Biocatalysis with Laccases: An Updated Overview



Istituto di Scienze e Tecnologie Chimiche “Giulio Natta” (SCITEC), CNR, Via Mario Bianco 9, 20131 Milano, Italy
*
Authors to whom correspondence should be addressed.
Catalysts 2021, 11(1), 26; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010026
Submission received: 4 December 2020
/
Revised: 21 December 2020
/
Accepted: 23 December 2020
/
Published: 28 December 2020
(This article belongs to the Special Issue Industrial Biocatalysis: Challenges and Opportunities)
Abstract
:Laccases are multicopper oxidases, which have been widely investigated in recent decades thanks to their ability to oxidize organic substrates to the corresponding radicals while producing water at the expense of molecular oxygen. Besides their successful (bio)technological applications, for example, in textile, petrochemical, and detoxifications/bioremediations industrial processes, their synthetic potentialities for the mild and green preparation or selective modification of fine chemicals are of outstanding value in biocatalyzed organic synthesis. Accordingly, this review is focused on reporting and rationalizing some of the most recent and interesting synthetic exploitations of laccases. Applications of the so-called laccase-mediator system (LMS) for alcohol oxidation are discussed with a focus on carbohydrate chemistry and natural products modification as well as on bio- and chemo-integrated processes. The laccase-catalyzed Csp2-H bonds activation via monoelectronic oxidation is also discussed by reporting examples of enzymatic C-C and C-O radical homo- and hetero-couplings, as well as of aromatic nucleophilic substitutions of hydroquinones or quinoids. Finally, the laccase-initiated domino/cascade synthesis of valuable aromatic (hetero)cycles, elegant strategies widely documented in the literature across more than three decades, is also presented.
1. Introduction
Laccases are enzymes belonging to the blue multicopper oxidase family that catalyze the oxidation of a wide range of substrates, such as phenols and aromatic or aliphatic amines, thereby reducing molecular oxygen to water. They are widely distributed in nature as they have been described in bacteria, fungi, higher plants and insects [1]. In eukaryotes, laccases are usually glycosylated and the carbohydrate component seems to ensure laccase conformational stability and to preserve enzymes from inactivation by radicals and proteolysis [2].
These enzymes exist in various forms. In fact, although most of them are monomeric, some have been reported to be homodimeric, heterodimeric, and multimeric, with molecular mass ranging from 50 to 140 kDa, including their sugar component. Their amino acid sequence could span from 220 to 800 amino acids and may contain 2 or 3 cupredoxin-like domains depending on the organism they belong to. These domains bind copper centers involved in inter-molecular electron transfer reactions and constitute the catalytic core of laccases. Despite the fact that laccases from each species exhibit peculiar catalytic characteristics and sequences, their molecular architecture is common for all multicopper oxidases showing a simple 3D structure mainly constituted of beta sheets and turns [2,3].
Since the discovery in 1896 of the first fungal laccase in a mushroom of the Boletus genus [2], laccases have been found in more than 60 fungal strains, and fungal laccases represent the most significant group of the blue multicopper oxidase family with regard to the number and extent of characterization [1,4,5]. Typical fungal laccases are 60–70 kDa monomeric glycoproteins containing three cupredoxin-like domains that bind four copper atoms [3].
The catalytic mechanism of laccases has been well characterized and involves the formation of radical species that can follow different pathways giving either dimers or polymers of their parent substrates by oxidative coupling reactions or yielding dead end products through intramolecular rearrangements. Furthermore, radical species may play as mediators by oxidizing non-phenolic compounds, and thus causing, i.a., bond cleavage [6].
In general, the physiological function of these enzymes is indeed related to their ability to catalyze polymerization or depolymerization processes. For example, in fungi these biocatalysts are involved in lignin degradation, in plants are key in the lignification process and cell wall formation [5], while in insects they seem to participate in the sclerotization of the cuticle [7].
Both the possibility to use laccases in synthetic or degradative processes and their wide substrate specificity make these enzymes suitable “green tools” for a plethora of applications in different fields, such as textile, paper, food, pharmaceutical and cosmetics [6,8]. In particular, their main technological applications are in the textile industry, e.g., for textile properties improvement and in processes related to fiber bleaching and dyeing, in the pulp and paper industries for the delignification of woody fibers, and in the food industry for food improvement [1,6,9]. The industrial interest toward laccases is also well documented by the large number of patents filed during the last years, as recently reviewed [10].
Besides these applications, laccases have gained wide attention in recent years for the one-pot synthesis of complex scaffolds, the selective modification of natural products by oxidation, as well as the biocatalytic activation of normally inert Csp2-H bonds. Presently, to the best of our knowledge, they have not found large scale (industrial) applications. However, as it is nowadays well documented in the scientific literature, these enzymes have indeed high potential for a significant impact in organic synthesis. In this review, we will describe the numerous reaction outcomes that can derive from their catalytic action.
2. Laccases Catalytic Cycle and Their Use in Non-Conventional Reaction Systems
The catalytic cycle of laccases starts with the mono-electronic oxidation of four equivalent suitable reducing substrates, such as phenols or aromatic and aliphatic amines to form organic radicals at the expense of molecular oxygen which is eventually reduced to two molecules of water. A four-membered copper cluster represents the catalytic machinery of these enzymes, being also the site of oxygen coordination and reduction as well as of water formation and release. In fungal laccases, three different sites are typically identified in the copper cluster depending on their role in the catalytic cycle: the so-called “blue site” or Type 1 (T1), “normal site” or Type 2 (T2) and the “binuclear site” or Type 3 (T3) (Scheme 1). Specifically, the T1 copper is involved in substrate oxidation, while T2 and T3 coppers, which define a trinuclear sub-cluster, catalyze the reduction in molecular oxygen [1,6,11].
From a chemical perspective, the action of laccases in synthetic processes is generally described by one of the simplified mechanisms reported below. All of them relay on the redox activity of the copper cluster located in the active site (Scheme 2).
When the substrate of interest possesses the proper redox potential, laccases can be used to directly oxidize it to the corresponding organic radical(s) (Scheme 2a). Instead, when a direct oxidation in not achievable due to steric hindrance/active site penetration and/or redox potentials incompatibility, the use of the so-called “chemical mediators”, acting as redox intermediates in a nature mimicking-fashion strategy (e.g., the cytochrome chain), is generally applied with success. Accordingly, the free radicals originating from the oxidations of these chemical mediators can both interact with bulky or high redox-potential substrate targets (Scheme 2b). This versatile redox apparatus, known in the literature by the name of “laccase-mediator-system” (LMS), represents one of the most reported laccase application both in organic synthesis, i.e., for the oxidation of activated hydroxyl groups to the corresponding carbonyls or acids, and in (bio)technological and manufacturing processes [12,13,14,15,16]. Moreover, the LMS can be also applied for cofactor regeneration in coupled, multienzymatic redox transformations (Scheme 2c): Haltrich and co-workers exploited the LMS to efficiently regenerate the FAD cofactor in a multienzymatic redox process [17]. Ferrandi et al., instead, applied LMS, in the form of laccases from Trametes pubescens and the redox intermediate Meldola Blue, to the regeneration of NAD+ in the multigram, biocatalytic preparation of 7-keto derivatives of cholic acid catalyzed by a 7α-hydroxysteroid dehydrogenase [18]. More recently, Hanefeld and coworkers expanded this topic using the LMS in the biocatalytic oxidation of the 12α-OH group of hydroxysteroids for the preparation of bile acids derivatives by employing a 12α-hydroxysteroid dehydrogenase in combination with laccases of different origins and a plethora of redox mediators [19].
From a practical point of view, the synthetic applications of enzymes very often require non-conventional reaction systems, including those applying organic solvents or other media capable of dissolving hydrophobic substrates, as well as those where the biocatalyst is immobilized/compartmentalized and the biotransformation is carried out in either batch-based or flow-based bioprocesses.
Since laccases substrates show in most cases a poor solubility in water, the use of water-miscible organic cosolvents is very common and generally well-tolerated by these enzymes. A systematic evaluation of the effects of various organic solvents on the activity of the plant laccase from Rhus vernicifera, in either free or immobilized form, was performed in 2010 by Wan and coauthors [20]. The results showed that the amounts of organic cosolvents that could be applied without a significant decrease in laccase performances vary depending on the specific solvent, but could be up to 50% v/v. A very good tolerance toward different water-miscible cosolvents, e.g., ethanol, methanol, dimethyl sulfoxide and acetone, was shown also by the alkaline laccase from Bacillus licheniformis LS04 [21]. Several examples showing the use of organic cosolvents and of water-organic solvent biphasic systems in biotransformations catalyzed by fungal laccases, such as the laccase from Myceliophthora thermophila [22,23,24], Trametes versicolor [25,26,27], and Agaricus bisporus [28,29,30], have been reported in the literature as well.
Recently, the possible application in laccase-catalyzed biotransformations of the so-called natural deep eutectic solvents (NADES), alternative non-toxic reaction media composed by mixtures of hydrogen bond donors, e.g., polyols, and hydrogen bond acceptors, e.g., ammonium salts, has been investigated by different research groups. Some preliminary studies on the influence of various NADES on the activity and stability of microbial laccases [31,32], showed that both choline- or betaine-based media could be applied as “green solvents” in place of organic solvents. Further investigations highlighted the need of a careful choice of NADES components. For instance, a strong inhibitory effect was observed when using choline chloride as hydrogen bond acceptor, but it could be easily overcome by replacing the chloride anion with either dihydrogen citrate or dihydrogen phosphate anions [33]. In addition, the possible effect of NADES on the overall reaction environments, e.g., on the reaction pH, has to be carefully taken into account since both laccase activity and stability could be significantly affected [34].
As recently reviewed [35], as far as laccase immobilization concerns, a wide number of supports, either inorganic, organic, or hybrids, have been investigated during the last few years, mostly prompted by the technological application of this class of enzymes in bioremediation and in the textile industry. The use of immobilized laccases in organic synthesis is documented as well [1,36]. For example, in 2005, immobilized samples of the M. thermophila laccase were obtained by adsorption on glass beads or celite and used in the oxidation of the model substrate 5,6,7,8-tetrahydronaphthlen-2-ol, thus studying the influence of different organic solvents on enzyme selectivity [23]. More recently, the T. versicolor laccase was instead covalently immobilized onto magnetic fibrous silica-based nanoparticles, carriers showing high surface/volume ratio and good mechanical properties [37]. The immobilized biocatalyst showed excellent stability by retaining about 80% of its initial activity after 15 reaction cycles and was successfully applied, in the presence of suitable chemical mediators, in the oxidation of different aromatic alcohols to the corresponding aldehydes, as well as in the one-pot synthesis of a set of chromene derivatives. Alternatively, the same laccase has been recently immobilized using, as support, a magnetic-graphene oxide-based nanocomposite [38]. The performances of this heterogeneous biocatalyst were tested in the synthesis of various sulfonamide derivatives, which could be obtained with up to 90% yields. Moreover, the immobilized laccase could be reused for eight reaction cycles, at the end of which the residual activity was about 85% of the starting one, thus showing quite good operational stability.
Surprisingly, despite the growing interest toward the use of immobilized enzymes in flow systems, i.e., the so-called flow biocatalysis [39,40], the examples with laccases in this field are still scarce and showing quite preliminary results. For instance, a flow bioreactor was recently prepared by immobilizing the T. versicolor laccase on gold-impregnated poplar wood samples and tested in continuous flow biocatalysis experiments [41]. The immobilized biocatalyst showed a remarkable stability by retaining about 90% of its starting activity after 25 cycles. However, the bioreactor performances were tested only in the oxidation of the artificial chemical mediator ABTS [2,20-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)] and not in other target reactions of synthetic interest. In another very recent example, laccases were included in a study of 3D-printed continuous flow bioreactors [42]. Unfortunately, when compared to other types of enzymes, e.g., alkaline phosphatase and glucose dehydrogenase, the results obtained with laccases showed very high levels of experimental error that hampered a definitive evaluation of this flow system. As stated by the authors, this variability could be possibly due to different oxygen diffusion rates in the flow system, thus suggesting the need of its further implementation to achieve a better control of reaction kinetics.
3. Synthetic Exploitation of Laccases
3.1. Laccase-Mediator Systems: From Alcohols Bio-Oxidation to Integrated Chemoenzymatic Systems
A selective and precise manipulation of the oxidation state of hydroxyl and carbonyl groups represents a versatile tool to access to (chiral) synthetic intermediates or to achieve late-stage modifications in complex (semi)synthetic compounds or natural products derivatives. Allowing the selective introduction of ketones or aldehydes in the framework of complex molecular skeletons (i.e., natural compounds and/or polyhydroxylated substrates) while working in mild and “green” conditions, the biocatalytic oxidation of primary and secondary alcohols is among the most documented enzymatic procedures applied in organic synthesis [43,44]. Among the plethora of different redox biocatalysts available to a synthetic chemist, the LMS offers the possibility of easily converting primary alcohols and activated (i.e., allylic and benzylic) secondary alcohols into the corresponding aldehydes, acids or ketones. A large number of reports are available in the literature on this topic and they have been collected in numerous review articles published through the years [5,12,13,14,15,16,36,45,46,47]. Some interesting applications of the LMS focusing on the preparation of bioactive compounds as well as the selective oxidation of valuable and bioactive naturally occurring compounds have also been described [48,49,50]. In addition, LMS has been successfully applied for the thermodynamically disfavored oxidation of β,β-dihalogenated secondary alcohols (i.e., 2,2-dihalo-1-phenylethanol derivatives) working in a biphasic medium and providing the corresponding ketones [51].
Given their ability to oxidize lignin or lignin models [52,53,54], more than one hundred compounds have been tested as component of LMS; among them, some of the most synthetically employed are reported in Figure 1. Both natural, i.e., small molecules involved in laccase-catalyzed lignin degradation in fungi such as 3-hydroxyanthranilic acid (HAA, Figure 1a), and artificial chemical mediators, e.g., ABTS [2,20-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid] or TEMPO (2,2,6,6-tetramethylpiperidine) (Figure 1b,c, respectively), have been employed in the LMS for alcohol oxidation [45,54,55,56].
The exploitation of TEMPO or ABTS and laccases for the oxidation of primary alcohols and even benzylic secondary alcohols was reported years ago [57,58]. Later on, the laccase from T. pubescens, together with TEMPO, was used to catalyze the regioselective oxidation of the primary hydroxyl groups in a panel of sugar derivatives to the corresponding carboxylic acids via the intermediate aldehydes (Scheme 3a). The efficiency of this system was initially tested with mono- and disaccharides (i.e., phenyl β-D-glucopyranoside), and then this LMS approach was also exploited to achieve the partial oxidation of a water soluble cellulose sample [59]. The same approach was exploited for the efficient and mild oxidation of more complex bioactive glycosides, e.g., thiocolchicoside (Scheme 3b) [48].
The oxidation of natural polysaccharides by TEMPO (regenerated in situ by different oxidants) is an old reaction and has been recently reviewed [60]. In comparison to other approaches, the use of laccases for the reoxidation of TEMPO proved to be very mild and, promoting a lower degree of oxidations, resulted in the formation of soft gels. The properties of these materials, specifically those obtained by the LMS oxidation of guar polygalactomannanes, was recently investigated by Galante and coworkers and reported in a series of publications [61,62,63,64,65,66,67].
The redox mechanism of TEMPO usually prevents the oxidations of secondary alcohols. Exceptions are given by benzylic, allylic and propargylic secondary alcohols. In an early example [68], Passarella, Riva and coworkers described the enzyme assisted enantioselective synthesis of the alkaloid (+)-aloperine exploiting the LMS for the oxidations of two alcoholic intermediates, as shown in Scheme 4:
The biocatalytic production of enantiomerically enriched alcohols is also of great synthetic interest. Clearly, laccases and LMS by themselves cannot fulfill this task, but the deracemization of chiral alcohols could be obtained by designing suitable multienzymatic and cascade processes as well as by exploiting integrated chemoenzymatic approaches [69,70,71,72].
