1–7. [Google Scholar] [CrossRef]
- Zhuang, C.; Jiang, W.; Huang, D.; Xu, L.; Yang, Q.; Zheng, L.; Wang, X.; Hu, L. Serum miR-21, miR-26a and miR-101 as Potential Biomarkers of Hepatocellular Carcinoma. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 386–396. [Google Scholar] [CrossRef]
- Clément, S.; Sobolewski, C.; Gomes, D.; Rojas, A.; Goossens, N.; Conzelmann, S.; Calo, N.; Negro, F.; Foti, M. Activation of the Oncogenic miR-21-5p Promotes HCV Replication and Steatosis Induced by the Viral Core 3a Protein. Liver Int. 2019, 39, 1226–1236. [Google Scholar] [CrossRef]
- Androsavich, J.R.; Chau, B.N.; Bhat, B.; Linsley, P.S.; Walter, N.G. Disease-Linked microRNA-21 Exhibits Drastically Reduced mRNA Binding and Silencing Activity in Healthy Mouse Liver. RNA 2012, 18, 1510–1526. [Google Scholar] [CrossRef] [Green Version]
- Nwadiugwu, M.C. Thyroid Tumor: Investigating MicroRNA-21 Gene Suppression in FTC and FTA. Cancer Inform. 2020, 19. [Google Scholar] [CrossRef]
- La Rocca, G.; Olejniczak, S.; González, A.J.; Briskin, D.; Vidigal, J.A.; Spraggon, L.; DeMatteo, R.G.; Radler, M.R.; Lindsten, T.; Ventura, A.; et al. In vivo, Argonaute-Bound microRNAs Exist Predominantly in a Reservoir of Low Molecular Weight Complexes Not Associated with mRNA. Proc. Natl. Acad. Sci. USA 2015, 112, 767–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, B.H.; Turchinovich, A.; Wang, Y.; Liao, Z.; Dar, M.A.; Rocca, G.L.; Umanah, G.E.; Zeiger, M.A.; Umbricht, C.B.; Witwer, K.W. Mir-21 Is Associated with Inactive Low Molecular Weight Argonaute Complexes in Thyroid Cancer Cell Lines. bioRxiv 2020. [Google Scholar] [CrossRef]
- Papatheofani, V.; Levidou, G.; Sarantis, P.; Koustas, E.; Karamouzis, M.; Pergaris, A.; Kouraklis, G.; Theocharis, S. HuR Protein in Hepatocellular Carcinoma: Implications in Development, Prognosis and Treatment. Biomedicines 2021, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Ren, W.; Zhang, L.; Li, S.; Kong, X.; Zhang, H.; Dong, J.; Cai, G.; Jin, C.; Zheng, D.; et al. PTENp1, a Natural Sponge of miR-21, Mediates PTEN Expression to Inhibit the Proliferation of Oral Squamous Cell Carcinoma. Mol. Carcinog. 2016, 56, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Zhu, Y.Q.; Lui, K.S.; Cai, Q.; Lu, P.; Poon, R.T. Aberrant Polo-Like Kinase 1-Cdc25A Pathway in Metastatic Hepatocellular Carcinoma. Clin. Cancer Res. 2008, 14, 6813–6820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Yamamoto, H.; Sakon, M.; Yasui, M.; Ngan, C.Y.; Fukunaga, H.; Morita, T.; Ogawa, M.; Nagano, H.; Nakamori, S.; et al. Overexpression of CDC25A Phosphatase Is Associated with Hypergrowth Activity and Poor Prognosis of Human Hepatocellular Carcinomas. Clin. Cancer Res. 2003, 9, 1764–1772. [Google Scholar] [PubMed]
- Liu, T.; Yu, X.; Li, G.; Yuan, R.; Wang, Q.; Tang, P.; Wu, L.; Liu, X.; Peng, X.; Shao, J. Rock2 Regulates Cdc25A through Ubiquitin Proteasome System in Hepatocellular Carcinoma Cells. Exp. Cell Res. 2012, 318, 1994–2003. [Google Scholar] [CrossRef]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB Functions as a Tumour Promoter in Inflammation-Associated Cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.-L.; Leffert, H.; Karin, M. IKKbeta Couples Hepatocyte Death to Cytokine-Driven Compensatory Proliferation That Promotes Chemical Hepa-to-Carcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, M.; Garlick, D.S.; Greiner, D.L.; Davis, R.J. The Role of JNK in the Development of Hepatocellular Carcinoma. Genes Dev. 2011, 25, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Zatloukal, k.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of Human HCC Cells and Chemically Induced Mouse Liver Cancers Requires JNK1-Dependent p21 Down-Regulation. J. Clin. Investig. 2008, 118, 3943–3953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bard-Chapeau, E.A.; Li, S.; Ding, J.; Zhang, S.S.; Zhu, H.H.; Princen, F.; Fang, D.D.; Han, T.; Bailly-Maitre, B.; Poli, V.; et al. Ptpn11/Shp2 Acts as a Tumor Suppressor in Hepatocellular Carcinogenesis. Cancer Cell 2011, 19, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Lafdil, F.; Wang, L.; Park, O.; Yin, S.; Niu, J.; Miller, A.M.; Sun, Z.; Gao, B. Hepatoprotective versus Oncogenic Functions of STAT3 in Liver Tumorigenesis. Am. J. Pathol. 2011, 179, 714–724. [Google Scholar] [CrossRef]
