
Molecular Subtyping and Survival Analysis of Osteosarcoma Reveals Prognostic Biomarkers and Key Canonical Pathways
 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
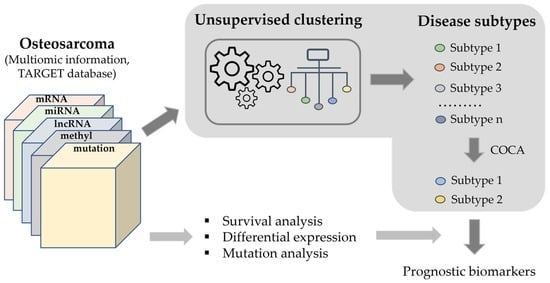
1. Introduction
2. Materials and Methods
3. Results
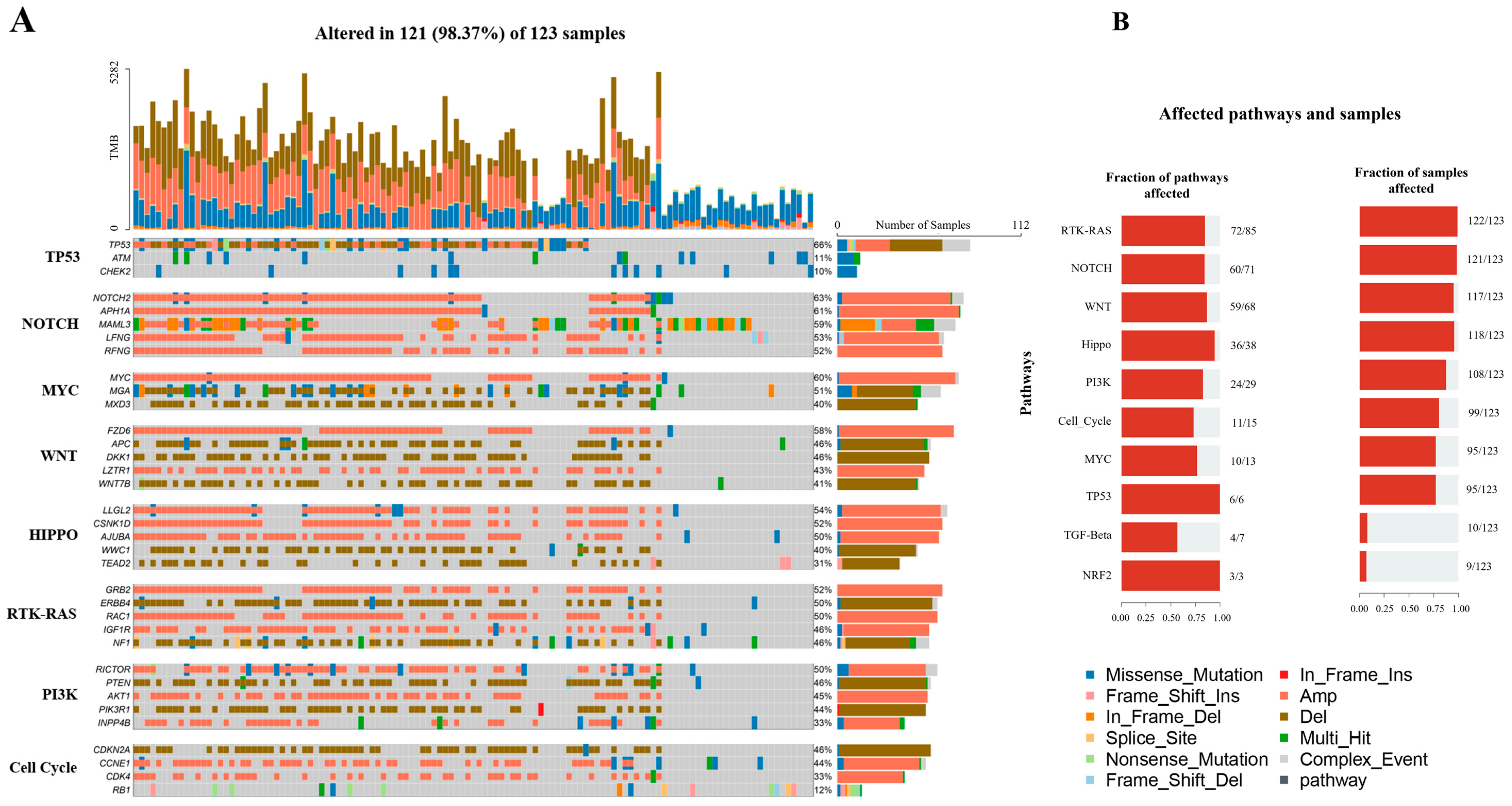
3.1. Mucin Family Genes Are Highly Mutated in OS
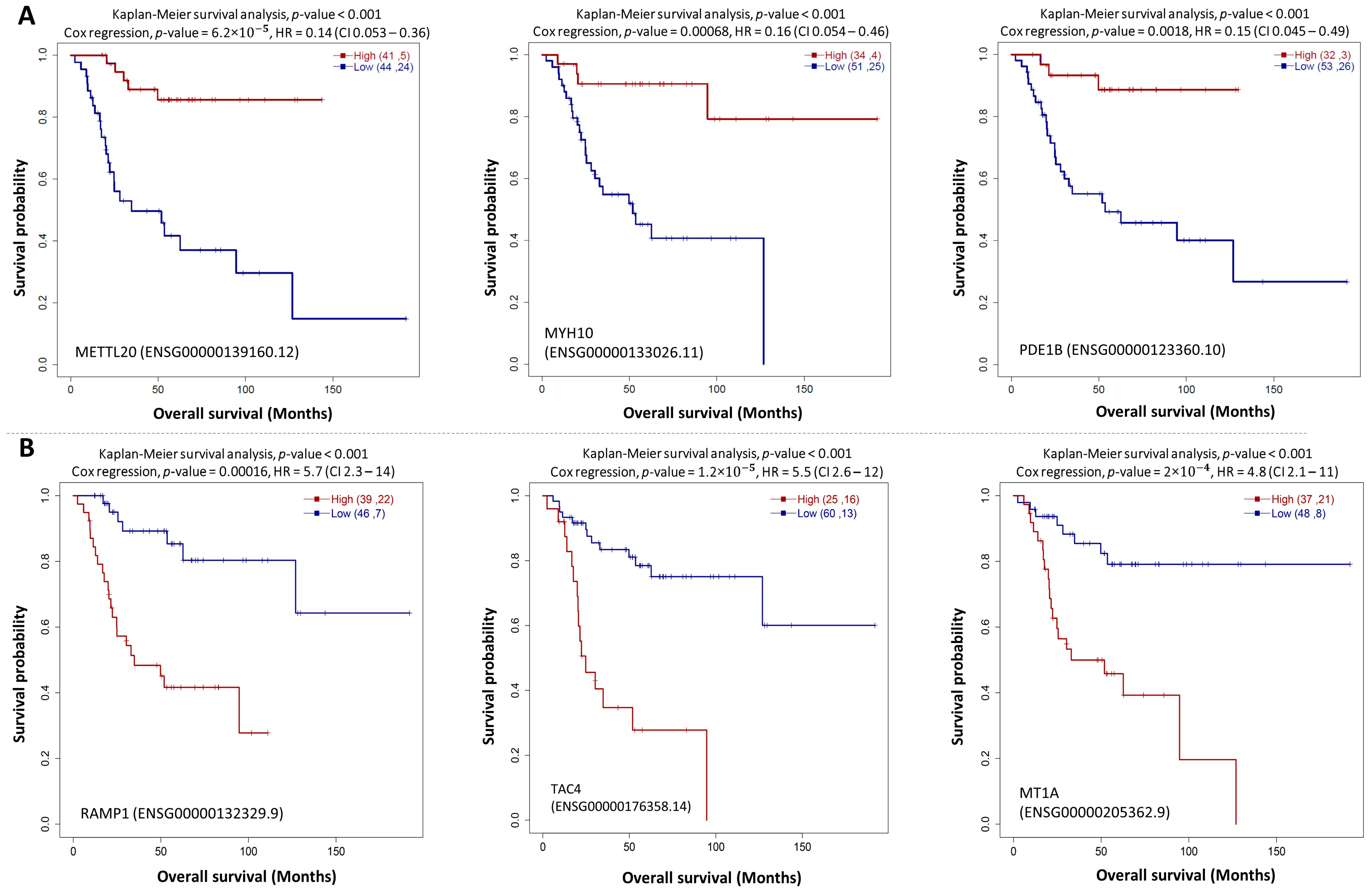
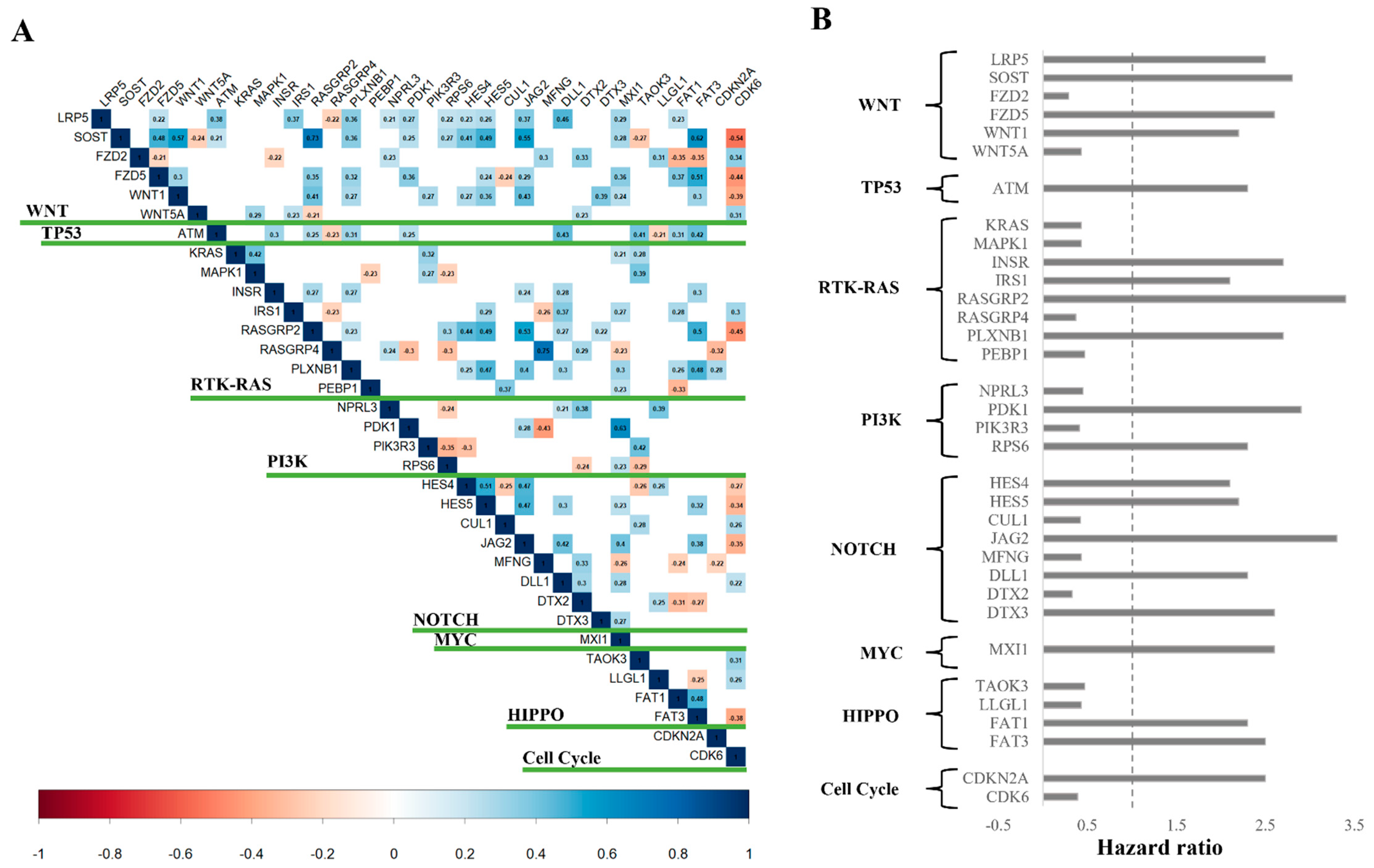
3.2. Survival Analysis Reveals Key Prognostic Pathway Genes
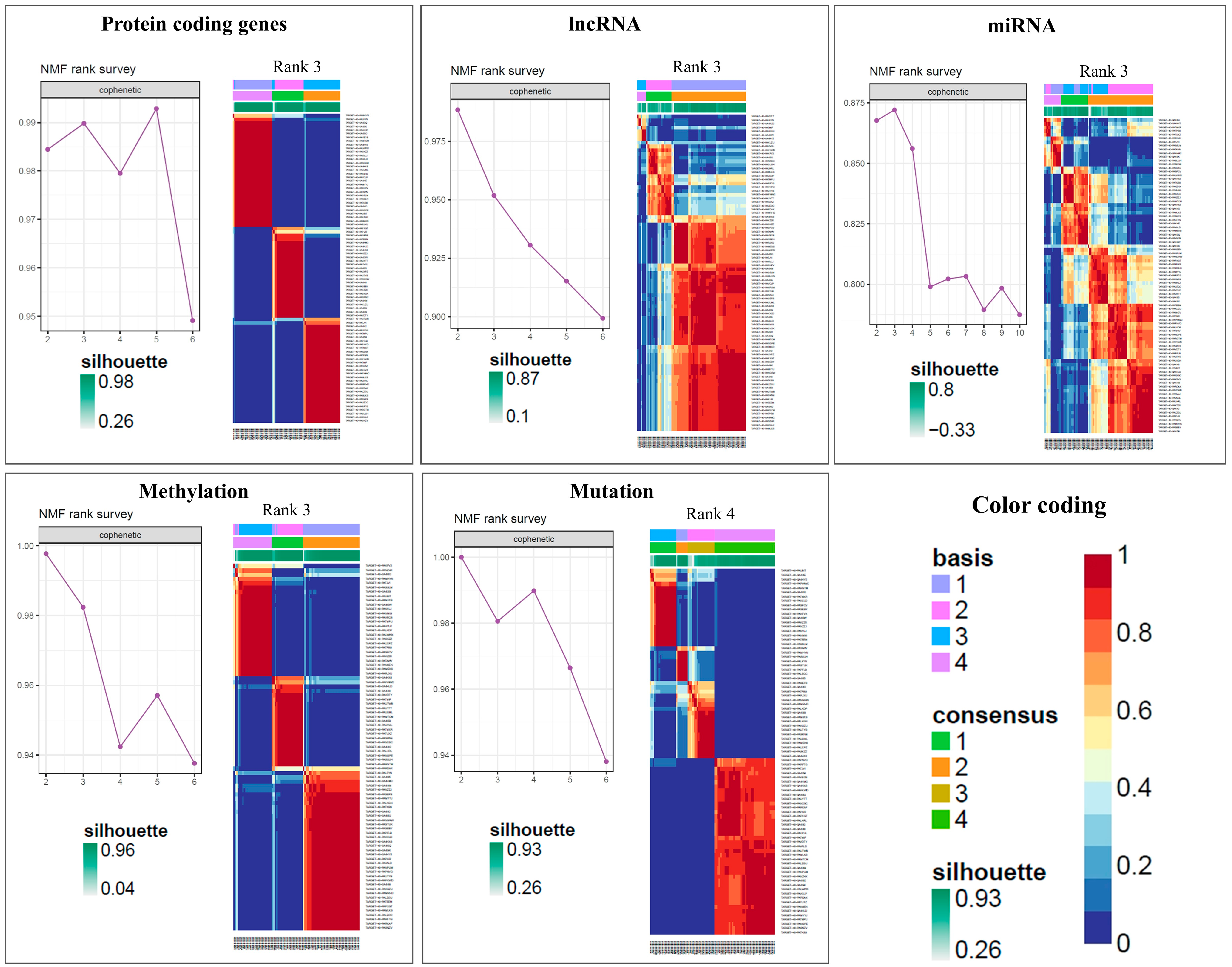
3.3. Clustering Analysis
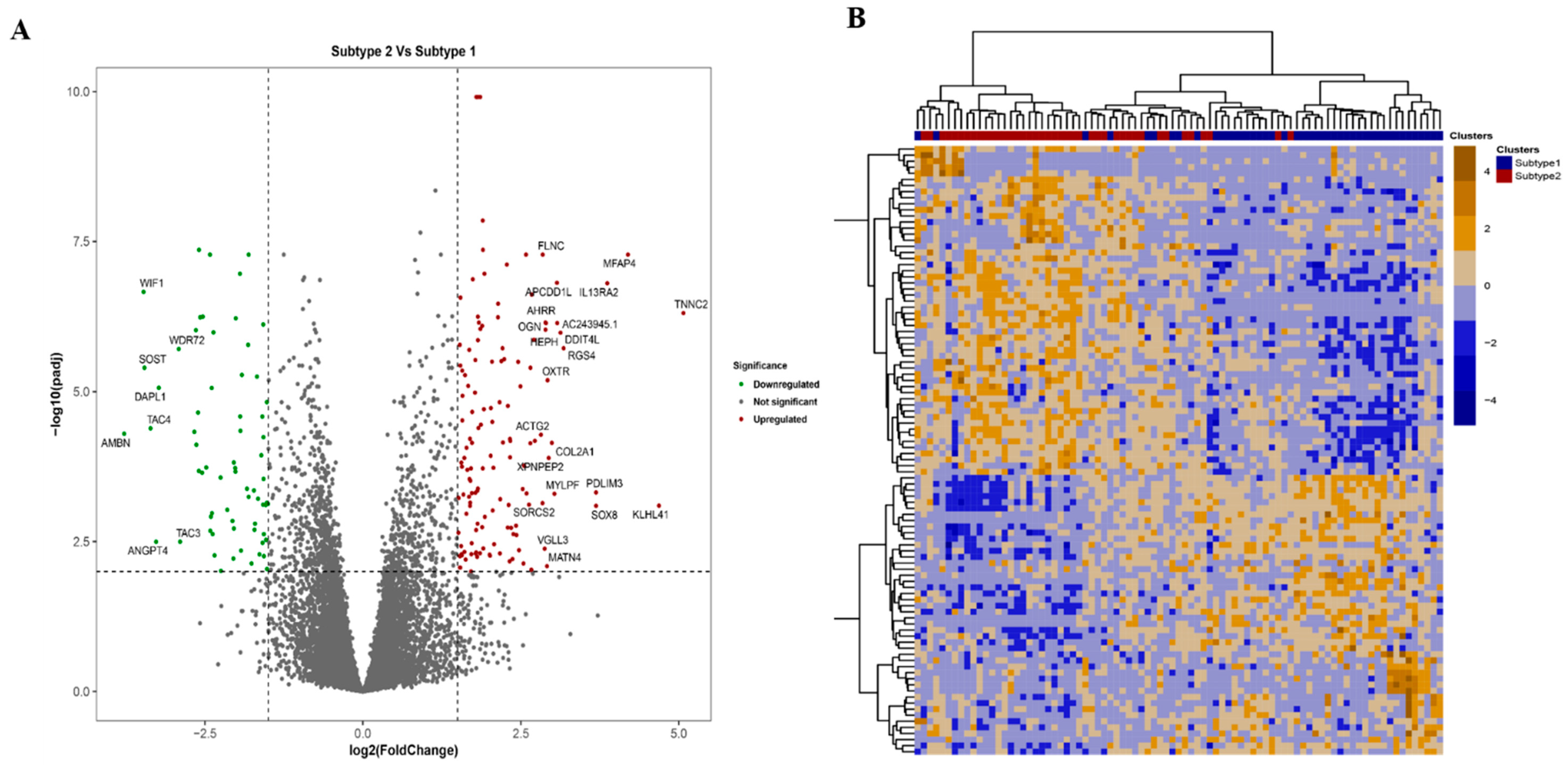
3.4. Comparison of the Identified Molecular Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lancia, C.; Anninga, J.K.; Sydes, M.R.; Spitoni, C.; Whelan, J.; Hogendoorn, P.C.W.; Gelderblom, H.; Fiocco, M. A novel method to address the association between received dose intensity and survival outcome: Benefits of approaching treatment intensification at a more individualised level in a trial of the European Osteosarcoma Intergroup. Cancer Chemother. Pharmacol. 2019, 83, 951–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menéndez, S.; Gallego, B.; Murillo, D.; Rodríguez, A.; Rodríguez, R. Cancer Stem Cells as a Source of Drug Resistance in Bone Sarcomas. J. Clin. Med. 2021, 10, 2621. [Google Scholar] [CrossRef] [PubMed]
- Cortini, M.; Baldini, N.; Avnet, S. New Advances in the Study of Bone Tumors: A Lesson From the 3D Environment. Front. Physiol. 2019, 10, 814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misaghi, A.; Goldin, A.; Awad, M.; Kulidjian, A.A. Osteosarcoma: A comprehensive review. SICOT-J 2018, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Bielack, S.S.; Kempf-Bielack, B.; Delling, G.; Exner, G.U.; Flege, S.; Helmke, K.; Kotz, R.; Salzer-Kuntschik, M.; Werner, M.; Winkelmann, W.; et al. Prognostic Factors in High-Grade Osteosarcoma of the Extremities or Trunk: An Analysis of 1,702 Patients Treated on Neoadjuvant Cooperative Osteosarcoma Study Group Protocols. J. Clin. Oncol. 2002, 20, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Luetke, A.; Meyers, P.A.; Lewis, I.; Juergens, H. Osteosarcoma treatment—Where do we stand? A state of the art review. Cancer Treat. Rev. 2014, 40, 523–532. [Google Scholar] [CrossRef]
- Jo, V.Y.; Fletcher, C.D. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 2014, 46, 95–104. [Google Scholar] [CrossRef]