For example, by sequentially applying the LMS-catalyzed alcohol oxidation and a stereoselective enzymatic transformation of the obtained prochiral carbonyl intermediate, racemic alcohols could be converted into enantiomerically enriched compounds, as shown in Scheme 5. In both cases TEMPO was the mediator used in combination with the laccase from T. versicolor. Martínez-Montero et al. were able to convert 17 differently substituted racemic (hetero)aromatic sec-alcohols into enantio-enriched amines. Enantioselectivity and conversions were up to 99%, depending on the substrate, by building a one-pot/two-step process combining (R)- or (S)-selective aminotransferases (ATAs) with the laccase (Scheme 5a) [73]. In a second example, González-Granda et al. combined the oxy-radical TEMPO-based LMS with a small library of NAD(P)H-dependent alcohol dehydrogenases (ADHs) for the deracemization of 1-arylprop-2-yn-1-ols through, again, a sequential one-pot two-step process. Propargylic ketones were obtained in quantitative conversions (87–99% yields), thus demonstrating the efficiency of LMS in comparison with traditional chemical oxidants, and their stereoselective reduction easily allowed the access to both (R) or (S) alcohol enantiomers depending on the aromatic pattern substitution (97–99% e.e.) (Scheme 5b) [74].
The synthesis of active pharmaceutical ingredients (APIs) is a fertile ground for the exploitation of multienzymatic synthetic methodologies. A three-step biocatalytic procedure for the conversion of methyl and ethyl cyclopentene- and cyclohexene-carboxylates into both the enantiomers of the corresponding chiral 3-oxoesters, useful intermediates en route to APIs, was described by Brenna et al. in 2017 [75]. In the designed synthetic pathway, the laccase/TEMPO system was applied to oxidize the intermediate allylic alcohols (obtained from the allylic hydroxylation of starting cycloalkene carboxylates catalyzed by Rhizopus oryzae resting cells entrapped in alginate beads) to the corresponding unsaturated ketones. The obtained products were then subjected to an ene reductase (ERED)-mediated hydrogenation of the alkene bond in the same reaction vessel in a sequential mode working at 30 °C in acetate buffer (Scheme 6).
In this framework, Martínez-Montero et al. also exploited the T. versicolor laccase and TEMPO to easily convert a series of racemic allylic sec-alcohols into the corresponding α,β-unsaturated ketones. These intermediate products could then be reduced by different commercially available EREDs, in concert with a glucose/glucose dehydrogenase (GDH) system for NADPH cofactor regeneration, into the corresponding saturated ketones as enantio-enriched species (Scheme 7) [76].
By combining chemical and biocatalytic strategies, integrated chemo-enzymatic processes offer virtually an endless resource of synthetic potency subjected to a chemist’s fantasy. The LMS catalyzed oxidation of allylic alcohols, when conducted on properly designed substrates, can trigger a series of subsequent structural rearrangements which allow to easily manipulate the carbon skeleton of a molecule via the promotion of intramolecular reactions. In 2018, Brenna et al. proposed a chemo-enzymatic processes to prepare enantio-enriched cis or trans 3-methylcyclohexan-1-ol starting from the corresponding endocyclic allylic tertiary alcohol. Specifically, the following catalytic steps were performed in a series: (i) a laccase-triggered [1,3]-oxidative rearrangement; (ii) a reduction of C=C double bond catalyzed by the ene reductase OYE1; (iii) a ADH catalyzed carbonyl reduction (Scheme 8) [77]. The feasibility of the LMS to catalyze the [1,3]-oxidative rearrangement using Bobbitt’s salt (TEMPO+ BF4-) in combination with T. versicolor laccase was successfully demonstrated and studied in detail. Two reaction media were engineered: a homogeneous aqueous system, highly efficient for the transposition of substrates devoid of electron withdrawing groups (EWGs), and a heterogeneous system exploiting an immobilized laccase preparation to perform the reaction on EWG-containing macrocyclic alkenols or tertiary alcohols in acetonitrile.
Interestingly, the LMS was also successfully applied by the Beifuss group in 2013 to catalyze the first-ever reported enzymatic Achmatowicz reaction, a rearrangement which transforms furfuryl alcohols into dihydropyrans (Scheme 9a) [78]. In detail, LMS was used to oxidize bulky, disubstituted (5-alkylfuran-2-yl)carbinols using aerial oxygen and TEMPO, and selectively afforded 6-hydroxy-(2H)-pyran-3(6H)-ones with yields up to 90% (Scheme 9b). In addition, starting from suitably substituted furan-2-yl carbinols as substrates, this procedure allowed the efficient preparation of (2H)-pyran-2,5(6H)-diones in a single step.
3.2. Laccases as Green Csp2-H Bonds Activators
Modern synthetic chemistry aims at proposing novel methods to construct chemical bonds in the framework of complex molecular structures with economical, efficient, and environmentally benign approaches. Since their outbreak discovery, transition-metal catalyzed organic reactions have been used in the construction of a variety of chemical bonds since they can minimize pre-functionalization steps and waste formation via the catalytic functionalization of unreactive C–H bonds [79,80,81,82,83]. In fact, the direct oxidation of an organic substrate to afford an organic radical is of particular interest. Organic radicals are indeed highly reactive species which undergo complex quenching mechanisms in the process of dissipating the excess in energy derived from the homolytic bond-breaking at the basis of their creation. By controlling the different quenching destines available to these unstable species, products of diverse chemical nature can theoretically be obtained.
In the framework of this review, focused on the role and application of laccases in organic synthesis for the preparation of fine chemicals, the reaction pathway previously described in Scheme 2a, that is the activation of normally inert Csp2-H bonds, is of particular interest. In this scenario, laccases offer a “green” alternative to transition-metal catalyzed organic reactions, allowing one to operate in a one-step reaction under mild, eco-friendly conditions.
Accordingly, different products can be obtained depending on the molecular skeleton of the radicals and the functional groups installed on them and substituents as well as the reaction media, conditions and partners (Scheme 10).
Specifically, when aromatic amines or phenols (substrates generally characterized by redox potentials suitable to be directly processed by laccase) are biocatalytically oxidized, radical formation occurs, at first, on the electronegative heteroatoms, i.e., nitrogen or oxygen. Thanks to mesomeric resonance, the formed radicals are then delocalized along all the carbonaceous skeleton of the fully conjugated π-system (i.e., the group of molecules shown in Scheme 11), de facto activating different ring positions and Csp2-H bonds toward the formation of new C-C or C-Het bonds. The use of specific, ad hoc designed substrates and the proper engineering of the operative reaction conditions allow us to “select” the quenching processes of the reactive species promoting different reaction mechanisms and the formation of different classes of products.
The use of laccase-catalyzed Csp2-H bonds activation for the synthesis of complex molecular skeletons is widely documented in the literature thanks to the synthetic knowledge acquired through the years in the fields of laccase-mediated formation of biaryl compounds [84,85,86,87], as well as of formal nucleophilic aromatic substitutions [88,89,90,91,92,93,94]. Selected examples will be discussed in the following.
3.2.1. Radical C-O and C-C Couplings
The development of innovative and, most importantly, mild catalytic synthetic entries to biaryl compounds is among the most investigated research field of methodological organic chemistry [95,96,97]. Being potential axially stereogenic and chiral due to atropoisomery, biaryl compounds find in fact wide (bio)technological and biological applications, which has made them versatile and highly cherished synthetic targets. Thus, the application of enzymatic protocols to enter biaryl compounds is of great interest and represents one of the most logical, even if not trivial at all, synthetic exploitation of direct laccase oxidation of anilines and phenols (see, for instance ref [22]).
In 2005, Ciecholewski et al. prepared a small library of biaryl compounds via laccase-catalyzed radical C-C homo-couplings (Scheme 12). A difficult control over the regiochemistry of the coupling and over the reaction proceedings itself (a high degree of polymerization was observed) were identified as two harming issues connected to some of the processes they investigated. As it is now generally well documented, the substitution pattern of the to-be-coupled aromatic rings appeared to be crucial, as the presence, for example, of electron-donating or withdrawing groups (EDGs, EWGs) in specific positions guides the process towards one of the possible C-H derivatized products by selectively stabilizing one activated radical over another [84].
Highlighting the potentialities of laccase-mediated biaryl synthesis, Beifuss and coworkers provided an elegant entry to 3-tert-butyl-1H-pyrazol-5(4H)-one containing biaryl compounds from the bio-oxidation of catechols which proceeded with very high isolated yields (Scheme 13) [98].
Moreover, Ncanana et al. demonstrated how good control over the reaction engineering can promote high degrees of selectivity in the laccase-catalyzed coupling of bioactive, natural products. The diterpene totarol was in fact successfully subjected to homocoupling reactions in which the use of different solvents, modulating the stabilizing effects of the C* radicals in place of the phenolic O* radicals, promoted the selective isolation of the target C-C dimer in excellent yield reducing the formation of the C-O dimeric byproduct (Scheme 14) [99].
A significant and unexpected solvent influence on the selectivity of laccase-catalyzed coupling of tetrahydro-2-naphthol derivatives was also previously observed [23].
Regardless of the presented examples and the number of reports available in the literature so far [84,85,86,87,98,99], the use of laccases for the preparation of biaryl compounds is still a challenging synthetic application, which appears difficult to be generalized as well as fully rationalized. As exemplified by the work of Constantin and coworkers in 2012, laccase-mediated bio-oxidations are quite prone to results in the formation of unexpected and unpredicted products since subtle differences in the structure of the oxidized substrates can largely influence the outcome of a reaction. In this specific work, sesamol, a natural compound component of sesame seeds, was subjected to laccase oxidation aiming at isolating the corresponding C-C dimer. Instead, the authors isolated and characterized for the first time a previously unknown trimeric compound which was obtained in high yield in a single step (Scheme 15) [85].
Even if they are often unpredictable by nature, radical couplings, when run of structurally complex and sterically hindered molecules, can allow highly selective and valuable late-stage derivatizations normally precluded to classical organic chemistry. As reported by Sagui et al. [100], laccase catalysis could be efficiently applied to promote the C-C coupling of the indolic alkaloids catharanthine and vindoline which resulted in the formation of a eniminium cationic intermediate (Scheme 16). Following NaBH4 reduction, the synthetically useful dimer anhydrovinblastine (AVBL) was isolated in a 56% yield and fully characterized. The practicability of this bioconversion on this class of bioactive alkaloids was further confirmed through the condensation of catharanthine with the vindoline analogue 11-methoxy-dihydrotabersonine.
3.2.2. Nucleophilic Aromatic Substitution
Scheme 17 depicts the operating mechanism, i.e., a 1,4-conjugate addition to a quinoid species formed in situ by enzymatic bio-oxidation, at the basis of laccase-catalyzed aromatic nucleophilic substitutions.
Different examples of laccase-catalyzed nucleophilic aromatic substitution dealing with different quinoid precursor as substrates and nucleophiles (nitrogen, oxygen and sulfur containing) can be found in the literature. In the framework of this review, some of the most interesting ones, either dealing with the manipulation of complex molecular skeletons or of bioactive compounds, will be discussed.
A first report of the use of aliphatic and, most interestingly, aromatic amines as nucleophiles to obtain derivatized substituted p-hydroquinones via nuclear amination was presented by Niedermeyer et al. in 2005 (Scheme 18) [88]. As stated in the previous paragraph, the yields and regioselectivities of the reaction presented in this study were strictly correlated to the substitution of the aromatic rings. The investigation on this topic was then widened to a systematic and mechanistic level in 2015 [101].
In subsequent works, laccase-catalyzed nuclear amination was exploited by Hahn and coworker to prepare bioactive morpholine derivatives [92,93], and by Mikolasch et al. to efficiently synthetize novel antibacterial derivatives from corollosporine [90] and penicillins [89,91,102], as shown in Scheme 19.
Several examples can be found in the literature describing an analogous synthetic sequence to produce sulfoquinones via the bio-oxidation of hydroquinones followed by a 1,4-conjugate Michael addition.
In 2012, Wellington et al. reported on the efficient and selective one-pot preparation of 1,4-naphthoquinone-2,3-bis-sulfide from 1,4-dihydroxy-2-naphthoic acid (Scheme 20) [94].
The 1,4-dihydroxy-2-naphthoic acid was a clever substrate ad hoc designed by the authors to guarantee high selectivity toward the formation of the target di-sulfurate products. Two reactive positions of the quinone are in fact blocked by the conjugation with the second aromatic ring, the presence of a EWG in the ortho position stabilizes and facilitates the formation of a radical in position 3 of the ring while the transient formation of a β-keto acid intermediate shifts the equilibrium toward a second step of 1,4-conjugate addition by irreversible decarboxylation (Scheme 21).
An interesting and outbreaking example of laccase-mediated quinone manipulation was presented by Cannatelli et al. in 2015 in the form of an enzymatic α-arylation of benzoyl acetonitrile with ortho-quinones. Specifically, the authors were able to selectively install a novel C-C bond between an aliphatic Csp3 and an aromatic Csp2, thus obtaining useful benzylic nitriles (Scheme 22) [87].
3.2.3. Csp2-H Bond Activation for Ring Closing Reactions
The laccase-mediated generation of reactive radical intermediates has been successfully applied to obtain domino, cascade and one-pot ring closure reactions for the synthesis of (hetero)cyclic compounds. Accordingly, by using rationally designed substrates and working under engineered and optimized reaction conditions, (pseudo)quinones formations, nucleophilic aromatic substitutions and C-O/C-C radical couplings can be merged to obtain laccase-mediated ring closure reactions [103]. The neat results of these transformations can be described as formal oxidative homo- and/or hetero-coupling couplings, that is, ring closing reactions that involve two molecules of the same substrate or two different partners.
It is noteworthy that no expensive, toxic and/or hazardous metal-based chemical catalysts, which usually require inert atmosphere and specific experimental conditions, are needed to perform this biocatalytic Csp2-H bond activation.
- Homocouplings
The first example of the preparation of heterocycles by laccase oxidation was the formation of phenoxazinones from 3-hydroxyanthranilic acid (3-HAA, Scheme 23), described by Eggert et al. in 1995 [55].
Regardless of the preliminary nature of this study and the fact that phenoxazinone formation was attested only by UV/Vis spectroscopy, this approach was soon validated by the synthesis of the antiproliferative molecule actinocin by Osiadacz et al. [104] (Scheme 24).
The synthetic utility of this protocol to obtain nitrogenous homocycles was further demonstrated by Sousa et al., who published three papers dealing with the rationally designed biosynthesis of substituted phenazines, phenoxazinones and benzocarbazoles by using the laccase from Bacillus subtilis (spore coat protein A, CotA, Scheme 25) [105,106,107].
A domino process can be built in order to afford oxygen-containing heterocyclic compounds, by exploiting the oxidative (homo)coupling of phenolic substrates [108,109,110]. Specifically, vinyl phenols and stilbenoids, molecules structurally related to laccases natural substrates, that is the mono-lignols composing lignin, can be mainly converted into three different groups of oxygenated heterocycles, as shown in Scheme 26: 2,3-dihydrobenzofurans (DHB, route i), dioxanes (route ii), and pinoresinol-like bicyclic compounds (hexahydrofuro[3,4-c]furans, route iii).
The formation of one of the proposed structures (which in some cases are obtained in mixture) is, again, guided by the structural features of the reacted substrates; all the novel stereocenters are built with no control over their absolute configuration, while, due to steric hindrance and thermodynamics, 2,3-DHBs and dioxanes are trans-configured.
In general, when R is both an alkyl or an aryl substituent and R’ is a second phenolic moiety (cathecol derivative as a substrate), a dioxane ring (structure ii) is usually obtained as the major product.
In the presence of an allylic alcohol (R = OH) and of an alkyl substituent as R’, the formation of racemic hexahydrofuro[3,4-c]furans (iii), the core of the natural product pinoresinol, is preferred [47,111].
Finally, 2,3-DHBs (i, β-5 type dimers in lignin framework) can be selectively obtained from ad hoc designed substrates. In these molecules, R’ is a “spectator group” (i.e., hydrogen, alkyl chains, protected phenols) and the R substituent is either an alkyl or an aryl group [27,110,112,113,114,115].
From a mechanistic point of view, as shown in Scheme 27, the ring closure occurs via a sequence of phenol oxidation, C-C/O radical coupling, and 1,4-conjugate addition. Two novel stereocenters are formed in this domino process.
These easy-to-handle enzymatic entries to complex oxygenated heterocycles were exploited to build a small library of substituted benzofurans, which was tested in vitro as potential modulators of the molecular chaperone Hsp90 in the quest for novel antiproliferative drugs [116].
More recently, this approach was successfully used to transform 4-hydroxy-chalcones of synthetic origin into the corresponding 2,3-DHB derivatives. Due to the presence of the conjugated system with the fully conjugated π-carbonyl group characterizing chalcones, a C-O dimer was obtained in mixture with the 2,3-DHB based products (Scheme 28) [117].
Nature has solved the lack of enantioselectivity in laccase-mediated synthesis of 2,3-DHBs by the action of the so-called “dirigent proteins” [118,119]. Since these enantiopure scaffolds represent valuable synthons for the preparation of the plethora of different naturally occurring bioactive compounds [120], alternative synthetic approaches have been investigated. An elegant strategy was proposed by Navarra et al. in two consecutive reports. The racemic mixtures of 2,3-DHB obtained from laccase catalysis were submitted to lipase-catalyzed kinetic resolutions in organic solvents. By remote center discrimination in alcoholysis reactions both enantiomers of 2,3-DHB could be obtained with e.e. up to the 98 % [121,122].
In an alternative approach, Gavezzotti et al. reported on the laccase-mediated Csp2-H activation, followed by the hydrolytic action of glycosidases and preparative RP-HPLC, to access both enantiomers of δ-viniferin, a natural 2,3-DHB-based polyphenol endowed with pharmaceutical potentialities, in a fully enzymatic approach starting from piceid, the α-glucoside of resveratrol [113]. The biological activities of these pure enantiomers were then carefully evaluated [123].