- Nejak-Bowen, K.N.; Thompson, M.D.; Singh, S.; Bowen, W.C., Jr.; Dar, M.J.; Khillan, J.; Dai, C.; Monga, S.P.S. Accelerated Liver Regeneration and Hepatocarcinogenesis in Mice Overexpressing Serine-45 Mutant Be-Ta-Catenin. Hepatology 2010, 51, 1603–1613. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.F.; Tan, X.; Zeng, G.; Misse, A.; Singh, S.; Kim, Y.; Klaunig, J.E.; Monga, S.P.S. Conditional Beta-Catenin Loss in Mice Promotes Chemical Hepatocarcinogenesis: Role of Oxidative Stress and Platelet-Derived Growth Factor Receptor Alpha/Phosphoinositide 3-Kinase Signaling. Hepatology 2010, 52, 954–965. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Yu, W.-N.; Chen, X.; Peng, X.-D.; Jeon, S.-M.; Birnbaum, M.J.; Guzman, G.; Hay, N. Spontaneous Hepatocellular Carcinoma after the Combined Deletion of Akt Isoforms. Cancer Cell 2016, 29, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Mühlberg, L.; Kühnemuth, B.; Costello, E.; Shaw, V.; Sipos, B.; Huber, M.; Griesmann, H.; Krug, S.; Schober, M.; Gress, T.M.; et al. miRNA Dynamics in Tumor-Infiltrating Myeloid Cells Modulating Tumor Progression in Pancreatic Cancer. OncoImmunology 2016, 5, e1160181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahraei, M.; Chaube, B.; Liu, Y.; Sun, J.; Kaplan, A.; Price, N.; Ding, W.; Oyaghire, S.; García-Milian, R.; Mehta, S.; et al. Suppressing miR-21 Activity in Tumor-Associated Macrophages Promotes an Antitumor Immune Response. J. Clin. Investig. 2019, 129, 5518–5536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, C.W.D.; Maxwell, J.P.; Ong, C.W.; Redmond, K.M.; McCann, C.; Neisen, J.; Ward, G.A.; Chessari, G.; Johnson, C.; Crawford, N.T.; et al. PTEN Deficiency Promotes Macrophage Infiltration and Hypersensitivity of Prostate Cancer to IAP An-Tagonist/Radiation Combination Therapy. Oncotarget 2016, 7, 7885–7898. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Cui, B.; Zhang, W.; Ma, W.; Zhao, G.; Xing, L. Exosomal miR-21 Secreted by IL-1β-Primed-Mesenchymal Stem Cells Induces Macrophage M2 Polarization and Ameliorates Sepsis. Life Sci. 2020, 264, 118658. [Google Scholar] [CrossRef] [PubMed]
- Simeoli, R.; Montague, K.; Jones, H.R.; Castaldi, L.; Chambers, D.; Kelleher, J.H.; Vacca, V.; Pitcher, T.; Grist, J.; Al-Ahdal, H.; et al. Exosomal Cargo Including microRNA Regulates Sensory Neuron to Macrophage Communication after Nerve Trauma. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Barbi, J.; Pan, F. The Regulation of Immune Tolerance by FOXP3. Nat. Rev. Immunol. 2017, 17, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Chaoul, N.; Mancarella, S.; Lupo, L.; Giannelli, G.; Dituri, F. Impaired Anti-Tumor T Cell Response in Hepatocellular Carcinoma. Cancers 2020, 12, 627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlessi, R.; Köhn-Gaone, J.; Olynyk, J.K.; Tirnitz-Parker, J.E.E. Mouse Models of Hepatocellular Carcinoma. In Hepatocellular Carcinoma; Tirnitz-Parker, J.E.E., Ed.; Codon Publications: Brisbane, Australia, 2019. [Google Scholar]
- Yim, S.Y.; Lee, J.-S. Genomic Perspective on Mouse Liver Cancer Models. Cancers 2019, 11, 1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Sanchez, P.; Lujambio, A. Experimental Models for Preclinical Research in Hepatocellular Carcinoma. In Hepatocelular Carcinoma: Translational Precision Medicine Approaches; Hoshida, Y., Ed.; Springer: Cham, Switzerland, 2019; pp. 333–358. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia de Sousa, M.; Calo, N.; Sobolewski, C.; Gjorgjieva, M.; Clément, S.; Maeder, C.; Dolicka, D.; Fournier, M.; Vinet, L.; Montet, X.; et al. Mir-21 Suppression Promotes Mouse Hepatocarcinogenesis. Cancers 2021, 13, 4983. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13194983
Correia de Sousa M, Calo N, Sobolewski C, Gjorgjieva M, Clément S, Maeder C, Dolicka D, Fournier M, Vinet L, Montet X, et al. Mir-21 Suppression Promotes Mouse Hepatocarcinogenesis. Cancers. 2021; 13(19):4983. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13194983
Chicago/Turabian StyleCorreia de Sousa, Marta, Nicolas Calo, Cyril Sobolewski, Monika Gjorgjieva, Sophie Clément, Christine Maeder, Dobrochna Dolicka, Margot Fournier, Laurent Vinet, Xavier Montet, and et al. 2021. "Mir-21 Suppression Promotes Mouse Hepatocarcinogenesis" Cancers 13, no. 19: 4983. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13194983