- Sbaraglia, M.; Bellan, E.; Tos, A.P.D. The 2020 WHO Classification of Soft Tissue Tumours: News and perspectives. Pathologica 2020, 113, 70–84. [Google Scholar] [CrossRef]
- Odri, G.A.; Tchicaya-Bouanga, J.; Yoon, D.J.Y.; Modrowski, D. Metastatic Progression of Osteosarcomas: A Review of Current Knowledge of Environmental versus Oncogenic Drivers. Cancers 2022, 14, 360. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Wang, D.; Niu, X.; Wang, Z.; Song, C.-L.; Huang, Z.; Chen, K.-N.; Duan, J.; Bai, H.; Xu, J.; Zhao, J.; et al. Multiregion Sequencing Reveals the Genetic Heterogeneity and Evolutionary History of Osteosarcoma and Matched Pulmonary Metastases. Cancer Res. 2019, 79, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wang, R.; Zhang, Y.; Wang, H.; Huang, Z.; Jin, T.; Yang, Y.; Sun, Y.; Cao, S.; Niu, X. Whole-exome sequencing in osteosarcoma with distinct prognosis reveals disparate genetic heterogeneity. Cancer Genet. 2021, 256, 149–157. [Google Scholar] [CrossRef]
- Zheng, Y.; Huang, Y.; Bi, G.; Du, Y.; Liang, J.; Zhao, M.; Chen, Z.; Zhan, C.; Xi, J.; Wang, Q. Multi-omics characterization and validation of MSI-related molecular features across multiple malignancies. Life Sci. 2021, 270, 119081. [Google Scholar] [CrossRef] [PubMed]
- Niemira, M.; Collin, F.; Szalkowska, A.; Bielska, A.; Chwialkowska, K.; Reszec, J.; Niklinski, J.; Kwasniewski, M.; Kretowski, A. Molecular Signature of Subtypes of Non-Small-Cell Lung Cancer by Large-Scale Transcriptional Profiling: Identification of Key Modules and Genes by Weighted Gene Co-Expression Network Analysis (WGCNA). Cancers 2019, 12, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwendenwein, A.; Megyesfalvi, Z.; Barany, N.; Valko, Z.; Bugyik, E.; Lang, C.; Ferencz, B.; Paku, S.; Lantos, A.; Fillinger, J.; et al. Molecular profiles of small cell lung cancer subtypes: Therapeutic implications. Mol. Ther. Oncolytics 2021, 20, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, G.; Wang, J.; Zain, A.M.; Guo, W. Lung cancer subtype diagnosis using weakly-paired multi-omics data. Bioinformatics 2022, 38, 5092–5099. [Google Scholar] [CrossRef]
- Valenzuela, G.; Canepa, J.; Simonetti, C.; de Zaldívar, L.S.; Marcelain, K.; González-Montero, J. Consensus molecular subtypes of colorectal cancer in clinical practice: A translational approach. World J. Clin. Oncol. 2021, 12, 1000–1008. [Google Scholar] [CrossRef]
- Zhou, B.; Yu, J.; Cai, X.; Wu, S. Constructing a molecular subtype model of colon cancer using machine learning. Front. Pharmacol. 2022, 13, 1008207. [Google Scholar] [CrossRef]