As largely highlighted during this discussion, laccase-mediated processes are far from being easily rationalized and the outcomes of the reactions they catalyze can often lead to unpredicted yet valuable chemical skeletons. As a noteworthy example, the oxidative homocoupling of tyrosol resulted in the unexpected formation of a 2-hydrobenzofuran-based tetracyclic, polyoxygenated product. As shown in Scheme 29, after a step of radical C-C coupling promoted by laccase oxidation, the obtained adduct immediately rearranged to form a C3 tetra-substituted benzofuran. This intermediate, due to the presence of a nucleophilic primary alcohol near to an electrophilic enone, spontaneously evolved to the tetracyclic product isolated in virtue of an intramolecular Michael addition [110].
- Heterocouplings
The aromatic substitution reactions discussed in the previous paragraph allowed us to obtain substituted quinones with different degrees of regioselectivity depending on both the hydroxyquinone partner and the nucleophile applied. When conducting the mentioned processes in the presence of a bidentate nucleophile, cyclic structures can be obtained via a sequence of one inter- and one intra-molecular laccase-mediated 1,4-additions. As an example, in 2015, Cannatelli et al. reported a biocatalyzed domino synthesis of 2,3-ethylenedithio-1,4-quinones based on the laccase-activation of the C2 and C3 positions in the presence of 1,2-ethanedithiol (Scheme 30) [124].
Hahn and coworkers studied the applicability of a domino process based on the laccase-activation of structurally complex hydroquinoid substrates to obtain poly(hetero)cyclic structures in the presence of different sulfurate or nitrogenous nucleophiles (Scheme 31). An extensive and very detailed analytical study was performed in order to characterize the complex mixture of products; difficulties in controlling the regiochemical outcome of the reactions was highlighted by the authors. Again, the substitution pattern of the substrates appeared as crucial [101,103].
Beifuss and coworkers have been studying the laccase-catalyzed domino/cascade synthesis of oxygenated heterocycles since 2005. Outstanding examples are represented by their works regarding the use of aromatic 1,3-dicarbonyls as carbon nucleophiles in combination with laccase-activated 1,2-dihydroquinones (Scheme 32) [125,126,127].
In their investigations, and in related publications from other authors [128], the possibility of using sterically demanding, hetero- and carba-cyclic/acyclic 1,3-dicarbonyls as nucleophiles in these biocatalyzed processes has been investigated and validated. The structurally diverse benzofurans obtained by the authors are shown in Figure 2:
Beifuss’ investigation was also expanded to the preparation of sulphurated and nitrogenous poly(hetero)cycles by the clever exploitation of S- and N-containing synthetic equivalents of 1,3-dicarbonyls. Their elegant biocatalytic entry to pyrimidinyl benzothiazoles, which was based on the use of 2-thioxo pyrimidine as nucleophiles, is presented in Scheme 33 [28]. Target products were obtained as enriched mixtures of regioisomers.
The complex regiochemical outcome of this domino process was further investigated by using cathecol and a sterically demanding 2-thioxo pyrimidine, as shown in Scheme 34.
- Laccase-catalyzed cycloadditions
When quinones are generated by the laccase-catalyzed oxidation of 1,4-dihydroquinones and catechols in the presence of a diene, Diels-Alder cycloadditions can occur. Ragauskas and coworkers extensively investigated this topic and published two reports dealing with the one-pot synthesis of 1,4-naphthoquinones (Scheme 35) [129,130].
3.3. Miscellanea
Scant reports have described the oxidation of aromatic methyl groups [57,131], alkenes [132], and even dibenzyl ethers [58], to the corresponding aldehydes catalyzed by the previously described LMS. However, with the last two groups of compounds, the conversions were generally low. In Table 1, representative examples of the oxidation of benzyl alcohols to the corresponding benzaldehydes are reported.
An interesting report in which the use of laccases in peptide synthesis has been proposed deserves a mention. In this study, laccases were used for the selective removal of phenylhydrazide-protecting groups in a mild process that caused neither oxidative modification nor destruction of methionine or tryptophan side chains [133].
Laccase-mediated C-H bond activation was also successfully employed in the selective hydroxylation of Csp3-H aliphatic bonds. In 2012, Chirivì et al. were able to unexpectedly but conveniently mono-hydroxylate a panel of ergot alkaloids at the C-4 benzylic position via laccases catalysis (Scheme 36) [134]. Quite notably, the proposed protocol allowed, for the first time, the regio- and stereoselective C-4 functionalization of the ergoline skeleton. When a C-10 methoxy substituent was not present, hydroxylation was stereoselective, thus furnishing C-4α OH derivatives.
3.4. Innovative Perspectives in Laccases Synthetic Exploitation
The last example of our overview on laccases synthetic applications is an interesting work by Sulci et al. in which laccase-mediated C-H activation was coupled with a stereoselective organocatalytic process [135]. The authors exploited a prolinol-based amine-based organocatalyst, i.e., the Hayachi-Jorgensen catalyst, suitable for the activation of the ⍺-position of carbonyl compounds [136,137], coupled with the previously discussed laccase-mediated activation of 1,2-hydro quinones to prepare enantiomerically enriched 2,3-DHBs in a organo-enzymatic integrated process (Scheme 37).
As shown in Scheme 37, the described multi-catalytic system gave products with high isolated yields and with moderate to high (65–92%) e.e., while it failed in promoting enantioselection in the case of the only reported example of tetrasubstituted stereocenter in position C3. The authors also proposed a catalytic mechanism, which is reported in Scheme 38.
This pioneering research paves the way to a wide range of synthetic applications for the laccase-mediated C-H activation assuming that the following issues might be addressed when assembling any integrated multi-catalysts chemo-enzymatic strategies: (1) reaction media compatibility (pH, solvents), (2) enzyme stability and activity, (3) stability and turnover of the organic catalyst, and (4) control over possible cross-reactivity.
4. Conclusions and Perspectives
As shown by the last presented example, the limits and the complete potentialities of laccase-mediated synthetic chemistry is still to be fully disclosed. These enzymes represent in fact an oxidative toolbox of extraordinary convenience in terms of mild reaction media and environmental impact. Being robust and solvent-compatible biocatalysts, laccases allow one to access complex molecular skeletons in one-pot, chemo-integrated or multistep/cascade redox processes by consuming the oxidizing power of molecular oxygen and producing water in exchange. By deeply understanding the redox features of these “radical forges” and by learning how to properly combine them with other chemo- and biocatalysts in more complex reaction systems, the number of possible laccase synthetic applications will be limited only by chemist imagination.
Author Contributions
All authors (I.B., E.E.F., S.R., and D.M.) have equally contributed to literature search and analysis and to manuscript preparation and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Riva, S. Laccases: Blue enzymes for green chemistry. Trends Biotechnol. 2006, 24, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Arregui, L.; Ayala, M.; Gómez-Gil, X.; Gutiérrez-Soto, G.; Hernández-Luna, C.E.; Herrera de los Santos, M.; Levin, L.; Rojo-Domínguez, A.; Romero-Martínez, D.; Saparrat, M.C.N.; et al. Laccases: Structure, function, and potential application in water bioremediation. Microb. Cell Fact. 2019, 18, 200. [Google Scholar] [CrossRef] [PubMed]
- Janusz, G.; Pawlik, A.; Świderska-Burek, U.; Polak, J.; Sulej, J.; Jarosz-Wilkołazka, A.; Paszczyński, A. Laccase properties, physiological functions, and evolution. Int. J. Mol. Sci. 2020, 21, 966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giardina, P.; Faraco, V.; Pezzella, C.; Piscitelli, A.; Vanhulle, S.; Sannia, G. Laccases: A never-ending story. Cell. Mol. Life Sci. 2010, 67, 369–385. [Google Scholar] [CrossRef]
- Mogharabi, M.; Faramarzi, M.A. Laccase and Laccase-Mediated Systems in the Synthesis of Organic Compounds. Adv. Synth. Catal. 2014, 356, 897–927. [Google Scholar] [CrossRef]
- Pezzella, C.; Guarino, L.; Piscitelli, A. How to enjoy laccases. Cell. Mol. Life Sci. 2015, 72, 923–940. [Google Scholar] [CrossRef]
- Binnington, K.C.; Barrett, F.M. Ultrastructural localization of phenoloxidases in cuticle and haemopoietic tissue of the blowfly Lucilia cuprina. Tissue Cell 1988, 20, 405–419. [Google Scholar] [CrossRef]
- Rodríguez Couto, S.; Toca Herrera, J.L. Industrial and biotechnological applications of laccases: A review. Biotechnol. Adv. 2006, 24, 500–513. [Google Scholar] [CrossRef]
- Kunamneni, A.; Plou, F.J.; Ballesteros, A.; Alcalde, M. Laccases and their applications: A patent review. Recent Pat. Biotechnol. 2008, 2, 10–24. [Google Scholar] [CrossRef] [Green Version]
- Zerva, A.; Simić, S.; Topakas, E.; Nikodinovic-Runic, J. Applications of Microbial Laccases: Patent Review of the Past Decade (2009–2019). Catalysts 2019, 9, 1023. [Google Scholar] [CrossRef] [Green Version]
- Quintanar, L.; Stoj, C.; Taylor, A.B.; Hart, P.J.; Kosman, D.J.; Solomon, E.I. Shall we dance? How a multicopper oxidase chooses its electron transfer partner. Acc. Chem. Res. 2007, 40, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yu, X.; Zhu, G.; Zheng, Z.; Feng, F.; Zhang, Z. Conditions Optimizing and Application of Laccase-mediator System (LMS) for the Laccase-catalyzed Pesticide Degradation. Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morozova, O.V.; Shumakovich, G.P.; Shleev, S.V.; Yaropolov, Y.I. Laccase-mediator systems and their applications: A review. Appl. Biochem. Microbiol. 2007, 43, 523–535. [Google Scholar] [CrossRef]
- Hilgers, R.; Vincken, J.P.; Gruppen, H.; Kabel, M.A. Laccase/Mediator Systems: Their Reactivity toward Phenolic Lignin Structures. ACS Sustain. Chem. Eng. 2018, 6, 2037–2046. [Google Scholar] [CrossRef] [Green Version]
- Mate, D.M.; Alcalde, M. Laccase: A multi-purpose biocatalyst at the forefront of biotechnology. Microb. Biotechnol. 2017, 10, 1457–1467. [Google Scholar] [CrossRef]
- Pankaj, K.C.; Shashi, L.B.; Manisha, S.; Sunil, K.S.; Rama, S.S.Y.; Sudha, Y. Fungal Laccases and their Biotechnological Significances in the Current Perspective: A Review. Curr. Org. Chem. 2015, 19, 1916–1934. [Google Scholar] [CrossRef]
- Ludwig, R.; Ozga, M.; Zámocky, M.; Peterbauer, C.; Kulbe, K.D.; Haltrich, D. Continuous Enzymatic Regeneration of Electron Acceptors Used by Flavoenzymes: Cellobiose Dehydrogenase-Catalyzed Production of Lactobionic Acid as an Example. Biocatal. Biotransform. 2004, 22, 97–104. [Google Scholar] [CrossRef]
- Ferrandi, E.E.; Monti, D.; Patel, I.; Kittl, R.; Haltrich, D.; Riva, S.; Ludwig, R. Exploitation of a Laccase/Meldola’s Blue System for NAD+ Regeneration in Preparative Scale Hydroxysteroid Dehydrogenase-Catalyzed Oxidations. Adv. Synth. Catal. 2012, 354, 2821–2828. [Google Scholar] [CrossRef]
- Tonin, F.; Martì, E.; Arends, I.W.C.E.; Hanefeld, U. Laccase Did It again: A Scalable and Clean Regeneration System for NAD+ and Its Application in the Synthesis of 12-oxo-Hydroxysteroids. Catalysts 2020, 10, 677. [Google Scholar] [CrossRef]
- Wan, Y.-Y.; Lu, R.; Xiao, L.; Du, Y.-M.; Miyakoshi, T.; Chen, C.-L.; Knill, C.J.; Kennedy, J.F. Effects of organic solvents on the activity of free and immobilised laccase from Rhus vernicifera. Int. J. Biol. Macromol. 2010, 47, 488–495. [Google Scholar] [CrossRef]
- Lu, L.; Zhao, M.; Wang, T.-N.; Zhao, L.-Y.; Du, M.-H.; Li, T.-L.; Li, D.-B. Characterization and dye decolorization ability of an alkaline resistant and organic solvents tolerant laccase from Bacillus licheniformis LS04. Bioresour. Technol. 2012, 115, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Nicotra, S.; Intra, A.; Ottolina, G.; Riva, S.; Danieli, B. Laccase-mediated oxidation of the steroid hormone 17β-estradiol in organic solvents. Tetrahedron Asymmetry 2004, 15, 2927–2931. [Google Scholar] [CrossRef]
- Intra, A.; Nicotra, S.; Riva, S.; Danieli, B. Significant and unexpected solvent influence on the selectivity of laccase-catalyzed coupling of tetrahydro-2-naphthol derivatives. Adv. Synth. Catal. 2005, 347, 973–977. [Google Scholar] [CrossRef]
- Wellington, K.W.; Kolesnikova, N.I. A laccase-catalysed one-pot synthesis of aminonaphthoquinones and their anticancer activity. Bioorg. Med. Chem. 2012, 20, 4472–4481. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.B.; Singhal, R.S. Laccase–gum Arabic conjugate for preparation of water-soluble oligomer of catechin with enhanced antioxidant activity. Food Chem. 2014, 150, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Lugaro, G.; Carrea, G.; Cremonesi, P.; Casellato, M.M.; Antonini, E. The oxidation of steroid hormones by fungal laccase in emulsion of water and organic solvents. Arch. Biochem. Biophys. 1973, 159, 1–6. [Google Scholar] [CrossRef]
- Ponzoni, C.; Beneventi, E.; Cramarossa, M.R.; Raimondi, S.; Trevisi, G.; Pagnoni, U.M.; Riva, S.; Forti, L. Laccase-Catalyzed Dimerization of Hydroxystilbenes. Adv. Synth. Catal. 2007, 349, 1497–1506. [Google Scholar] [CrossRef]
- Abdel-Mohsen, H.T.; Conrad, J.; Beifuss, U. Laccase-Catalyzed Domino Reaction between Catechols and 6-Substituted 1,2,3,4-Tetrahydro-4-oxo-2-thioxo-5-pyrimidinecarbonitriles for the Synthesis of Pyrimidobenzothiazole Derivatives. J. Org. Chem. 2013, 78, 7986–8003. [Google Scholar] [CrossRef]
- Abdel-Mohsen, H.T.; Conrad, J.; Beifuss, U. Laccase-catalyzed synthesis of catechol thioethers by reaction of catechols with thiols using air as an oxidant. Green Chem. 2014, 16, 90–95. [Google Scholar] [CrossRef]
- Abdel-Mohsen, H.T.; Conrad, J.; Harms, K.; Nohr, D.; Beifuss, U. Laccase-catalyzed green synthesis and cytotoxic activity of novel pyrimidobenzothiazoles and catechol thioethers. RSC Adv. 2017, 7, 17427–17441. [Google Scholar] [CrossRef] [Green Version]
- Ezgi Ünlü, A.; Prasad, B.; Anavekar, K.; Bubenheim, P.; Liese, A. Investigation of a green process for the polymerization of catechin. Prep. Biochem. Biotechnol. 2017, 47, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Khodaverdian, S.; Dabirmanesh, B.; Heydari, A.; Dashtban-moghadam, E.; Khajeh, K.; Ghazi, F. Activity, stability and structure of laccase in betaine based natural deep eutectic solvents. Int. J. Biol. Macromol. 2018, 107, 2574–2579. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M.L.; Pereira, M.M.; Freire, M.G.; Silva, J.P.A.; Coutinho, J.A.P.; Tavares, A.P.M. Laccase Activation in Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2019, 7, 11806–11814. [Google Scholar] [CrossRef]
- Delorme, A.E.; Andanson, J.-M.; Verney, V. Improving laccase thermostability with aqueous natural deep eutectic solvents. Int. J. Biol. Macromol. 2020, 163, 919–926. [Google Scholar] [CrossRef]
- Daronch, N.A.; Kelbert, M.; Pereira, C.S.; de Araújo, P.H.H.; de Oliveira, D. Elucidating the choice for a precise matrix for laccase immobilization: A review. Chem. Eng. J. 2020, 397, 125506. [Google Scholar] [CrossRef]
- Romero-Guido, C.; Baez, A.; Torres, E. Dioxygen Activation by Laccases: Green Chemistry for Fine Chemical Synthesis. Catalysts 2018, 8, 223. [Google Scholar] [CrossRef] [Green Version]
- Mogharabi-Manzari, M.; Ghahremani, M.H.; Sedaghat, T.; Shayan, F.; Faramarzi, M.A. A Laccase Heterogeneous Magnetic Fibrous Silica-Based Biocatalyst for Green and One-Pot Cascade Synthesis of Chromene Derivatives. European J. Org. Chem. 2019, 2019, 1741–1747. [Google Scholar] [CrossRef]
- Rouhani, S.; Azizi, S.; Kibechu, R.W.; Mamba, B.B.; Msagati, T.A.M. Laccase Immobilized Fe3O4-Graphene Oxide Nanobiocatalyst Improves Stability and Immobilization Efficiency in the Green Preparation of Sulfa Drugs. Catalysts 2020, 10, 459. [Google Scholar] [CrossRef]
- Tamborini, L.; Fernandes, P.; Paradisi, F.; Molinari, F. Flow Bioreactors as Complementary Tools for Biocatalytic Process Intensification. Trends Biotechnol. 2018, 36, 73–88. [Google Scholar] [CrossRef]
- Bolivar, J.M.; López-Gallego, F. Characterization and evaluation of immobilized enzymes for applications in flow reactors. Curr. Opin. Green Sustain. Chem. 2020, 25, 100349. [Google Scholar] [CrossRef]
- Goldhahn, C.; Taut, J.A.; Schubert, M.; Burgert, I.; Chanana, M. Enzyme immobilization inside the porous wood structure: A natural scaffold for continuous-flow biocatalysis. RSC Adv. 2020, 10, 20608–20619. [Google Scholar] [CrossRef]
- Spano, M.B.; Tran, B.H.; Majumdar, S.; Weiss, G.A. 3D-Printed Labware for High-Throughput Immobilization of Enzymes. J. Org. Chem. 2020, 85, 8480–8488. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.; Ottolina, G.; Carrea, G.; Riva, S. Redox Reactions Catalyzed by Isolated Enzymes. Chem. Rev. 2011, 111, 4111–4140. [Google Scholar] [CrossRef] [PubMed]
- Puetz, H.; Puchľová, E.; Vranková, K.; Hollmann, F. Biocatalytic Oxidation of Alcohols. Catalysts 2020, 10, 952. [Google Scholar] [CrossRef]
- Díaz-Rodríguez, A.; Martínez-Montero, L.; Lavandera, I.; Gotor, V.; Gotor-Fernández, V. Laccase/2,2,6,6-tetramethylpiperidinoxyl radical (TEMPO): An efficient catalytic system for selective oxidations of primary hydroxy and amino groups in aqueous and biphasic media. Adv. Synth. Catal. 2014, 356, 2321–2329. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, Z.; Ding, W.; Xie, J.; Chen, Y.; Wu, J.; Chen, X.; Ying, H. A mild and highly efficient laccase-mediator system for aerobic oxidation of alcohols. Green Chem. 2014, 16, 1131–1138. [Google Scholar] [CrossRef]
- Wan, Y.; Lu, R.; Akiyama, K.; Miyakoshi, T.; Du, Y. Enzymatic synthesis of bioactive compounds by Rhus laccase from Chinese Rhus vernicifera. Sci. China Ser. B Chem. 2007, 50, 179–182. [Google Scholar] [CrossRef]
- Baratto, L.; Candido, A.; Marzorati, M.; Sagui, F.; Riva, S.; Danieli, B. Laccase-mediated oxidation of natural glycosides. J. Mol. Catal. B Enzym. 2006, 39, 3–8. [Google Scholar] [CrossRef]
- Kudanga, T.; Nemadziva, B.; Le Roes-Hill, M. Laccase catalysis for the synthesis of bioactive compounds. Appl. Microbiol. Biotechnol. 2017, 101, 13–33. [Google Scholar] [CrossRef]
- Patel, H.; Gupte, A. Laccase catalysis: A green approach in bioactive compound synthesis. In Research Advancements in Pharmaceutical, Nutritional, and Industrial Enzymology; IGI Global: Hershey, PA, USA, 2018; pp. 178–212. ISBN 9781522552383. [Google Scholar]
- Kȩdziora, K.; Díaz-Rodríguez, A.; Lavandera, I.; Gotor-Fernández, V.; Gotor, V. Laccase/TEMPO-mediated system for the thermodynamically disfavored oxidation of 2,2-dihalo-1-phenylethanol derivatives. Green Chem. 2014, 16, 2448–2453. [Google Scholar] [CrossRef] [Green Version]
- Barreca, A.M.; Fabbrini, M.; Galli, C.; Gentili, P.; Ljunggren, S. Laccase/mediated oxidation of a lignin model for improved delignification procedures. J. Mol. Catal. B Enzym. 2003, 26, 105–110. [Google Scholar] [CrossRef]
- Call, H.P.; Mücke, I. History, overview and applications of mediated lignolytic systems, especially laccase-mediator-systems (Lignozym®-process). J. Biotechnol. 1997, 53, 163–202. [Google Scholar] [CrossRef]
- Bourbonnais, R.; Paice, M.G. Oxidation of non-phenolic substrates. An expanded role for laccase in lignin biodegradation. FEBS Lett. 1990, 267, 99–102. [Google Scholar] [CrossRef] [Green Version]