- Eliyatkin, N.; Yalcin, E.; Zengel, B.; Aktaş, S.; Vardar, E. Molecular Classification of Breast Carcinoma: From Traditional, Old-Fashioned Way to A New Age, and A New Way. J. Breast Health 2015, 11, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef]
- Ochoa, S.; Hernández-Lemus, E. Functional impact of multi-omic interactions in breast cancer subtypes. Front. Genet. 2023, 13, 1078609. [Google Scholar] [CrossRef]
- Martini, G.; Dienstmann, R.; Ros, J.; Baraibar, I.; Cuadra-Urteaga, J.L.; Salva, F.; Ciardiello, D.; Mulet, N.; Argiles, G.; Tabernero, J.; et al. Molecular subtypes and the evolution of treatment management in metastatic colorectal cancer. Ther. Adv. Med Oncol. 2020, 12, 1758835920936089. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Inaki, K.; Shibutani, T.; Maeda, N.; Eppenberger-Castori, S.; Nicolet, S.; Kaneda, Y.; Koyama, K.; Qiu, Y.; Wakita, K.; Murakami, M. Pan-cancer gene expression analysis of tissue microarray using EdgeSeq oncology biomarker panel and a cross-comparison with HER2 and HER3 immunohistochemical analysis. PLoS ONE 2022, 17, e0274140. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Chen, Z.; Xue, Q.; Sun, P.; Wang, Y.; Zhu, C.; Shi, W. Identification of molecular subtypes and a novel prognostic model of diffuse large B-cell lymphoma based on a metabolism-associated gene signature. J. Transl. Med. 2022, 20, 186. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Huang, C.-S.; Phan, N.-N.; Lu, T.-P.; Liu, C.-Y.; Huang, C.-J.; Chiu, J.-H.; Tseng, L.-M. Molecular subtyping of breast cancer intrinsic taxonomy with oligonucleotide microarray and NanoString nCounter. Biosci. Rep. 2021, 41. [Google Scholar] [CrossRef]
- Kulkarni, M.M. Digital Multiplexed Gene Expression Analysis Using the NanoString nCounter System. Curr. Protoc. Mol. Biol. 2011, 94, 25B-10. [Google Scholar] [CrossRef]
- Raymond, A.K.; Jaffe, N. Osteosarcoma Multidisciplinary Approach to the Management from the Pathologist’s Perspective. Pediatr. Adolesc. Osteosarcoma 2009, 152, 63–84. [Google Scholar] [CrossRef]
- Zhu, L.; McManus, M.M.; Hughes, D.P.M. Understanding the Biology of Bone Sarcoma from Early Initiating Events through Late Events in Metastasis and Disease Progression. Front. Oncol. 2013, 3, 230. [Google Scholar] [CrossRef] [Green Version]