- Eggert, C.; Temp, U.; Dean, J.F.D.; Eriksson, K.E.L. Laccase-mediated formation of the phenoxazinone derivative, cinnabarinic acid. FEBS Lett. 1995, 376, 202–206. [Google Scholar] [CrossRef] [Green Version]
- Rochefort, D.; Leech, D.; Bourbonnais, R. Electron transfer mediator systems for bleaching of paper pulp. Green Chem. 2004, 6, 14–24. [Google Scholar] [CrossRef]
- Potthast, A.; Rosenau, T.; Chen, C.-L.; Gratzl, J.S. Selective Enzymic Oxidation of Aromatic Methyl Groups to Aldehydes. J. Org. Chem. 1995, 60, 4320–4321. [Google Scholar] [CrossRef]
- Fabbrini, M.; Galli, C.; Gentili, P.; Macchitella, D. An oxidation of alcohols by oxygen with the enzyme laccase and mediation by TEMPO. Tetrahedron Lett. 2001, 42, 7551–7553. [Google Scholar] [CrossRef]
- Marzorati, M.; Danieli, B.; Haltrich, D.; Riva, S. Selective laccase-mediated oxidation of sugars derivatives. Green Chem. 2005, 7, 310–315. [Google Scholar] [CrossRef]
- Pierre, G.; Punta, C.; Delattre, C.; Melone, L.; Dubessay, P.; Fiorati, A.; Pastori, N.; Galante, Y.M.; Michaud, P. TEMPO-mediated oxidation of polysaccharides: An ongoing story. Carbohydr. Polym. 2017, 165, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Lavazza, M.; Formantici, C.; Langella, V.; Monti, D.; Pfeiffer, U.; Galante, Y.M. Oxidation of galactomannan by laccase plus TEMPO yields an elastic gel. J. Biotechnol. 2011, 156, 108–116. [Google Scholar] [CrossRef]
- Merlini, L.; Boccia, A.C.; Mendichi, R.; Galante, Y.M. Enzymatic and chemical oxidation of polygalactomannans from the seeds of a few species of leguminous plants and characterization of the oxidized products. J. Biotechnol. 2015, 198, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Rossi, B.; Campia, P.; Merlini, L.; Brasca, M.; Pastori, N.; Farris, S.; Melone, L.; Punta, C.; Galante, Y.M. An aerogel obtained from chemo-enzymatically oxidized fenugreek galactomannans as a versatile delivery system. Carbohydr. Polym. 2016, 144, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Campia, P.; Ponzini, E.; Rossi, B.; Farris, S.; Silvetti, T.; Merlini, L.; Brasca, M.; Grandori, R.; Galante, Y.M. Aerogels of enzymatically oxidized galactomannans from leguminous plants: Versatile delivery systems of antimicrobial peptides and enzymes. Carbohydr. Polym. 2017, 158, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, B.; Ponzini, E.; Merlini, L.; Grandori, R.; Galante, Y.M. Characterization of aerogels from chemo-enzymatically oxidized galactomannans as novel polymeric biomaterials. Eur. Polym. J. 2017, 93, 347–357. [Google Scholar] [CrossRef]
- Silvetti, T.; Merlini, L.; Brasca, M.; Galante, Y.M. Aerogel from chemo-enzymatically oxidized fenugreek gum: An innovative delivery system of isothiazolinones biocides. Appl. Microbiol. Biotechnol. 2018, 102, 2683–2692. [Google Scholar] [CrossRef]
- Galante, Y.M.; Merlini, L.; Silvetti, T.; Campia, P.; Rossi, B.; Viani, F.; Brasca, M. Enzyme oxidation of plant galactomannans yielding biomaterials with novel properties and applications, including as delivery systems. Appl. Microbiol. Biotechnol. 2018, 102, 4687–4702. [Google Scholar] [CrossRef]
- Barilli, A.; Belinghieri, F.; Passarella, D.; Lesma, G.; Riva, S.; Silvani, A.; Danieli, B. Enzyme assisted enantioselective synthesis of the alkaloid (+)-aloperine. Tetrahedron Asymmetry 2004, 15, 2921–2925. [Google Scholar] [CrossRef]
- García-Junceda, E.; Lavandera, I.; Rother, D.; Schrittwieser, J.H. (Chemo)enzymatic cascades—Nature’s synthetic strategy transferred to the laboratory. J. Mol. Catal. B Enzym. 2015, 114, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Gruber, C.C.; Lavandera, I.; Faber, K.; Kroutil, W. From a Racemate to a Single Enantiomer: Deracemization by Stereoinversion. Adv. Synth. Catal. 2006, 348, 1789–1805. [Google Scholar] [CrossRef]
- Diaz-Rodriguez, A.; Lavandera, I.; Gotor, V. Why Leave a Job Half Done? Recent Progress in Enzymatic Deracemizations. Curr. Green Chem. 2015, 2, 192–211. [Google Scholar] [CrossRef] [Green Version]
- France, S.P.; Hepworth, L.J.; Turner, N.J.; Flitsch, S.L. Constructing Biocatalytic Cascades: In Vitro and in Vivo Approaches to de Novo Multi-Enzyme Pathways. ACS Catal. 2017, 7, 710–724. [Google Scholar] [CrossRef]
- Martínez-Montero, L.; Gotor, V.; Gotor-Fernández, V.; Lavandera, I. Stereoselective amination of racemic sec-alcohols through sequential application of laccases and transaminases. Green Chem. 2017, 19, 474–480. [Google Scholar] [CrossRef] [Green Version]
- González-Granda, S.; Méndez-Sánchez, D.; Lavandera, I.; Gotor-Fernández, V. Laccase-mediated Oxidations of Propargylic Alcohols. Application in the Deracemization of 1-arylprop-2-yn-1-ols in Combination with Alcohol Dehydrogenases. ChemCatChem 2020, 12, 520–527. [Google Scholar] [CrossRef]
- Brenna, E.; Crotti, M.; Gatti, F.G.; Monti, D.; Parmeggiani, F.; Pugliese, A.; Tentori, F. Biocatalytic synthesis of chiral cyclic γ-oxoesters by sequential C-H hydroxylation, alcohol oxidation and alkene reduction. Green Chem. 2017, 19, 5122–5130. [Google Scholar] [CrossRef]
- Martínez-Montero, L.; Gotor, V.; Gotor-Fernández, V.; Lavandera, I. Mild Chemoenzymatic Oxidation of Allylic sec-Alcohols. Application to Biocatalytic Stereoselective Redox Isomerizations. ACS Catal. 2018, 8, 2413–2419. [Google Scholar] [CrossRef]
- Brenna, E.; Crotti, M.; De Pieri, M.; Gatti, F.G.; Manenti, G.; Monti, D. Chemo-Enzymatic Oxidative Rearrangement of Tertiary Allylic Alcohols: Synthetic Application and Integration into a Cascade Process. Adv. Synth. Catal. 2018, 360, 3677–3686. [Google Scholar] [CrossRef]
- Asta, C.; Schmidt, D.; Conrad, J.; Förster-Fromme, B.; Tolasch, T.; Beifuss, U. The first enzymatic Achmatowicz reaction: Selective laccase-catalyzed synthesis of 6-hydroxy-(2H)-pyran-3(6H)-ones and (2H)-pyran-2,5(6H)-diones. RSC Adv. 2013, 3, 19259. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, J.; Zimmerman, S.C. Designed transition metal catalysts for intracellular organic synthesis. Chem. Soc. Rev. 2018, 47, 1811–1821. [Google Scholar] [CrossRef]
- Dursch, T.J. Transition-Metal Complexes: Simple(r) Solutions to Complex Chemistry. Trends Chem. 2019, 1, 455–456. [Google Scholar] [CrossRef]
- Cheng, W.-M.; Shang, R. Transition Metal-Catalyzed Organic Reactions under Visible Light: Recent Developments and Future Perspectives. ACS Catal. 2020, 10, 9170–9196. [Google Scholar] [CrossRef]
- Nájera, C.; Beletskaya, I.P.; Yus, M. Metal-catalyzed regiodivergent organic reactions. Chem. Soc. Rev. 2019, 48, 4515–4618. [Google Scholar] [CrossRef]
- Zhou, Q.-L. Transition-Metal Catalysis and Organocatalysis: Where Can Progress Be Expected? Angew. Chemie Int. Ed. 2016, 55, 5352–5353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciecholewski, S.; Hammer, E.; Manda, K.; Bose, G.; Nguyen, V.T.H.; Langer, P.; Schauer, F. Laccase-catalyzed carbon–carbon bond formation: Oxidative dimerization of salicylic esters by air in aqueous solution. Tetrahedron 2005, 61, 4615–4619. [Google Scholar] [CrossRef]
- Constantin, M.-A.; Conrad, J.; Beifuss, U. An unprecedented oxidative trimerization of sesamol catalyzed by laccases. Tetrahedron Lett. 2012, 53, 3254–3258. [Google Scholar] [CrossRef]
- Abdel-Mohsen, H.T.; Sudheendran, K.; Conrad, J.; Beifuss, U. Synthesis of disulfides by laccase-catalyzed oxidative coupling of heterocyclic thiols. Green Chem. 2013, 15, 1490. [Google Scholar] [CrossRef]
- Cannatelli, M.D.; Ragauskas, A.J. Laccase-catalyzed α-arylation of benzoylacetonitrile with substituted hydroquinones. Chem. Eng. Res. Des. 2015, 97, 128–134. [Google Scholar] [CrossRef]
- Niedermeyer, T.H.J.; Mikolasch, A.; Lalk, M. Nuclear Amination Catalyzed by Fungal Laccases: Reaction Products of p -Hydroquinones and Primary Aromatic Amines. J. Org. Chem. 2005, 70, 2002–2008. [Google Scholar] [CrossRef] [PubMed]
- Mikolasch, A.; Niedermeyer, T.H.J.; Lalk, M.; Witt, S.; Seefeldt, S.; Hammer, E.; Schauer, F.; Gesell, M.; Hessel, S.; Jülich, W.-D.; et al. Novel Penicillins Synthesized by Biotransformation Using Laccase from Trametes spec. Chem. Pharm. Bull. (Tokyo) 2006, 54, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Mikolasch, A.; Hessel, S.; Salazar, M.G.; Neumann, H.; Manda, K.; Gōrdes, D.; Schmidt, E.; Thurow, K.; Hammer, E.; Lindequist, U.; et al. Synthesis of New N-Analogous Corollosporine Derivatives with Antibacterial Activity by Laccase-Catalyzed Amination. Chem. Pharm. Bull. (Tokyo) 2008, 56, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Mikolasch, A.; Hildebrandt, O.; Schlüter, R.; Hammer, E.; Witt, S.; Lindequist, U. Targeted synthesis of novel β-lactam antibiotics by laccase-catalyzed reaction of aromatic substrates selected by pre-testing for their antimicrobial and cytotoxic activity. Appl. Microbiol. Biotechnol. 2016, 100, 4885–4899. [Google Scholar] [CrossRef]
- Hahn, V.; Mikolasch, A.; Wende, K.; Bartrow, H.; Lindequist, U.; Schauer, F. Synthesis of model morpholine derivatives with biological activities by laccase-catalysed reactions. Biotechnol. Appl. Biochem. 2009, 54, 187–195. [Google Scholar] [CrossRef]
- Hahn, V.; Mikolasch, A.; Schauer, F. Cleavage and synthesis function of high and low redox potential laccases towards 4-morpholinoaniline and aminated as well as chlorinated phenols. Appl. Microbiol. Biotechnol. 2014, 98, 1609–1620. [Google Scholar] [CrossRef]
- Wellington, K.W.; Bokako, R.; Raseroka, N.; Steenkamp, P. A one-pot synthesis of 1,4-naphthoquinone-2,3-bis-sulfides catalysed by a commercial laccase. Green Chem. 2012, 14, 2567. [Google Scholar] [CrossRef]
- Bringmann, G.; Walter, R.; Weirich, R. The Directed Synthesis of Biaryl Compounds: Modern Concepts and Strategies. Angew. Chemie Int. Ed. English 1990, 29, 977–991. [Google Scholar] [CrossRef]
- Wencel-Delord, J.; Panossian, A.; Leroux, F.R.; Colobert, F. Recent advances and new concepts for the synthesis of axially stereoenriched biaryls. Chem. Soc. Rev. 2015, 44, 3418–3430. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Chang, J.; Yu, B. Construction of Biologically Important Biaryl Scaffolds through Direct C–H Bond Activation: Advances and Prospects. Top. Curr. Chem. 2020, 378, 23. [Google Scholar] [CrossRef]
- Emirdağ-Öztürk, S.; Hajdok, S.; Conrad, J.; Beifuss, U. Laccase-catalyzed reaction of 3-tert-butyl-1H-pyrazol-5(4H)-one with substituted catechols using air as an oxidant. Tetrahedron 2013, 69, 3664–3668. [Google Scholar] [CrossRef]
- Ncanana, S.; Baratto, L.; Roncaglia, L.; Riva, S.; Burton, S.G. Laccase-mediated oxidation of totarol. Adv. Synth. Catal. 2007, 349, 1507–1513. [Google Scholar] [CrossRef]
- Sagui, F.; Chirivì, C.; Fontana, G.; Nicotra, S.; Passarella, D.; Riva, S.; Danieli, B. Laccase-catalyzed coupling of catharanthine and vindoline: An efficient approach to the bisindole alkaloid anhydrovinblastine. Tetrahedron 2009, 65, 312–317. [Google Scholar] [CrossRef]
- Hahn, V.; Mikolasch, A.; Kuhlisch, C.; Schauer, F. Laccase-mediated multi-step homo- and heteromolecular reactions of ortho -dihydroxylated aromatic compounds and mono- or diaminated substances resulting in C C, C O and C N bonds. J. Mol. Catal. B Enzym. 2015, 122, 56–63. [Google Scholar] [CrossRef]
- Mikolasch, A.; Manda, K.; Schlüter, R.; Lalk, M.; Witt, S.; Seefeldt, S.; Hammer, E.; Schauer, F.; Jülich, W.-D.; Lindequist, U. Comparative analyses of laccase-catalyzed amination reactions for production of novel β-lactam antibiotics. Biotechnol. Appl. Biochem. 2012, 59, 295–306. [Google Scholar] [CrossRef]
- Hahn, V.; Davids, T.; Lalk, M.; Schauer, F.; Mikolasch, A. Enzymatic cyclizations using laccases: Multiple bond formation between dihydroxybenzoic acid derivatives and aromatic amines. Green Chem. 2010, 12, 879. [Google Scholar] [CrossRef]
- Osiadacz, J.; Al-Adhami, A.J.; Bajraszewska, D.; Fischer, P.; Peczyñska-Czoch, W. On the use of Trametes versicolor laccase for the conversion of 4-methyl-3-hydroxyanthranilic acid to actinocin chromophore. J. Biotechnol. 1999, 72, 141–149. [Google Scholar] [CrossRef]
- Sousa, A.C.; Oliveira, M.C.; Martins, L.O.; Robalo, M.P. Towards the rational biosynthesis of substituted phenazines and phenoxazinones by laccases. Green Chem. 2014, 16, 4127–4136. [Google Scholar] [CrossRef] [Green Version]
- Sousa, A.C.; Piedade, M.F.M.M.; Martins, L.O.; Robalo, M.P. An enzymatic route to a benzocarbazole framework using bacterial CotA laccase. Green Chem. 2015, 17, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Sousa, A.C.; Conceição Oliveira, M.; Martins, L.O.; Robalo, M.P. A Sustainable Synthesis of Asymmetric Phenazines and Phenoxazinones Mediated by CotA-Laccase. Adv. Synth. Catal. 2018, 360, 575–583. [Google Scholar] [CrossRef]
- Constantin, M.-A.; Conrad, J.; Beifuss, U. Laccase-catalyzed oxidative phenolic coupling of vanillidene derivatives. Green Chem. 2012, 14, 2375. [Google Scholar] [CrossRef]
- Ricklefs, E.; Girhard, M.; Urlacher, V.B. Three-steps in one-pot: Whole-cell biocatalytic synthesis of enantiopure (+)- and (−)-pinoresinol via kinetic resolution. Microb. Cell Fact. 2016, 15, 78. [Google Scholar] [CrossRef] [Green Version]
- Navarra, C.; Goodwin, C.; Burton, S.; Danieli, B.; Riva, S. Laccase-mediated oxidation of phenolic derivatives. J. Mol. Catal. B Enzym. 2010, 65, 52–57. [Google Scholar] [CrossRef]
- Ricklefs, E.; Girhard, M.; Koschorreck, K.; Smit, M.S.; Urlacher, V.B. Two-Step One-Pot Synthesis of Pinoresinol from Eugenol in an Enzymatic Cascade. ChemCatChem 2015, 7, 1857–1864. [Google Scholar] [CrossRef]
- Beneventi, E.; Conte, S.; Cramarossa, M.R.; Riva, S.; Forti, L. Chemo-enzymatic synthesis of new resveratrol-related dimers containing the benzo[b]furan framework and evaluation of their radical scavenger activities. Tetrahedron 2015, 71, 3052–3058. [Google Scholar] [CrossRef] [Green Version]
- Gavezzotti, P.; Bertacchi, F.; Fronza, G.; Křen, V.; Monti, D.; Riva, S. Laccase-Catalyzed Dimerization of Piceid, a Resveratrol Glucoside, and its Further Enzymatic Elaboration. Adv. Synth. Catal. 2015, 357, 1831–1839. [Google Scholar] [CrossRef]
- Witayakran, S.; Gelbaum, L.; Ragauskas, A.J. Cascade synthesis of benzofuran derivatives via laccase oxidation-Michael addition. Tetrahedron 2007, 63, 10958–10962. [Google Scholar] [CrossRef]
- Bassanini, I.; Gavezzotti, P.; Monti, D.; Krejzová, J.; Křen, V.; Riva, S. Laccase-catalyzed dimerization of glycosylated lignols. J. Mol. Catal. B Enzym. 2016, 134, 295–301. [Google Scholar] [CrossRef]
- Bassanini, I.; D’Annessa, I.; Costa, M.; Monti, D.; Colombo, G.; Riva, S. Chemo-enzymatic synthesis of (: E)-2,3-diaryl-5-styryl- trans -2,3-dihydrobenzofuran-based scaffolds and their in vitro and in silico evaluation as a novel sub-family of potential allosteric modulators of the 90 kDa heat shock protein (Hsp90). Org. Biomol. Chem. 2018, 16. [Google Scholar] [CrossRef]
- Grosso, S.; Radaelli, F.; Fronza, G.; Passarella, D.; Monti, D.; Riva, S. Studies on the Laccase-Catalyzed Oxidation of 4-Hydroxy-Chalcones. Adv. Synth. Catal. 2019, 361, adsc.201900190. [Google Scholar] [CrossRef] [Green Version]
- Davin, L.B.; Wang, H.-B.; Crowell, A.L.; Bedgar, D.L.; Martin, D.M.; Sarkanen, S.; Lewis, N.G. Stereoselective Bimolecular Phenoxy Radical Coupling by an Auxiliary (Dirigent) Protein Without an Active Center. Science 1997, 275, 362–367. [Google Scholar] [CrossRef]
- Pickel, B.; Constantin, M.-A.; Pfannstiel, J.; Conrad, J.; Beifuss, U.; Schaller, A. An Enantiocomplementary Dirigent Protein for the Enantioselective Laccase-Catalyzed Oxidative Coupling of Phenols. Angew. Chemie Int. Ed. 2010, 49, 202–204. [Google Scholar] [CrossRef]
- Chen, Z.; Pitchakuntla, M.; Jia, Y. Synthetic approaches to natural products containing 2,3-dihydrobenzofuran skeleton. Nat. Prod. Rep. 2019, 36, 666–690. [Google Scholar] [CrossRef]
- Gavezzotti, P.; Navarra, C.; Caufin, S.; Danieli, B.; Magrone, P.; Monti, D.; Riva, S. Synthesis of enantiomerically enriched dimers of vinylphenols by tandem action of laccases and lipases. Adv. Synth. Catal. 2011. [Google Scholar] [CrossRef]
- Navarra, C.; Gavezzotti, P.; Monti, D.; Panzeri, W.; Riva, S. Biocatalyzed synthesis of enantiomerically enriched β-5-like dimer of 4-vinylphenol. J. Mol. Catal. B Enzym. 2012, 84, 115–120. [Google Scholar] [CrossRef]
- Ficarra, S.; Tellone, E.; Pirolli, D.; Russo, A.; Barreca, D.; Galtieri, A.; Giardina, B.; Gavezzotti, P.; Riva, S.; De Rosa, M.C. Insights into the properties of the two enantiomers of trans-δ-viniferin, a resveratrol derivative: Antioxidant activity, biochemical and molecular modeling studies of its interactions with hemoglobin. Mol. Biosyst. 2016, 12, 1276–1286. [Google Scholar] [CrossRef]
- Cannatelli, M.D.; Ragauskas, A.J. Laccase-catalyzed synthesis of 2,3-ethylenedithio-1,4-quinones. J. Mol. Catal. B Enzym. 2015, 119, 85–89. [Google Scholar] [CrossRef]
- Leutbecher, H.; Conrad, J.; Klaiber, I.; Beifuss, U. O-Heterocycles via Laccase-Catalyzed Domino Reactions with O2 as the Oxidant. Synlett 2005, 3126–3130. [Google Scholar] [CrossRef]
- Hajdok, S.; Leutbecher, H.; Greiner, G.; Conrad, J.; Beifuss, U. Laccase initiated oxidative domino reactions for the efficient synthesis of 3,4-dihydro-7,8-dihydroxy-2H-dibenzofuran-1-ones. Tetrahedron Lett. 2007, 48, 5073–5076. [Google Scholar] [CrossRef]
- Leutbecher, H.; Hajdok, S.; Braunberger, C.; Neumann, M.; Mika, S.; Conrad, J.; Beifuss, U. Combined action of enzymes: The first domino reaction catalyzed by Agaricus bisporus. Green Chem. 2009, 11, 676. [Google Scholar] [CrossRef]
- Wellington, K.W.; Qwebani-Ogunleye, T.; Kolesnikova, N.I.; Brady, D.; de Koning, C.B. One-Pot Laccase-Catalysed Synthesis of 5,6-Dihydroxylated Benzo[ b ]furans and Catechol Derivatives, and Their Anticancer Activity. Arch. Pharm. (Weinheim) 2013, 346, 266–277. [Google Scholar] [CrossRef]
- Witayakran, S.; Ragauskas, A.J. One-pot synthesis of 1,4-naphthoquinones and related structures with laccase. Green Chem. 2007, 9, 475. [Google Scholar] [CrossRef]
- Witayakran, S.; Zettili, A.; Ragauskas, A.J. Laccase-generated quinones in naphthoquinone synthesis via Diels-Alder reaction. Tetrahedron Lett. 2007, 48, 2983–2987. [Google Scholar] [CrossRef]
- Fritz-Langhals, E.; Kunath, B. Synthesis of aromatic aldehydes by laccase-mediator assisted oxidation. Tetrahedron Lett. 1998, 39, 5955–5956. [Google Scholar] [CrossRef]
- Niku-Paavola, M.-L.; Viikari, L. Enzymatic oxidation of alkenes. J. Mol. Catal. B Enzym. 2000, 10, 435–444. [Google Scholar] [CrossRef]
- Semenov, A.N.; Lomonsova, I.V.; Berezin, V.I.; Titov, M.I. Peroxidase and laccase as catalysts for removal of the phenylhydrazide protecting group under mild conditions. Biotechnol. Bioeng. 1993, 42, 1137–1141. [Google Scholar] [CrossRef]
- Chirivì, C.; Fontana, G.; Monti, D.; Ottolina, G.; Riva, S.; Danieli, B. The quest for new mild and selective modifications of natural structures: Laccase-catalysed oxidation of ergot alkaloids leads to unexpected stereoselective C-4 hydroxylation. Chem. A Eur. J. 2012, 18, 10355–10361. [Google Scholar] [CrossRef]
- Suljić, S.; Pietruszka, J.; Worgull, D. Asymmetric Bio- and Organocatalytic Cascade Reaction—Laccase and Secondary Amine-Catalyzed α-Arylation of Aldehydes. Adv. Synth. Catal. 2015, 1822–1830. [Google Scholar] [CrossRef]
- Gaunt, M.J.; Johansson, C.C.C.; McNally, A.; Vo, N.T. Enantioselective organocatalysis. Drug Discov. Today 2007, 12, 8–27. [Google Scholar] [CrossRef]
- List, B. Proline-catalyzed asymmetric reactions. Tetrahedron 2002, 58, 5573–5590. [Google Scholar] [CrossRef]
Scheme 1.
Schematic representation of the active site of the laccase from Trametes versicolor and of the reactions catalyzed in a redox cycle [1].
Scheme 1.
Schematic representation of the active site of the laccase from Trametes versicolor and of the reactions catalyzed in a redox cycle [1].