- Kurt, A.M.; Unni, K.K.; McLeod, R.A.; Pritchard, D.J. Low-grade intraosseous osteosarcoma. Cancer 1990, 65, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Guo, M.; Wang, K.; Zhang, C.; Liu, X. Identification of Pan-Cancer Prognostic Biomarkers Through Integration of Multi-Omics Data. Front. Bioeng. Biotechnol. 2020, 8, 268. [Google Scholar] [CrossRef]
- Zamborsky, R.; Kokavec, M.; Harsanyi, S.; Danisovic, L. Identification of Prognostic and Predictive Osteosarcoma Biomarkers. Med. Sci. 2019, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; He, R.; Zhuang, Z.; Ren, J.; Wang, Z.; Liu, Y.; Wu, J.; Jiang, S.; Wang, K. A risk signature-based on metastasis-associated genes to predict survival of patients with osteosarcoma. J. Cell. Biochem. 2020, 121, 3479–3490. [Google Scholar] [CrossRef]
- Qi, W.; Yan, Q.; Lv, M.; Song, D.; Wang, X.; Tian, K. Prognostic Signature of Osteosarcoma Based on 14 Autophagy-Related Genes. Pathol. Oncol. Res. 2021, 27, 1609782. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Krause, L.; Nones, K.; Loffler, K.A.; Nancarrow, D.; Oey, H.; Tang, Y.H.; Wayte, N.J.; Patch, A.M.; Patel, K.; Brosda, S.; et al. Identification of the CIMP-like subtype and aberrant methylation of members of the chromosomal segregation and spindle assembly pathways in esophageal adenocarcinoma. Carcinogenesis 2016, 37, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Triche, T.J.; Laird, P.W.; Shen, H. SeSAMe: Reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 2018, 46, e123. [Google Scholar] [CrossRef] [Green Version]
- Di Lena, P.; Sala, C.; Prodi, A.; Nardini, C. Missing value estimation methods for DNA methylation data. Bioinformatics 2019, 35, 3786–3793. [Google Scholar] [CrossRef]
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 2014, 30, 428–430. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zhou, H.; Xu, H.; You, H.; Cheng, H. Construction of an Immune-Related lncRNA Signature That Predicts Prognosis and Immune Microenvironment in Osteosarcoma Patients. Front. Oncol. 2022, 12, 769202. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Chen, J.; Liu, X.; Gong, W.; Zheng, J.; Guo, X.; Liu, Y.; Liu, L.; Ma, J.; Wang, P.; et al. PVT1 regulates the malignant behaviors of human glioma cells by targeting miR-190a-5p and miR-488-3p. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 1783–1794. [Google Scholar] [CrossRef]