Scheme 2.
Laccase-catalyzed redox cycles for substrate oxidation: (a) direct substrate oxidation; (b) substrate oxidation in the presence of a chemical mediator; (c) cofactor regeneration in multienzymatic coupled redox biotransformations [1].
Scheme 2.
Laccase-catalyzed redox cycles for substrate oxidation: (a) direct substrate oxidation; (b) substrate oxidation in the presence of a chemical mediator; (c) cofactor regeneration in multienzymatic coupled redox biotransformations [1].

Figure 1.
Examples of chemical mediators employed in laccase-mediator system (LMS). (a) 3-Hydroxyanthranilic acid (HAA); (b) 2,20-azino-bis-(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS); (c) 2,2,6,6-tetramethyl piperidine-1-yloxy (TEMPO); (d) N-hydroxybenzo-triazole (HBT); (e) violuric acid (VLA); (f) methyl ester of 4-hydroxy-3,5-dimethoxy-benzoic acid (syringic acid).
Figure 1.
Examples of chemical mediators employed in laccase-mediator system (LMS). (a) 3-Hydroxyanthranilic acid (HAA); (b) 2,20-azino-bis-(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS); (c) 2,2,6,6-tetramethyl piperidine-1-yloxy (TEMPO); (d) N-hydroxybenzo-triazole (HBT); (e) violuric acid (VLA); (f) methyl ester of 4-hydroxy-3,5-dimethoxy-benzoic acid (syringic acid).

Scheme 3.
Exploitation of laccase-mediator systems for the regioselective oxidation of primary hydroxyl groups. (a) Oxidation of glucosides to the corresponding glucuronic acids; (b) oxidation of the bioactive glucoside thiocolchicoside [48].
Scheme 3.
Exploitation of laccase-mediator systems for the regioselective oxidation of primary hydroxyl groups. (a) Oxidation of glucosides to the corresponding glucuronic acids; (b) oxidation of the bioactive glucoside thiocolchicoside [48].

Scheme 4.
Exploitation of LMS in the synthesis of the alkaloid (+)-aloperine [68].
Scheme 4.
Exploitation of LMS in the synthesis of the alkaloid (+)-aloperine [68].

Scheme 5.
Applications of LMS in multienzymatic and cascade processes aimed at the preparation of enantioenriched amines (route (a)) or alcohols (route (b)) [73,74].

Scheme 6.
Multienzymatic synthesis of chiral cyclic γ-oxoesters by (i) allylic hydroxylation catalyzed by Rhizopus oryzae whole cells, (ii) oxidation of the allylic alcohols by LMS, and (iii) alkenes reduction by ene reductases [75].
Scheme 6.
Multienzymatic synthesis of chiral cyclic γ-oxoesters by (i) allylic hydroxylation catalyzed by Rhizopus oryzae whole cells, (ii) oxidation of the allylic alcohols by LMS, and (iii) alkenes reduction by ene reductases [75].

Scheme 7.
Use of LMS in the multienzymatic synthesis of enantiomerically enriched saturated ketones from the corresponding racemic allylic sec-alcohols [76].
Scheme 7.
Use of LMS in the multienzymatic synthesis of enantiomerically enriched saturated ketones from the corresponding racemic allylic sec-alcohols [76].

Scheme 8.
Chemoenzymatic preparation of enantio-enriched cis or trans 3-methylcyclohexan-1-ols from a starting tertiary allylic alcohol: (i) [1,3]-oxidative rearrangement by a laccase/TEMPO+BF4- system; (ii) stereoselective reduction of the activated ketone by an ene reductase; (iii) ADH-catalyzed stereoselective carbonyl reduction. Alternative cascade processes were developed by coupling the ene reductase with either a pro(R)-ADH (iv) or a pro(S)-ADH (v) [77].
Scheme 8.
Chemoenzymatic preparation of enantio-enriched cis or trans 3-methylcyclohexan-1-ols from a starting tertiary allylic alcohol: (i) [1,3]-oxidative rearrangement by a laccase/TEMPO+BF4- system; (ii) stereoselective reduction of the activated ketone by an ene reductase; (iii) ADH-catalyzed stereoselective carbonyl reduction. Alternative cascade processes were developed by coupling the ene reductase with either a pro(R)-ADH (iv) or a pro(S)-ADH (v) [77].

Scheme 9.
(a) The Achmatowicz reaction; (b) LMS-catalyzed oxidation of furan-2-yl carbinols [78].
Scheme 9.
(a) The Achmatowicz reaction; (b) LMS-catalyzed oxidation of furan-2-yl carbinols [78].

Scheme 10.
Schematic representation of the possible destinies of laccase-formed organic radicals.

Scheme 11.
Csp2-H radical activation of fully conjugated aromatic phenols and anilines.

Scheme 12.
Overview of the oxidative homocoupling of salicylic esters conducted by Ciecholewski [84].
Scheme 12.
Overview of the oxidative homocoupling of salicylic esters conducted by Ciecholewski [84].

Scheme 13.
The 3-tert-butyl-1H-pyrazol-5(4H)-one containing biaryl compounds obtained by laccase-catalyzed oxidation of catechols [98].
Scheme 13.
The 3-tert-butyl-1H-pyrazol-5(4H)-one containing biaryl compounds obtained by laccase-catalyzed oxidation of catechols [98].

Scheme 14.
C-O and C-C dimers isolated from laccase-catalyzed oxidation of totarol [99].
Scheme 14.
C-O and C-C dimers isolated from laccase-catalyzed oxidation of totarol [99].

Scheme 15.
Formation of a trimeric C-C adduct from sesamol in the presence of laccase [85].
Scheme 15.
Formation of a trimeric C-C adduct from sesamol in the presence of laccase [85].

Scheme 16.
Laccase-based chemoenzymatic entry to anhydrovinblastine (AVBL) from catharanthine and vindoline [100].
Scheme 16.
Laccase-based chemoenzymatic entry to anhydrovinblastine (AVBL) from catharanthine and vindoline [100].

Scheme 17.
Operating mechanism for the laccase-mediated aromatic nucleophilic substitution reactions.
Scheme 17.
Operating mechanism for the laccase-mediated aromatic nucleophilic substitution reactions.

Scheme 18.
Representative examples of laccase-mediated nuclear amination with anilines [101].
Scheme 18.
Representative examples of laccase-mediated nuclear amination with anilines [101].


Scheme 20.
Laccase-catalyzed synthesis of 2,3-bis-sulfides derivatives [94].
Scheme 20.
Laccase-catalyzed synthesis of 2,3-bis-sulfides derivatives [94].

Scheme 21.
Operating mechanism for the laccase-mediated preparation of 2,3- bis-sulfides 1,4-dihydroxy-2-naphthoic acid sulphurated derivatives [94].
Scheme 21.
Operating mechanism for the laccase-mediated preparation of 2,3- bis-sulfides 1,4-dihydroxy-2-naphthoic acid sulphurated derivatives [94].

Scheme 22.
Laccase-based entry to benzylic nitriles from 1,4-hydroxyquinones [87].
Scheme 22.
Laccase-based entry to benzylic nitriles from 1,4-hydroxyquinones [87].

Scheme 23.
Laccase-catalyzed oxidation of 3-hydroxyanthranilic acid (3-HAA) to the corresponding phenoxazinone [55].
Scheme 23.
Laccase-catalyzed oxidation of 3-hydroxyanthranilic acid (3-HAA) to the corresponding phenoxazinone [55].

Scheme 24.
Laccase-catalyzed synthesis of actinocin by oxidative homocoupling [104].
Scheme 24.
Laccase-catalyzed synthesis of actinocin by oxidative homocoupling [104].

Scheme 25.
Substituted phenazines, phenoxazinones (a) obtained from laccase from Bacillus subtilis (spore coat protein A, CotA). Mechanistic insights on phenazines (b) and benzocarbazoles (c) formation catalyzed by laccases CotA. [105,106,107].

Scheme 26.
Oxygenated heterocyclic products that can be obtained via laccase-mediated homocouplings.
Scheme 26.
Oxygenated heterocyclic products that can be obtained via laccase-mediated homocouplings.

Scheme 27.
Domino process to 2,3-DHBs via formal oxidative homocoupling.

Scheme 28.
Laccase-catalyzed dimerization of 4-hydroxy-chalcones.

Scheme 29.
Laccase-catalyzed oxidation of tyrosol [110].
Scheme 29.
Laccase-catalyzed oxidation of tyrosol [110].

Scheme 30.
Laccase-mediated cyclization reaction in the presence of 1,2-ethanedithiol [124].
Scheme 30.
Laccase-mediated cyclization reaction in the presence of 1,2-ethanedithiol [124].


Scheme 32.
Aromatic 1,3-dicarbonyls as carbon nucleophiles toward laccase-activated 1,2-dihydroquinones [125,126,127].


Scheme 33.
Biocatalytic entry to pyrimidinyl benzothiazoles [28].
Scheme 33.
Biocatalytic entry to pyrimidinyl benzothiazoles [28].

Scheme 34.
Laccase-initiated domino process to access pyrimidinyl benzothiazoles [28].
Scheme 34.
Laccase-initiated domino process to access pyrimidinyl benzothiazoles [28].

Scheme 35.
Overview of the obtained cyclic products from laccase-initiated Dies-Alder cycloadditions.
Scheme 35.
Overview of the obtained cyclic products from laccase-initiated Dies-Alder cycloadditions.

Scheme 36.
Example of the laccase-mediated stereoselective hydroxylation of an Ergot alkaloid [134].
Scheme 36.
Example of the laccase-mediated stereoselective hydroxylation of an Ergot alkaloid [134].

Scheme 37.
Laccase-initiated organocatalytic synthesis of enantioenriched 2,3-DHB derivatives.

Scheme 38.
Catalytic cycle for the laccase-initiated asymmetric 2,3-DHB formation.


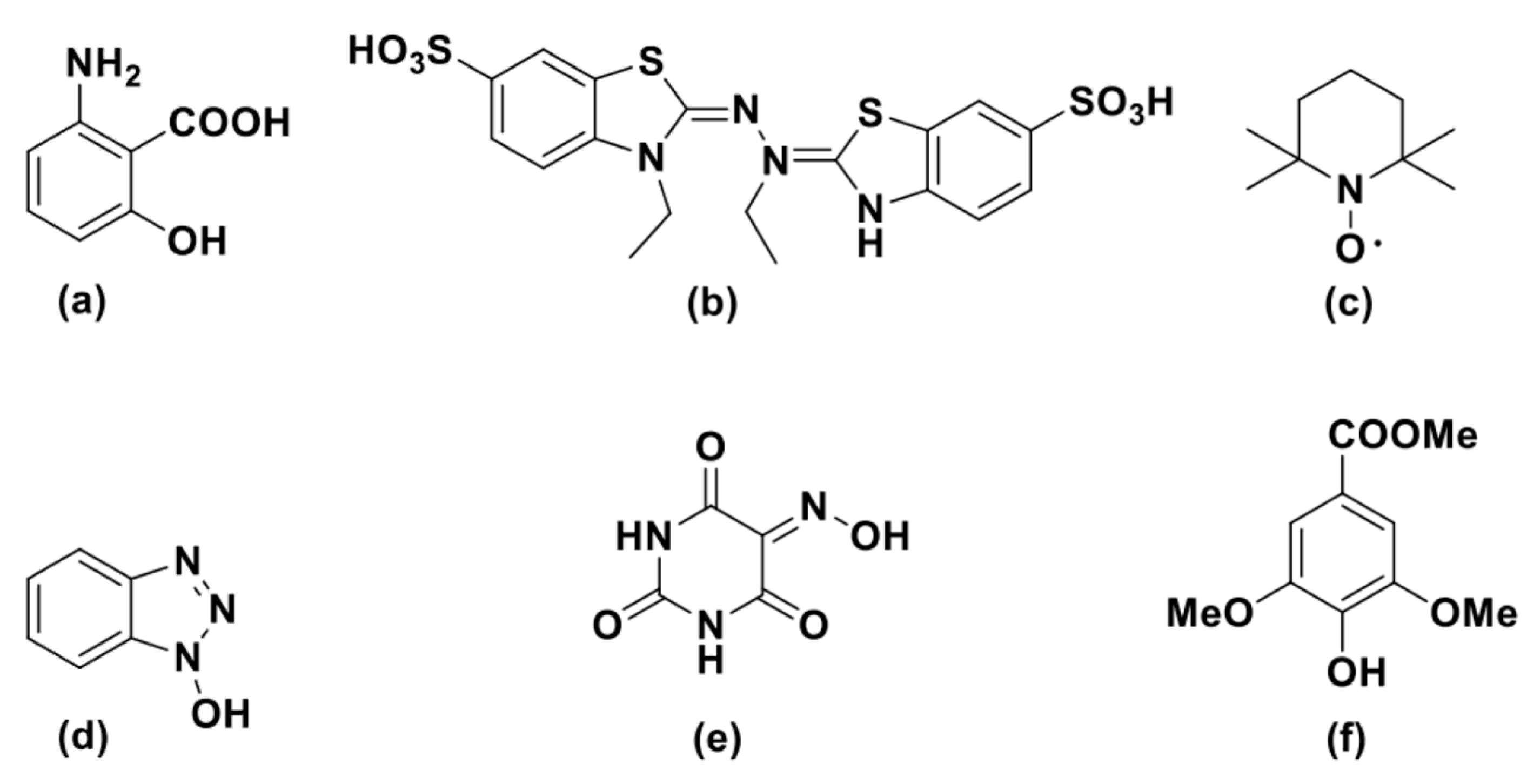{kind=link}
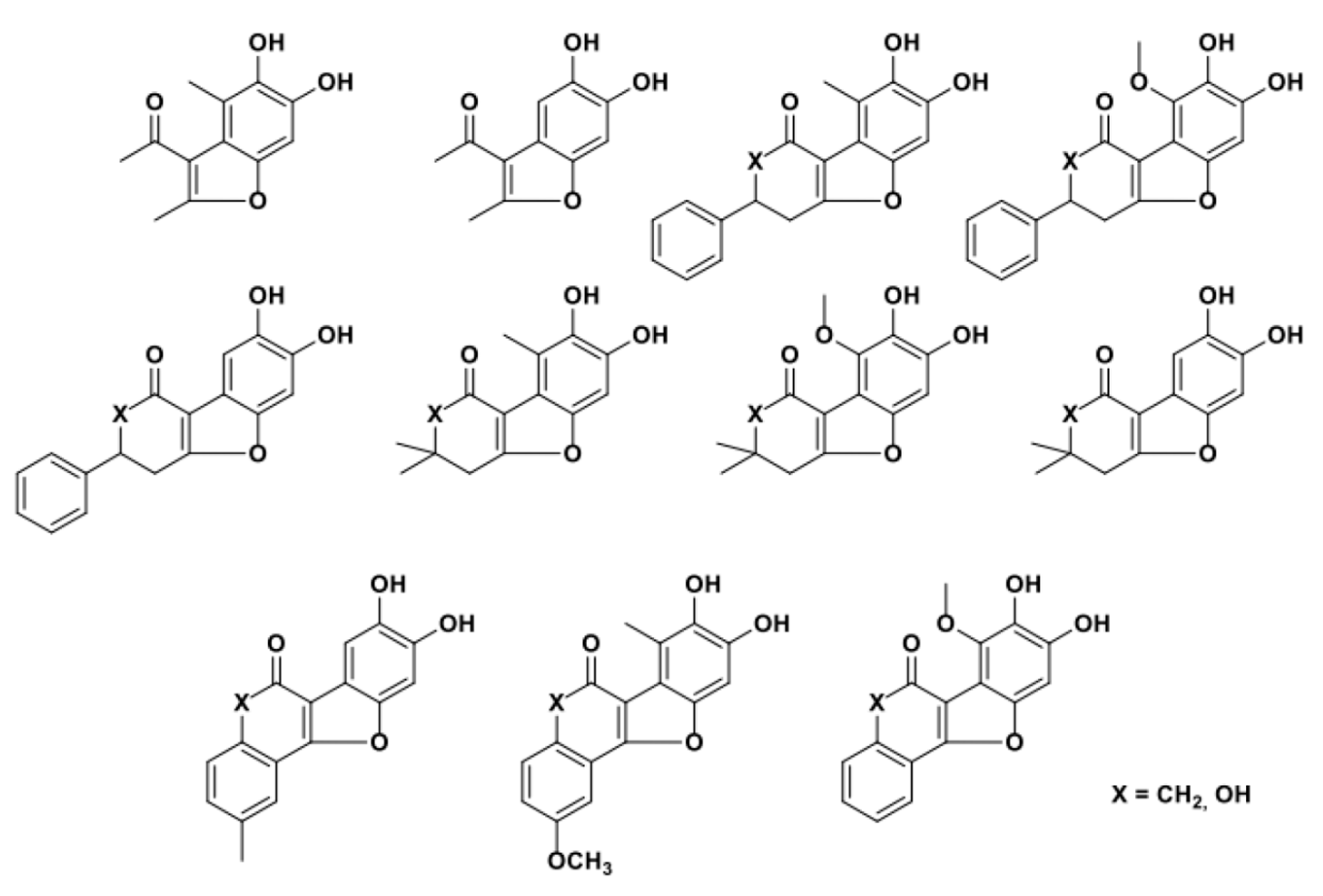{kind=link}
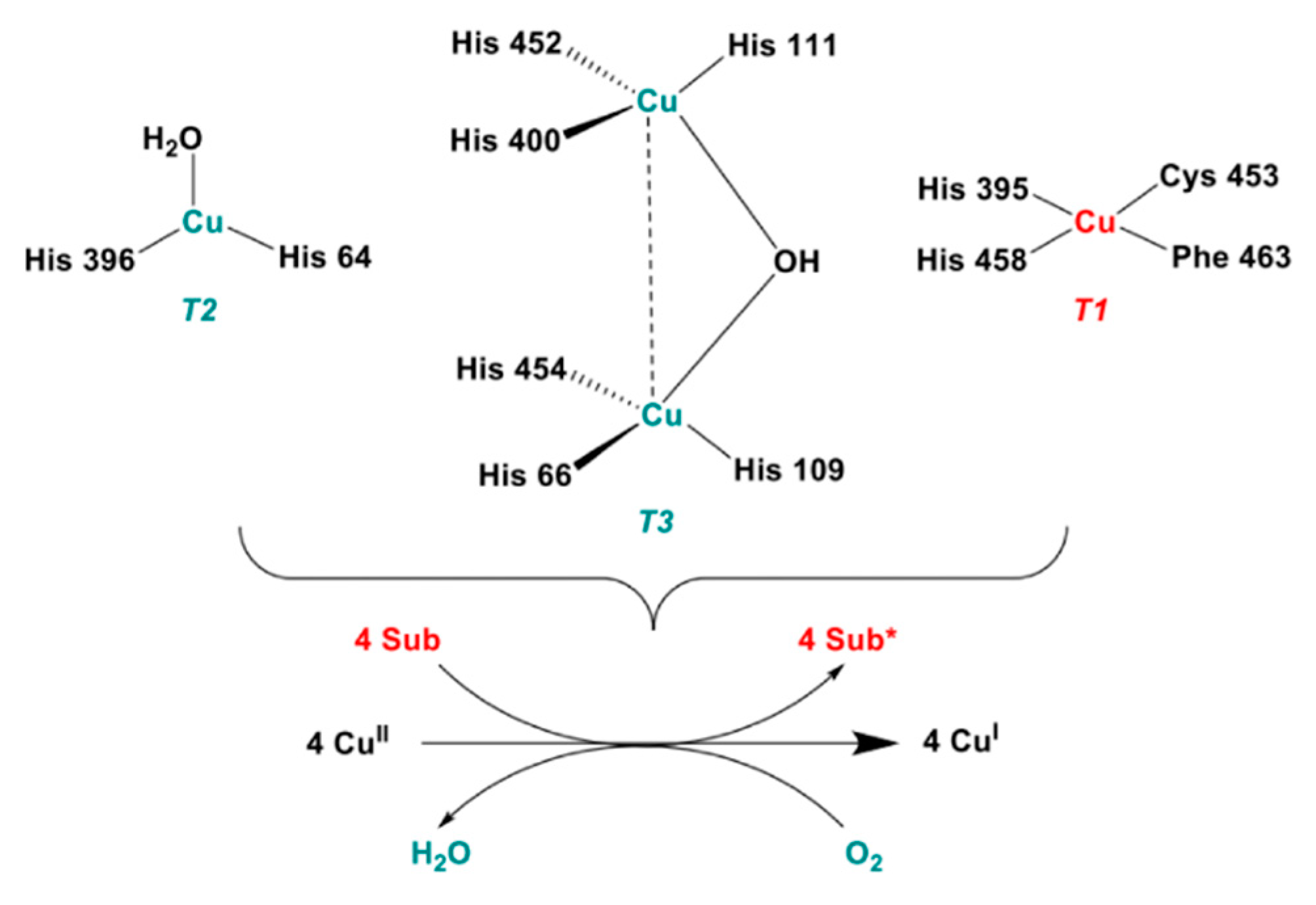{kind=link}
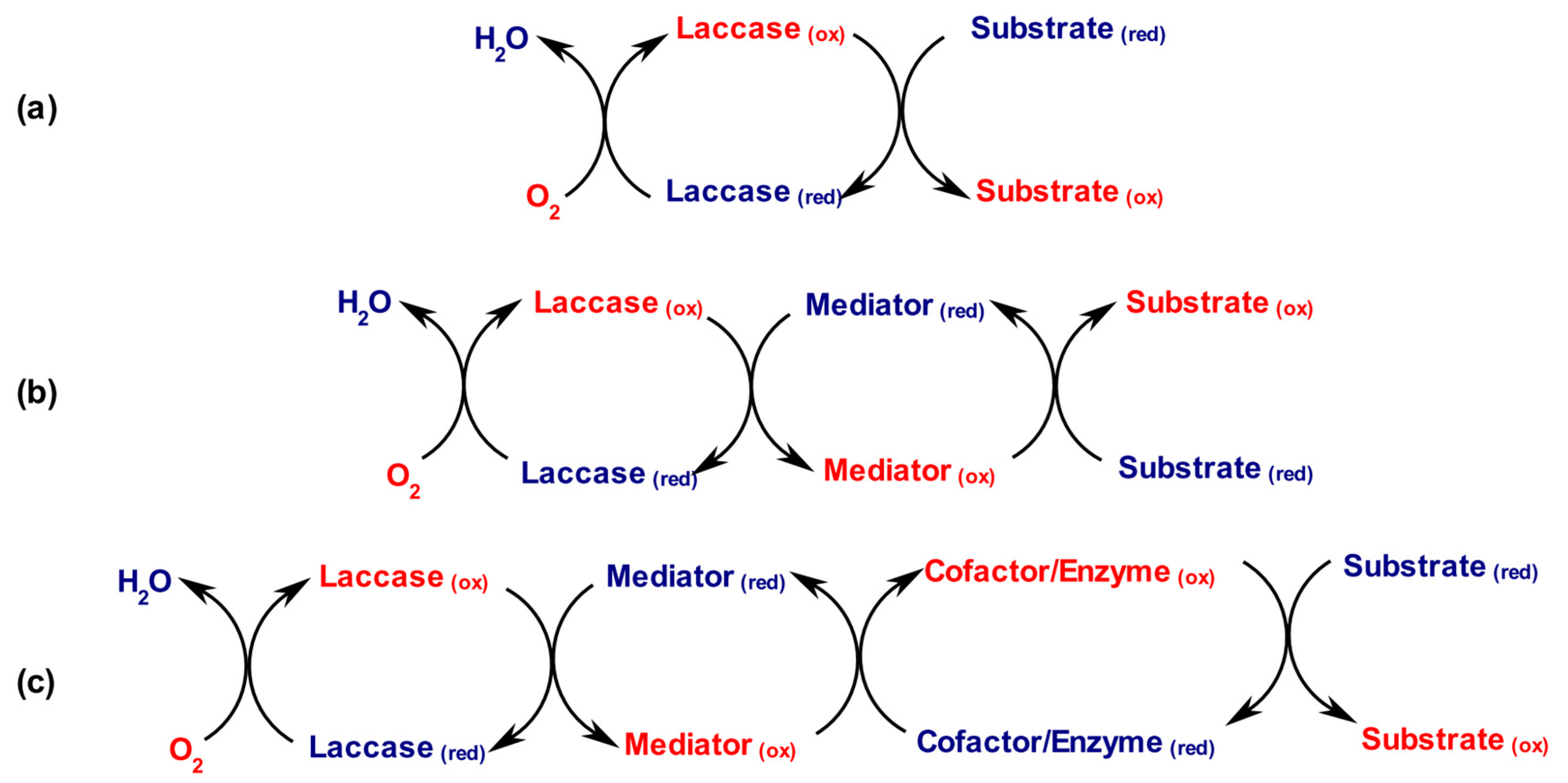{kind=link}
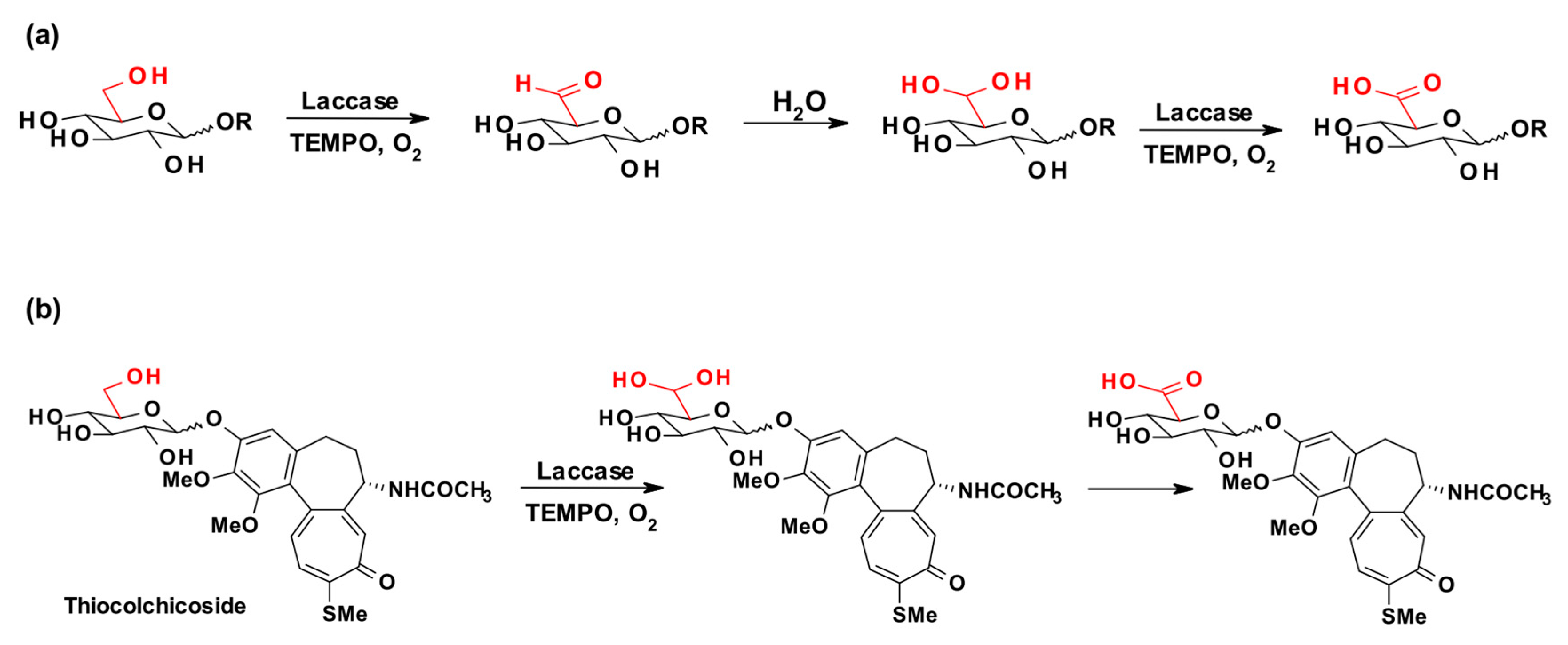{kind=link}
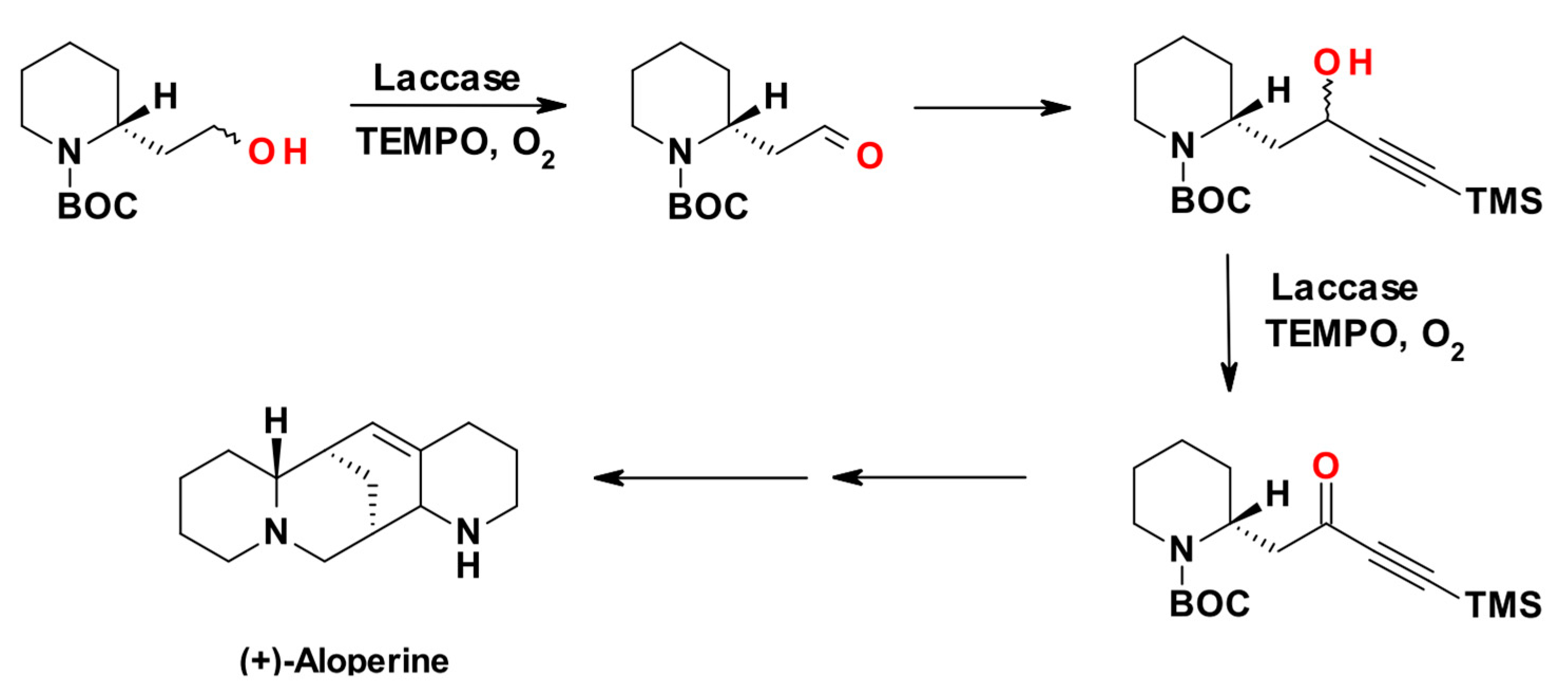{kind=link}
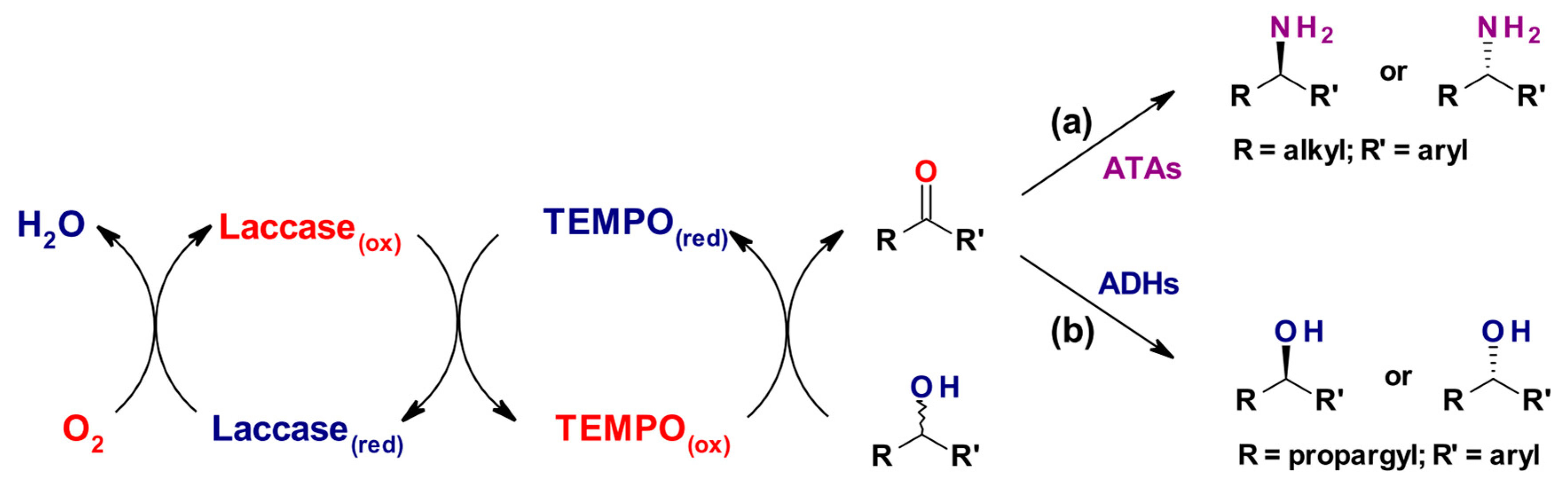{kind=link}