- Xu, S.; Wang, T.; Song, W.; Jiang, T.; Zhang, F.; Yin, Y.; Jiang, S.-W.; Wu, K.; Yu, Z.; Wang, C.; et al. The inhibitory effects of AR/miR-190a/YB-1 negative feedback loop on prostate cancer and underlying mechanism. Sci. Rep. 2015, 5, 13528. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Cao, X.-C. miR-190-5p in human diseases. Cancer Cell Int. 2019, 19, 257. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Jin, Z.; Yin, T.; Duan, C.; Sun, J.; Xiong, R.; Li, Z. MiR-122 Promotes the Development of Colon Cancer by Targeting ALDOA In Vitro. Technol. Cancer Res. Treat. 2019, 18, 1533033819871300. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Han, L.; Gu, C.; Lai, Y.; Lai, Q.; Li, Q.; He, C.; Meng, Y.; Pan, L.; Liu, S.; et al. MiR-452-5p promotes colorectal cancer progression by regulating an ERK/MAPK positive feedback loop. Aging 2021, 13, 7608–7626. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Singla, A.; Wang, J.; Yang, R.; Geller, D.S.; Loeb, D.M.; Hoang, B.H. Wnt Signaling in Osteosarcoma. Adv. Exp. Med. Biol. 2020, 8, 125–139. [Google Scholar] [CrossRef]
- Hensel, J.; Wetterwald, A.; Temanni, R.; Keller, I.; Riether, C.; van der Pluijm, G.; Cecchini, M.G.; Thalmann, G.N. Osteolytic cancer cells induce vascular/axon guidance processes in the bone/bone marrow stroma. Oncotarget 2018, 9, 28877–28896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ghedalia-Peled, N.; Vago, R. Wnt Signaling in the Development of Bone Metastasis. Cells 2022, 11, 3934. [Google Scholar] [CrossRef]
- Jalanko, T.; de Jong, J.J.; Gibb, E.A.; Seiler, R.; Black, P.C. Genomic Subtyping in Bladder Cancer. Curr. Urol. Rep. 2020, 21, 9. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Song, Y.-J.; Xu, Y.; Deng, C.; Zhu, X.; Fu, J.; Chen, H.; Lu, J.; Xu, H.; Song, G.; Tang, Q.; et al. Gene Expression Classifier Reveals Prognostic Osteosarcoma Microenvironment Molecular Subtypes. Front. Immunol. 2021, 12, 623762. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, F.; Creighton, C.J. Pan-cancer molecular subtypes of metastasis reveal distinct and evolving transcriptional programs. Cell Rep. Med. 2023, 4, 100932. [Google Scholar] [CrossRef]
- Kong, C.; Hansen, M.F. Biomarkers in osteosarcoma. Expert Opin. Med Diagn. 2009, 3, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Tirabosco, R.; Berisha, F.; Ye, H.; Halai, D.; Amary, M.F.; Flanagan, A.M. Assessment of MUC4 expression in primary bone tumours. Histopathology 2013, 63, 142–143. [Google Scholar] [CrossRef]
- Liu, W.; Huang, Z.; Ding, Y.; Yang, Y.; Jin, T.; Deng, Z.; Xu, H.; Wang, X.; Zhang, Y.; Wang, H.; et al. Comprehensive genomic profiling of patients with favorable prognosis in osteosarcoma. J. Clin. Oncol. 2019, 37, e22501. [Google Scholar] [CrossRef]