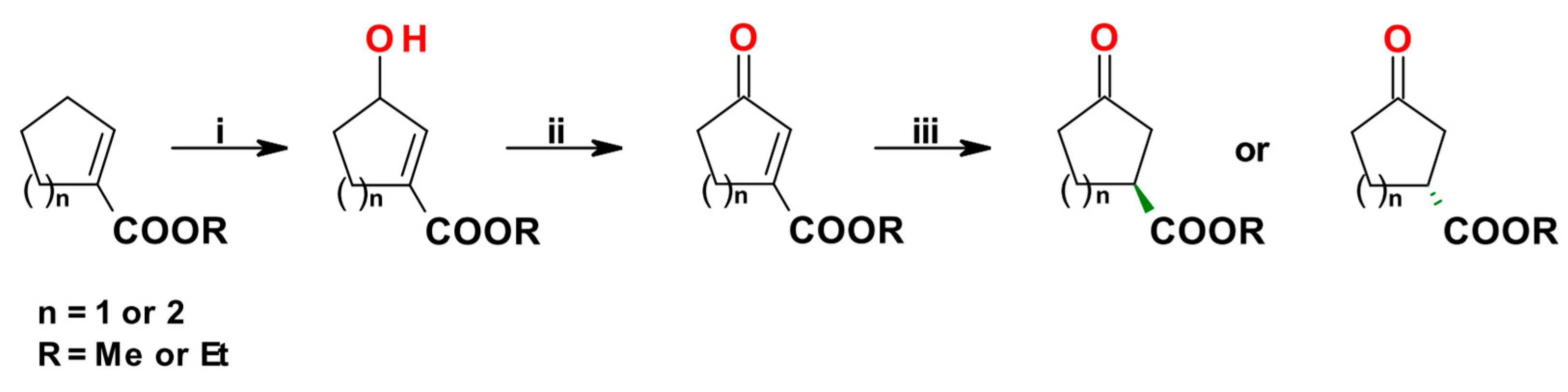{kind=link}
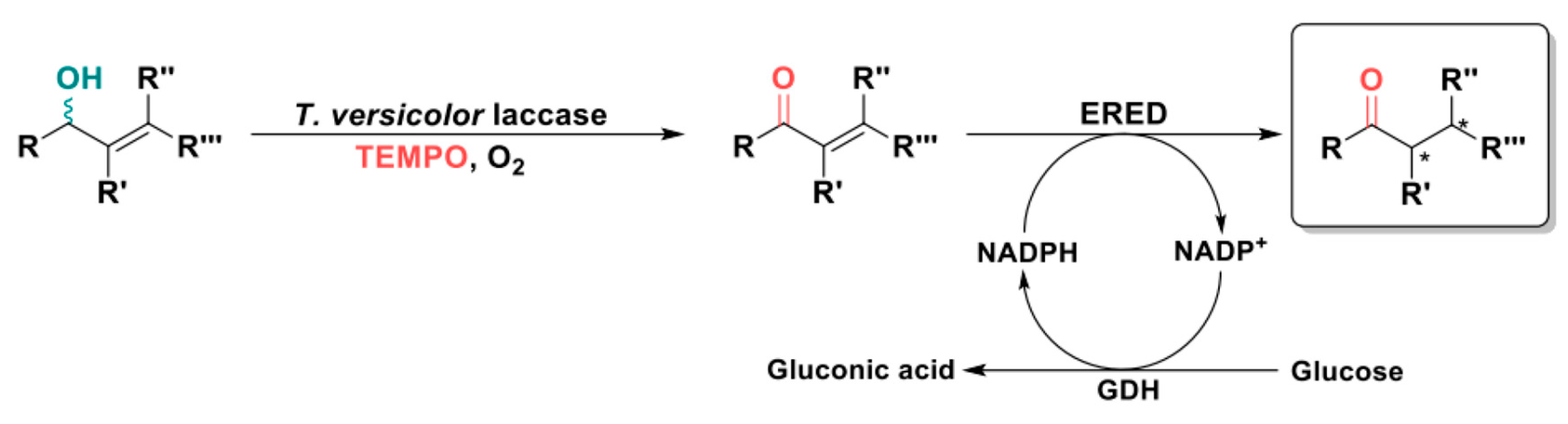{kind=link}
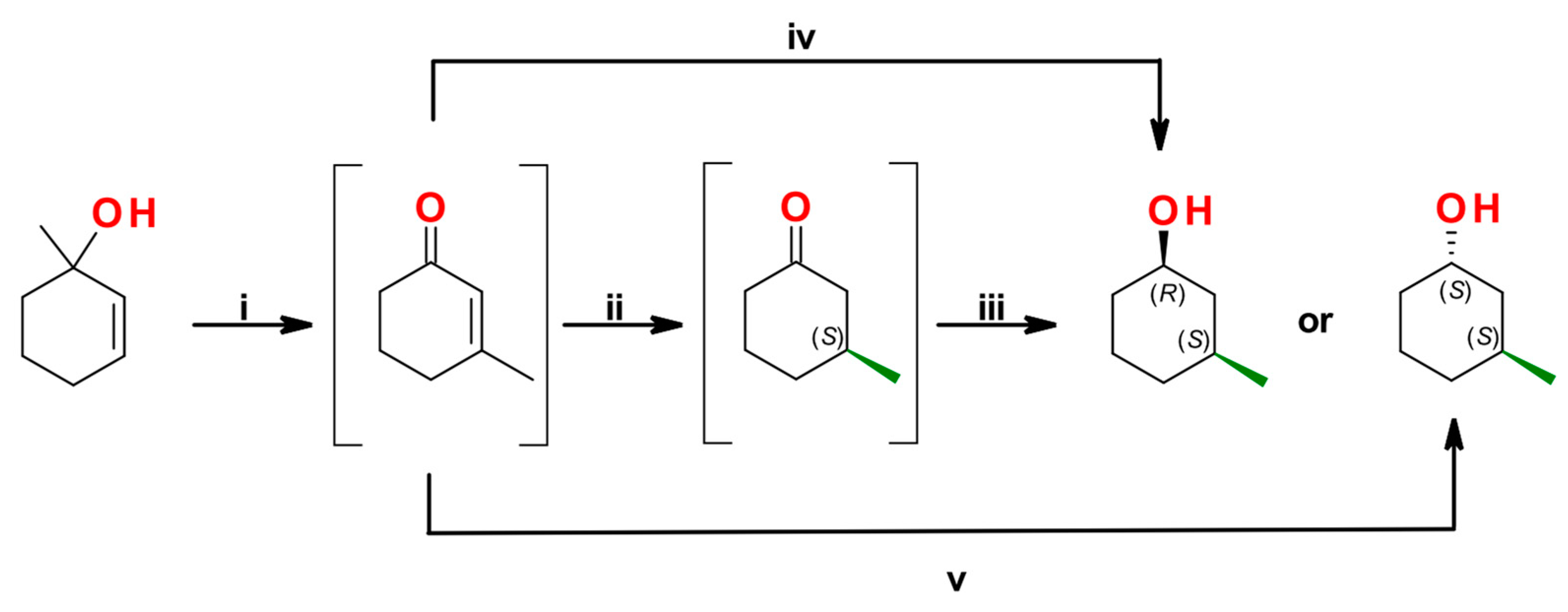{kind=link}
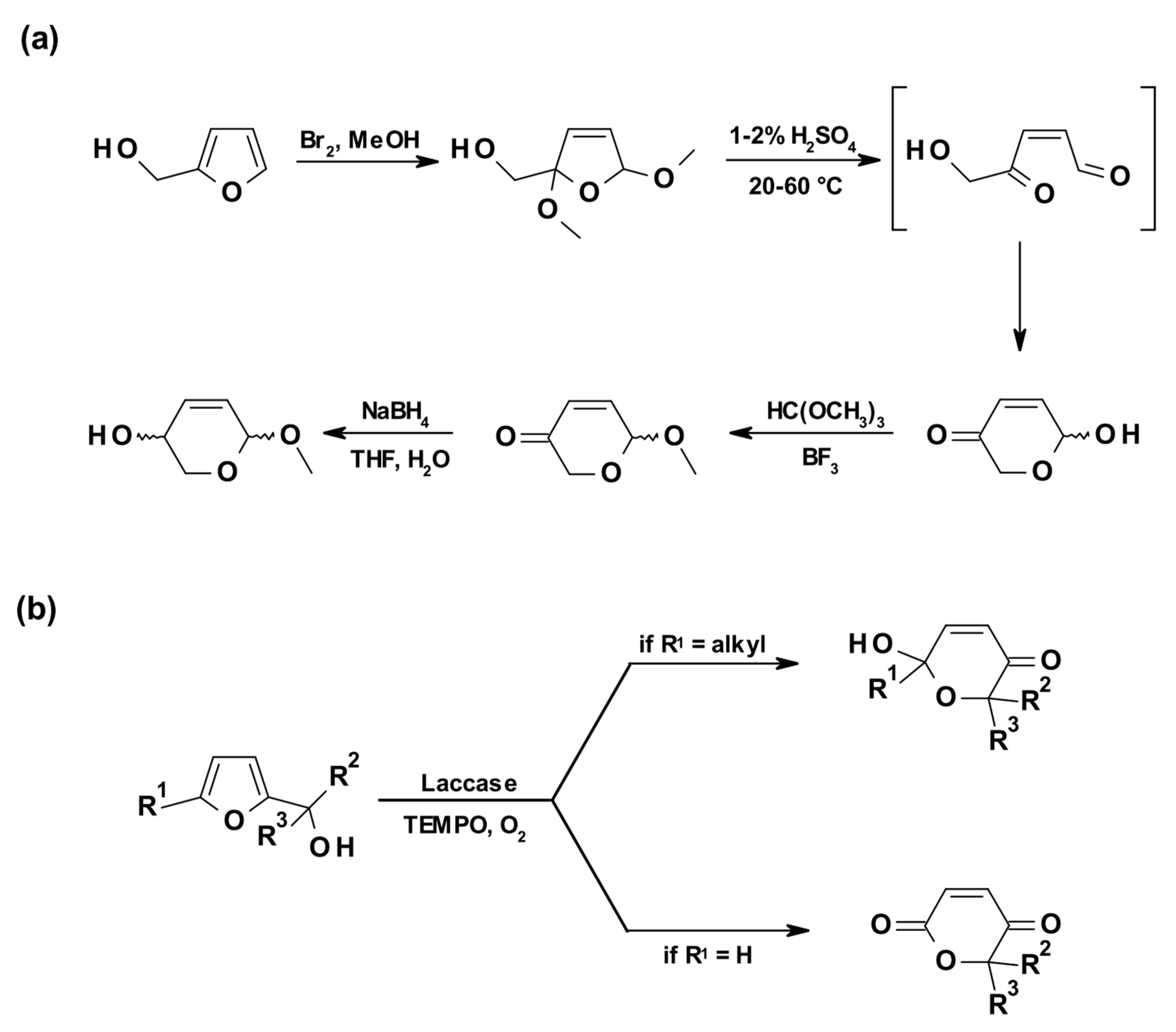{kind=link}
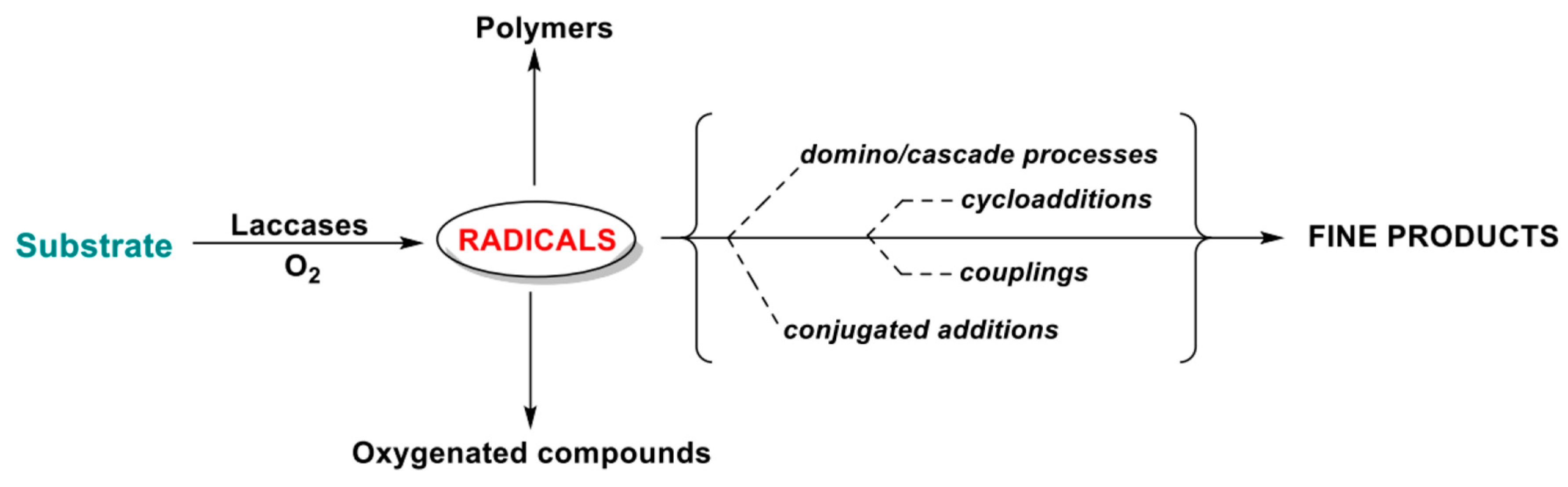{kind=link}
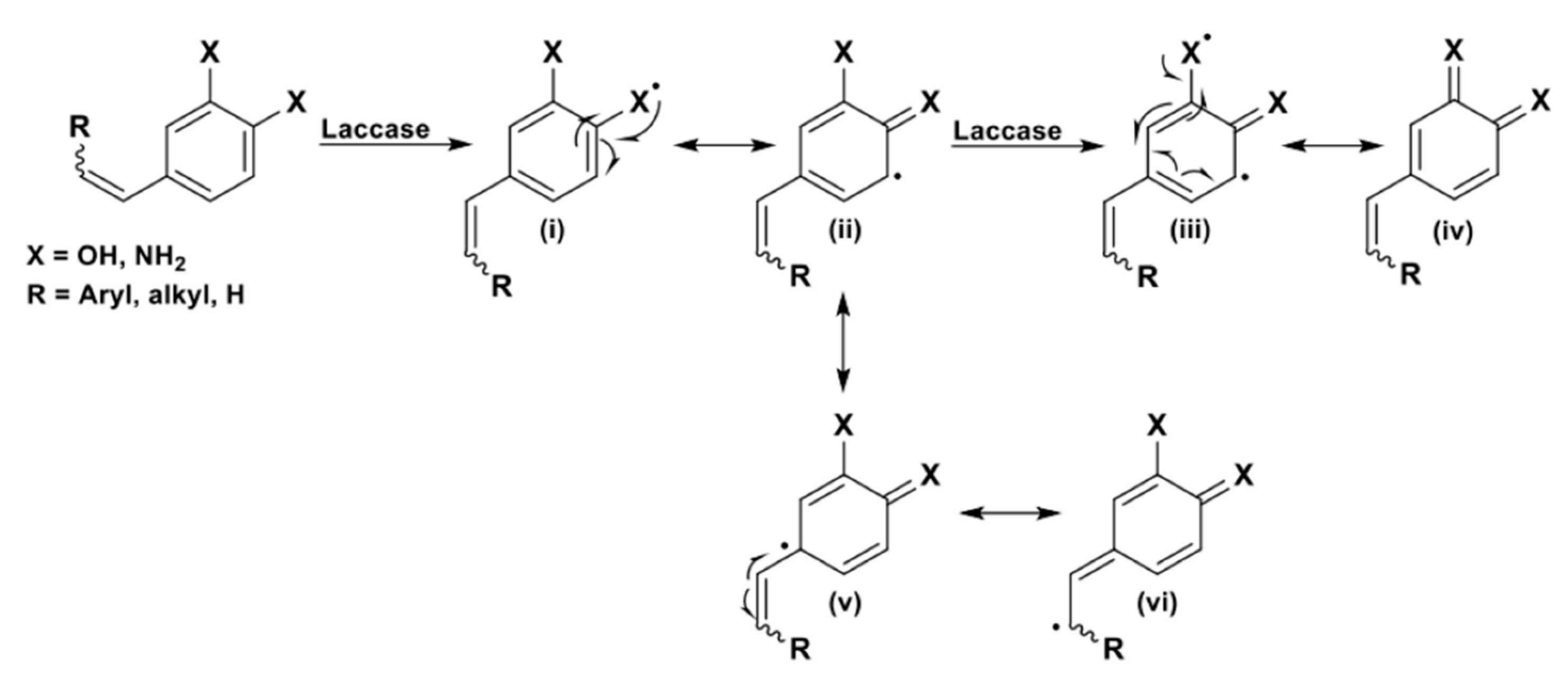{kind=link}
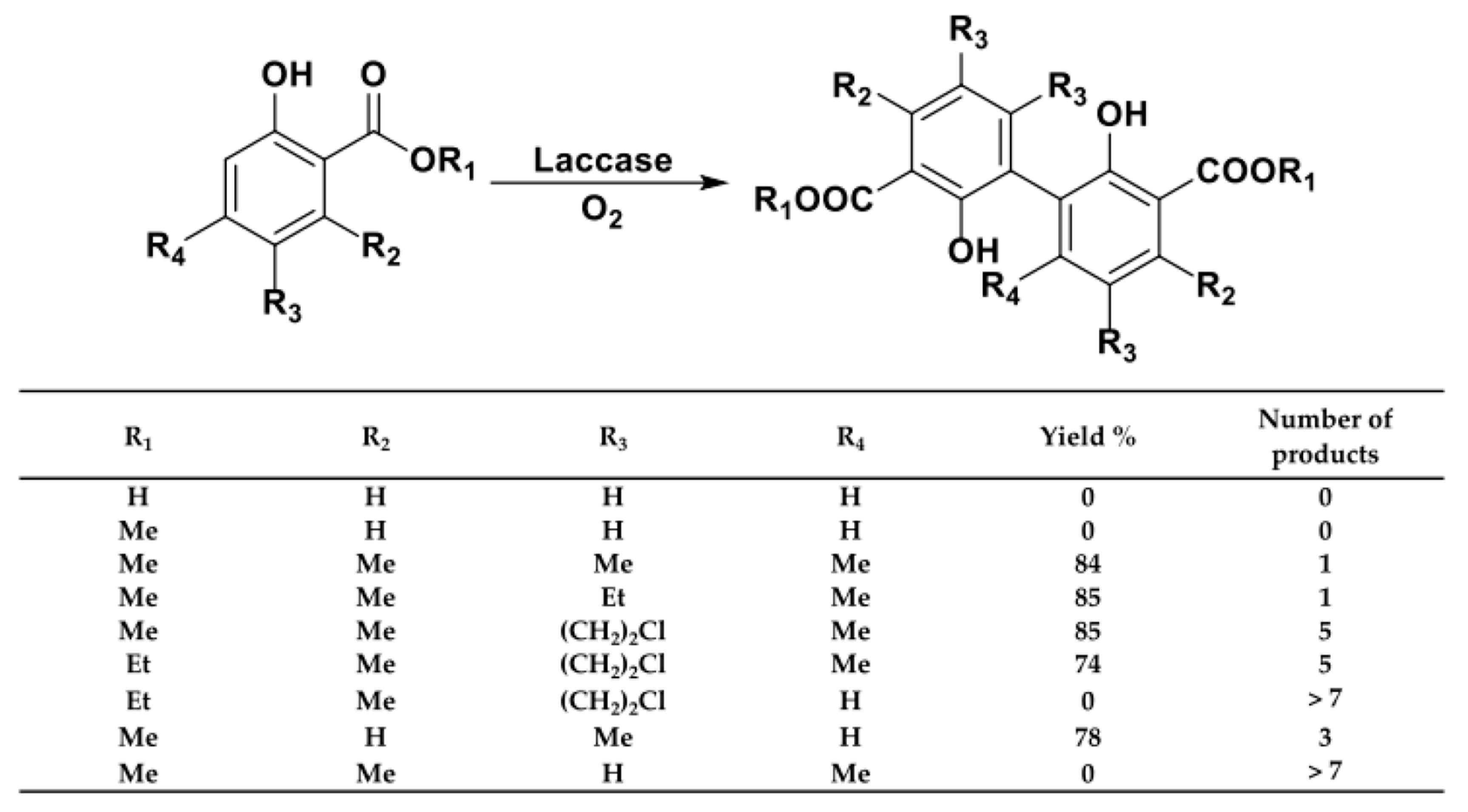{kind=link}