- Chen, T.; Chen, Z.; Lian, X.; Wu, W.; Chu, L.; Zhang, S.; Wang, L. MUC 15 Promotes Osteosarcoma Cell Proliferation, Migration and Invasion through Livin, MMP-2/MMP-9 and Wnt/β-Catenin Signal Pathway. J. Cancer 2021, 12, 467–473. [Google Scholar] [CrossRef]
- Chiappetta, C.; Puggioni, C.; Carletti, R.; Petrozza, V.; Della Rocca, C.; Di Cristofano, C. The nuclear-cytoplasmic trafficking of a chromatin-modifying and remodelling protein (KMT2C), in osteosarcoma. Oncotarget 2018, 9, 30624–30634. [Google Scholar] [CrossRef] [Green Version]
- Chiappetta, C.; Mancini, M.; Lessi, F.; Aretini, P.; De Gregorio, V.; Puggioni, C.; Carletti, R.; Petrozza, V.; Civita, P.; Franceschi, S.; et al. Whole-exome analysis in osteosarcoma to identify a personalized therapy. Oncotarget 2017, 8, 80416–80428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Yang, J.; Yang, D.; Tian, W.; Zhu, Z. The genetic basis for inactivation of Wnt pathway in human osteosarcoma. BMC Cancer 2014, 14, 450. [Google Scholar] [CrossRef]
- Matsuoka, K.; Bakiri, L.; Wolff, L.I.; Linder, M.; Mikels-Vigdal, A.; Patiño-García, A.; Lecanda, F.; Hartmann, C.; Sibilia, M.; Wagner, E.F. Wnt signaling and Loxl2 promote aggressive osteosarcoma. Cell Res. 2020, 30, 885–901. [Google Scholar] [CrossRef]
- Wang, L.; Jin, F.; Qin, A.; Hao, Y.; Dong, Y.; Ge, S.; Dai, K. Targeting Notch1 signaling pathway positively affects the sensitivity of osteosarcoma to cisplatin by regulating the expression and/or activity of Caspase family. Mol. Cancer 2014, 13, 139. [Google Scholar] [CrossRef] [Green Version]
- McManus, M.M.; Weiss, K.R.; Hughes, D.P.M. Understanding the Role of Notch in Osteosarcoma. Adv. Exp. Med. Biol. 2014, 804, 67–92. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Wu, Y.-N.; Huang, J.-Q.; Wang, W.; Xu, M.; Jia, J.-P.; Han, G.; Mao, B.-B.; Bi, W.-Z. Hippo/YAP signaling pathway is involved in osteosarcoma chemoresistance. Chin. J. Cancer 2016, 35, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, F.; VanCleave, A.; Helmuth, R.; Torres, H.; Rickel, K.; Wollenzien, H.; Sun, H.; Zeng, E.; Zhao, J.; Tao, J. Targeting the Wnt/β-catenin pathway in human osteosarcoma cells. Oncotarget 2018, 9, 36780–36792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallmayer, M.; Li, J.; Ohmura, S.; Rubio, R.A.; Baldauf, M.C.; Hölting, T.L.B.; Musa, J.; Knott, M.M.L.; Stein, S.; Cidre-Aranaz, F.; et al. Targeting the CALCB/RAMP1 axis inhibits growth of Ewing sarcoma. Cell Death Dis. 2019, 10, 116. [Google Scholar] [CrossRef] [Green Version]
- Logan, M.; Anderson, P.D.; Saab, S.T.; Hameed, O.; Abdulkadir, S.A. RAMP1 Is a Direct NKX3.1 Target Gene Up-Regulated in Prostate Cancer that Promotes Tumorigenesis. Am. J. Pathol. 2013, 183, 951–963. [Google Scholar] [CrossRef] [Green Version]
- Fu, P.; Shi, Y.; Chen, G.; Gao, Z. Establishment of key genes and associated outcomes in osteosarcoma patients using bioinformatics methods. Int. J. Clin. Exp. Med. 2021, 14, 2242–2249. [Google Scholar]
- Shi, D.; Mu, S.; Pu, F.; Liu, J.; Zhong, B.; Hu, B.; Ni, N.; Wang, H.; Luu, H.H.; Haydon, R.C.; et al. Integrative analysis of immune-related multi-omics profiles identifies distinct prognosis and tumor microenvironment patterns in osteosarcoma. Mol. Oncol. 2022, 16, 2174–2194. [Google Scholar] [CrossRef] [PubMed]
- Baumhoer, D.; Elsner, M.; Smida, J.; Zillmer, S.; Rauser, S.; Schoene, C.; Balluff, B.; Bielack, S.; Jundt, G.; Walch, A.; et al. CRIP1 expression is correlated with a favorable outcome and less metastases in osteosarcoma patients. Oncotarget 2011, 2, 970–975. [Google Scholar] [CrossRef] [Green Version]
- Cassoni, P.; Allia, E.; Marrocco, T.; Ghè, C.; Ghigo, E.; Muccioli, G.; Papotti, M. Ghrelin and cortistatin in lung cancer: Expression of peptides and related receptors in human primary tumors and in vitro effect on the H345 small cell carcinoma cell line. J. Endocrinol. Investig. 2006, 29, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Cassoni, P.; Muccioli, G.; Marrocco, T.; Volante, M.; Allia, E.; Ghigo, E.; Deghenghi, R.; Papotti, M. Cortistatin-14 inhibits cell proliferation of human thyroid carcinoma cell lines of both follicular and parafollicular origin. J. Endocrinol. Investig. 2002, 25, 362–368. [Google Scholar] [CrossRef]
- Wu, Z.-L.; Deng, Y.-J.; Zhang, G.-Z.; Ren, E.-H.; Yuan, W.-H.; Xie, Q.-Q. Development of a novel immune-related genes prognostic signature for osteosarcoma. Sci. Rep. 2020, 10, 18402. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | HR | HR_low95 | HR_up95 | Beta | Gene | HR | HR_low95 | HR_up95 | Beta |
|---|---|---|---|---|---|---|---|---|---|
| RAMP1 | 5.7 | 2.3 | 14 | 1.7 | LY86 | 0.24 | 0.1 | 0.56 | −1.4 |
| TAC4 | 5.5 | 2.6 | 12 | 1.7 | SHISA5 | 0.24 | 0.1 | 0.57 | −1.4 |
| TIMM23B | 4.7 | 2.1 | 11 | 1.6 | FOLR2 | 0.25 | 0.11 | 0.56 | −1.4 |
| COL5A2 | 4.7 | 2 | 11 | 1.6 | C11orf45 | 0.25 | 0.11 | 0.57 | −1.4 |
| MT1A | 4.8 | 2.1 | 11 | 1.6 | UBE2L3 | 0.24 | 0.11 | 0.54 | −1.4 |
| CRIP1 | 4.8 | 1.9 | 12 | 1.6 | CYFIP1 | 0.24 | 0.1 | 0.55 | −1.4 |
| UFC1 | 4.6 | 1.9 | 11 | 1.5 | SNX1 | 0.22 | 0.091 | 0.55 | −1.5 |
| PROSER2 | 4.4 | 1.9 | 10 | 1.5 | ACTB | 0.22 | 0.095 | 0.5 | −1.5 |
| NHEJ1 | 4.7 | 2 | 11 | 1.5 | MPP1 | 0.22 | 0.091 | 0.55 | −1.5 |
| CORT | 4.6 | 2.1 | 10 | 1.5 | CD180 | 0.23 | 0.092 | 0.56 | −1.5 |
| CKMT2 | 4.1 | 2 | 8.8 | 1.4 | TPMT | 0.22 | 0.09 | 0.55 | −1.5 |
| CHD1L | 3.9 | 1.7 | 8.9 | 1.4 | BBS4 | 0.22 | 0.088 | 0.53 | −1.5 |
| LGR6 | 4.1 | 1.9 | 9.1 | 1.4 | ITGAM | 0.22 | 0.091 | 0.56 | −1.5 |
| IFFO2 | 4.2 | 1.8 | 9.7 | 1.4 | TCN2 | 0.23 | 0.093 | 0.56 | −1.5 |
| MAFK | 4.1 | 1.8 | 9.7 | 1.4 | SIRPA | 0.21 | 0.087 | 0.53 | −1.5 |
| PGAM4 | 3.9 | 1.8 | 8.5 | 1.4 | SETD9 | 0.19 | 0.079 | 0.48 | −1.6 |
| SNAP91 | 3.8 | 1.8 | 8 | 1.3 | SNTB2 | 0.21 | 0.085 | 0.53 | −1.6 |
| PDE4C | 3.5 | 1.7 | 7.5 | 1.3 | MITF | 0.21 | 0.079 | 0.55 | −1.6 |
| KRT2 | 3.6 | 1.7 | 7.6 | 1.3 | NUBP1 | 0.17 | 0.073 | 0.41 | −1.8 |
| GCSAM | 3.7 | 1.7 | 8 | 1.3 | IRF2BPL | 0.17 | 0.069 | 0.42 | −1.8 |
| BAI1 | 3.7 | 1.7 | 7.8 | 1.3 | ERCC4 | 0.17 | 0.066 | 0.46 | −1.8 |
| CFAP44 | 3.8 | 1.8 | 8 | 1.3 | PDE1B | 0.15 | 0.045 | 0.49 | −1.9 |
| FAM166B | 3.5 | 1.7 | 7.3 | 1.3 | MYH10 | 0.16 | 0.054 | 0.46 | −1.9 |
| APBB1IP | 0.24 | 0.11 | 0.55 | −1.4 | METTL20 | 0.14 | 0.053 | 0.36 | −2 |
| COMMD9 | 0.24 | 0.11 | 0.55 | −1.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Southekal, S.; Shakyawar, S.K.; Bajpai, P.; Elkholy, A.; Manne, U.; Mishra, N.K.; Guda, C. Molecular Subtyping and Survival Analysis of Osteosarcoma Reveals Prognostic Biomarkers and Key Canonical Pathways. Cancers 2023, 15, 2134. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15072134
Southekal S, Shakyawar SK, Bajpai P, Elkholy A, Manne U, Mishra NK, Guda C. Molecular Subtyping and Survival Analysis of Osteosarcoma Reveals Prognostic Biomarkers and Key Canonical Pathways. Cancers. 2023; 15(7):2134. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15072134
Chicago/Turabian StyleSouthekal, Siddesh, Sushil Kumar Shakyawar, Prachi Bajpai, Amr Elkholy, Upender Manne, Nitish Kumar Mishra, and Chittibabu Guda. 2023. "Molecular Subtyping and Survival Analysis of Osteosarcoma Reveals Prognostic Biomarkers and Key Canonical Pathways" Cancers 15, no. 7: 2134. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15072134