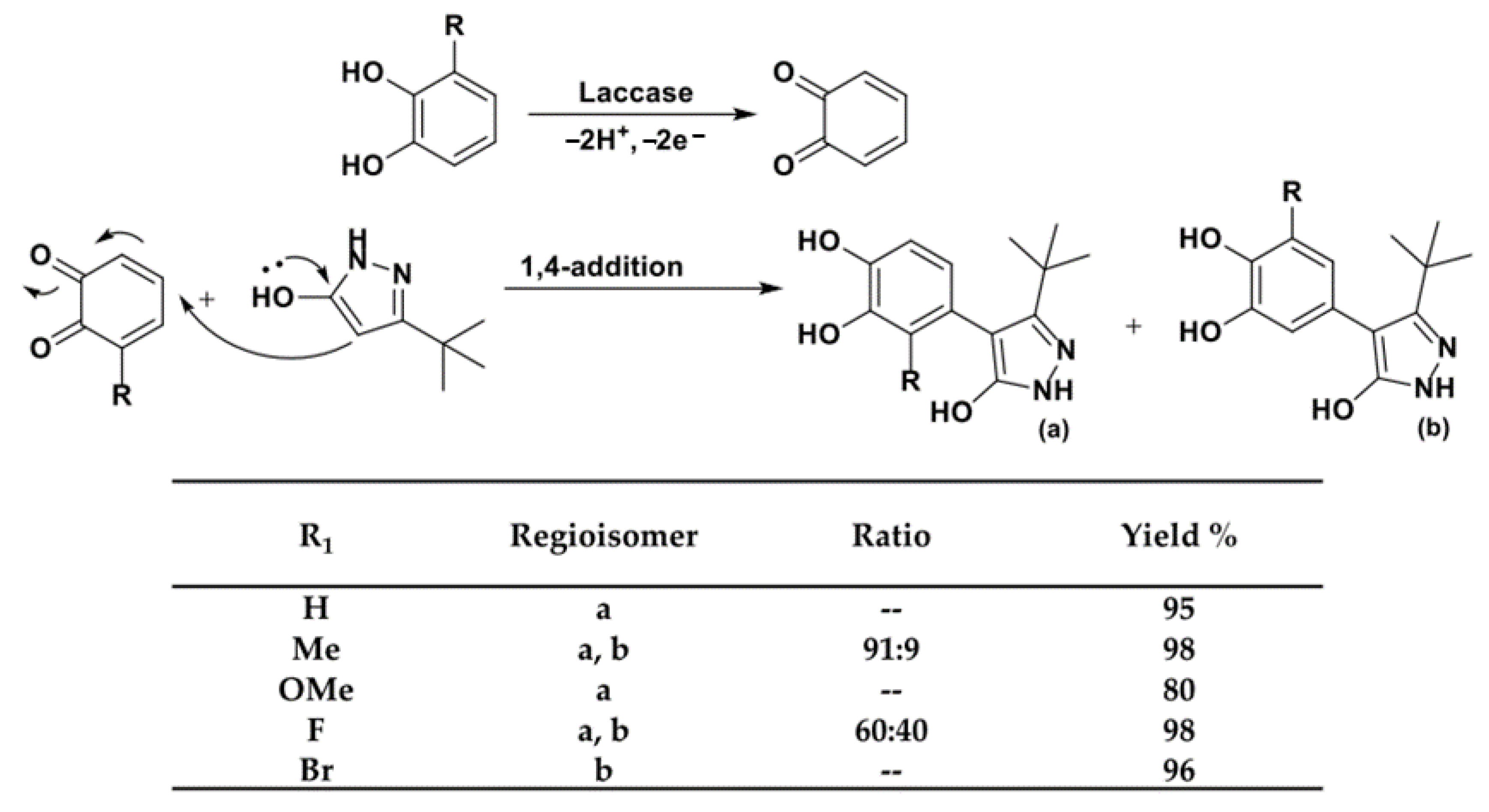{kind=link}
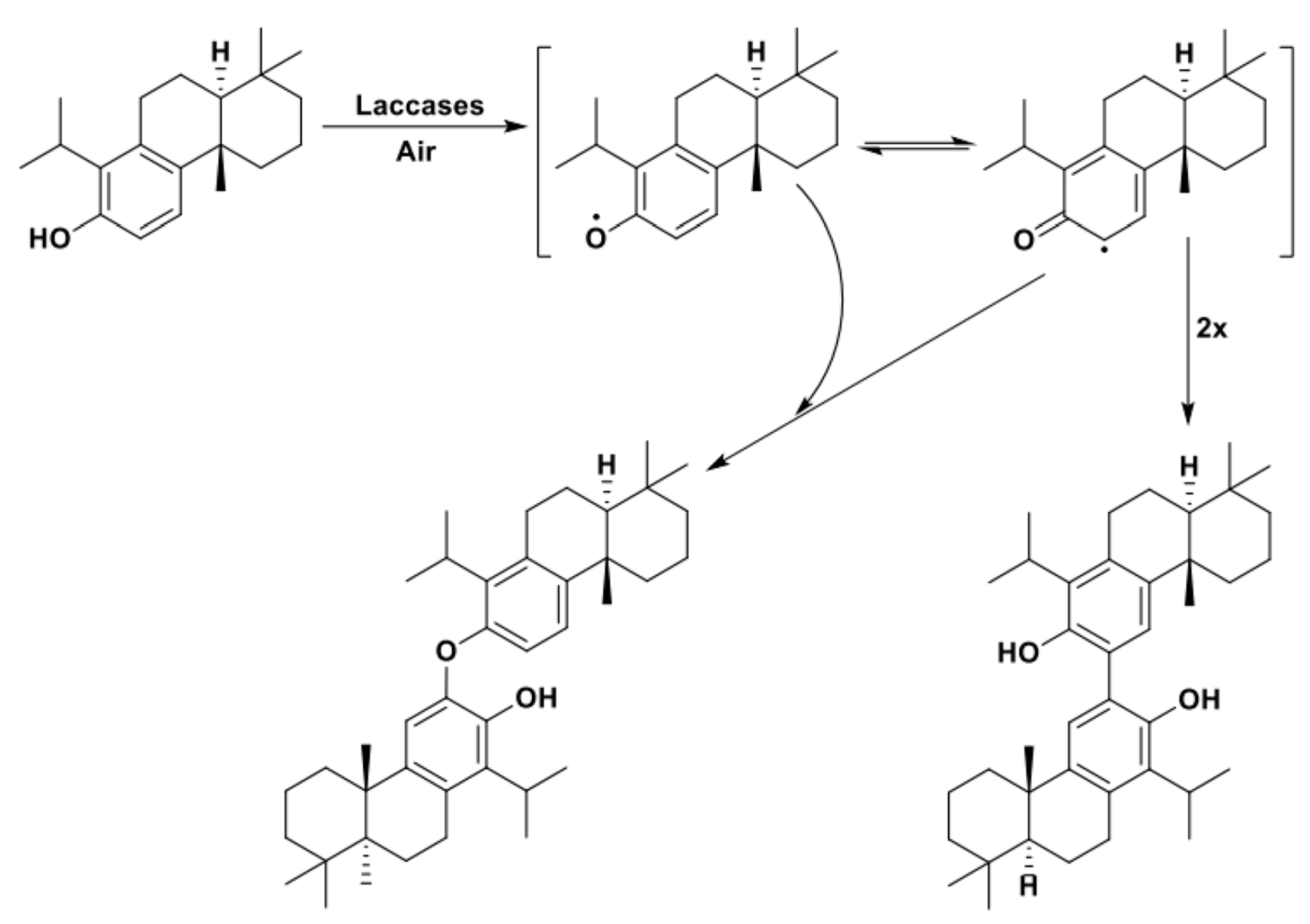{kind=link}
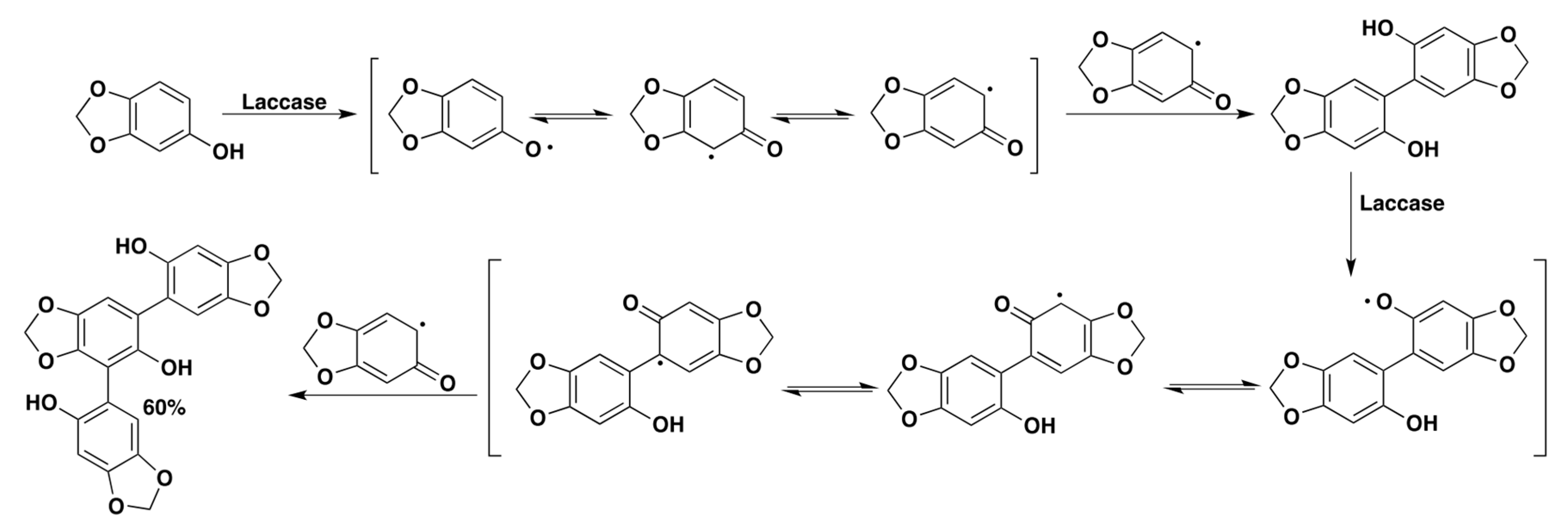{kind=link}
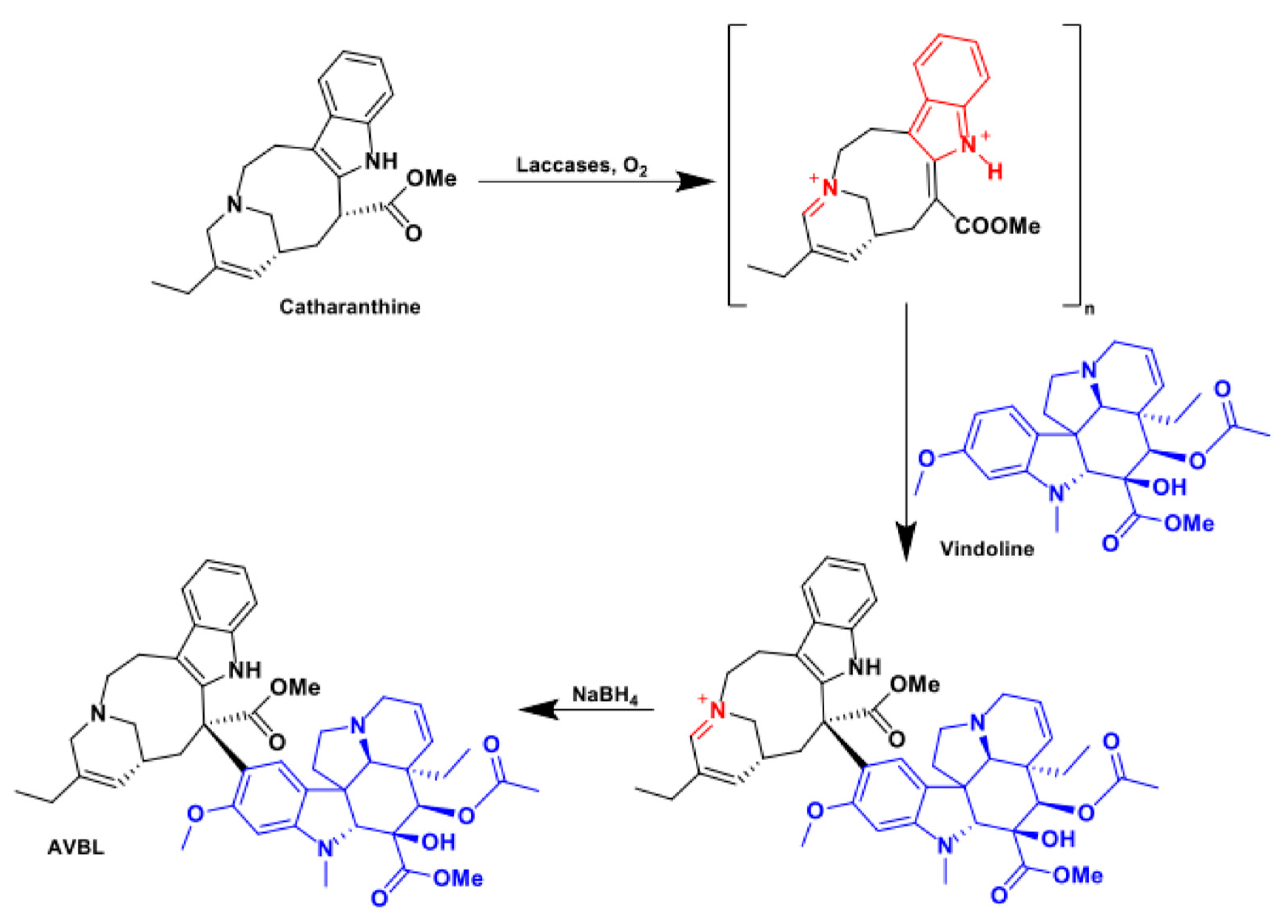{kind=link}
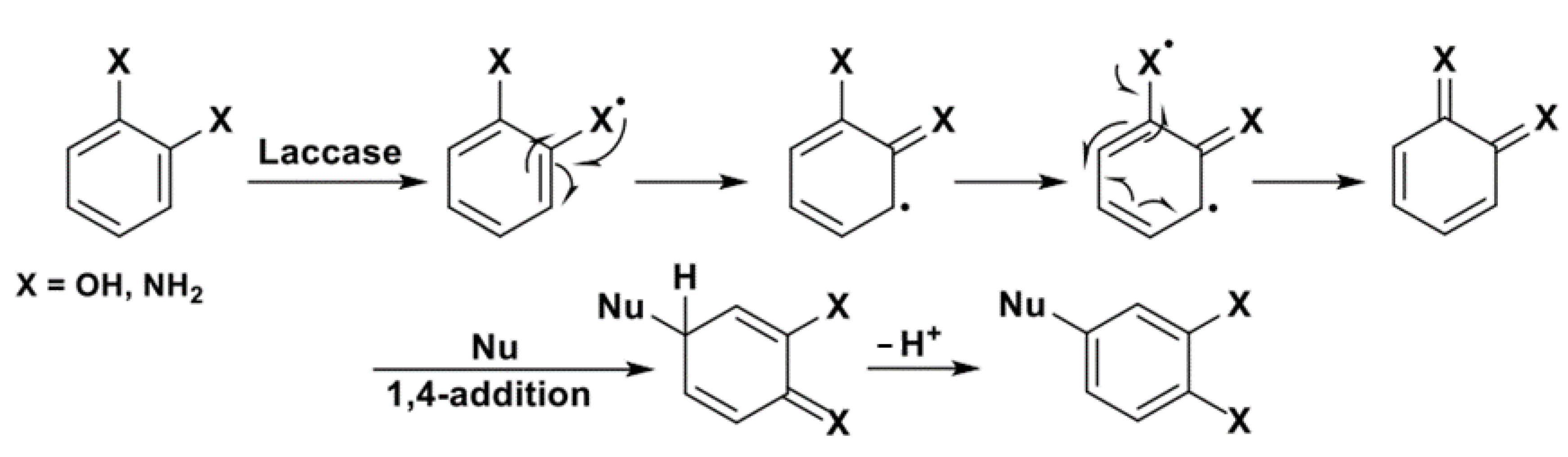{kind=link}
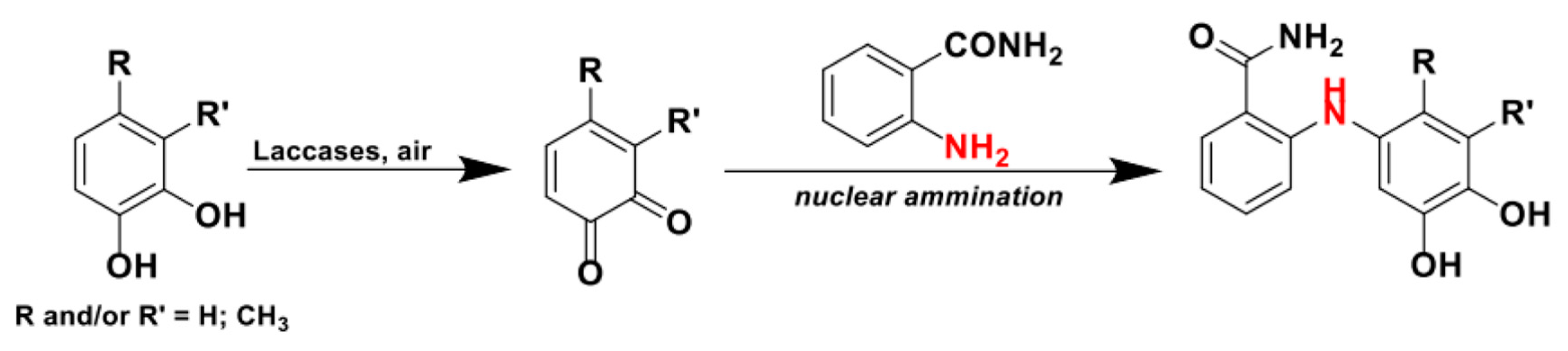{kind=link}
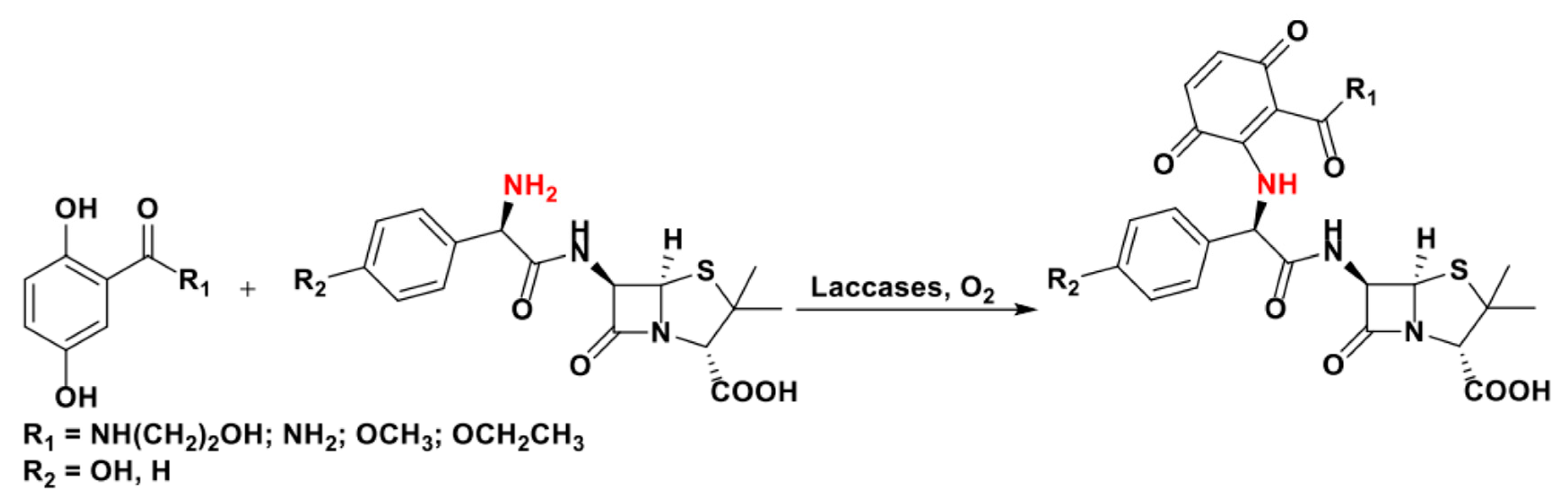{kind=link}
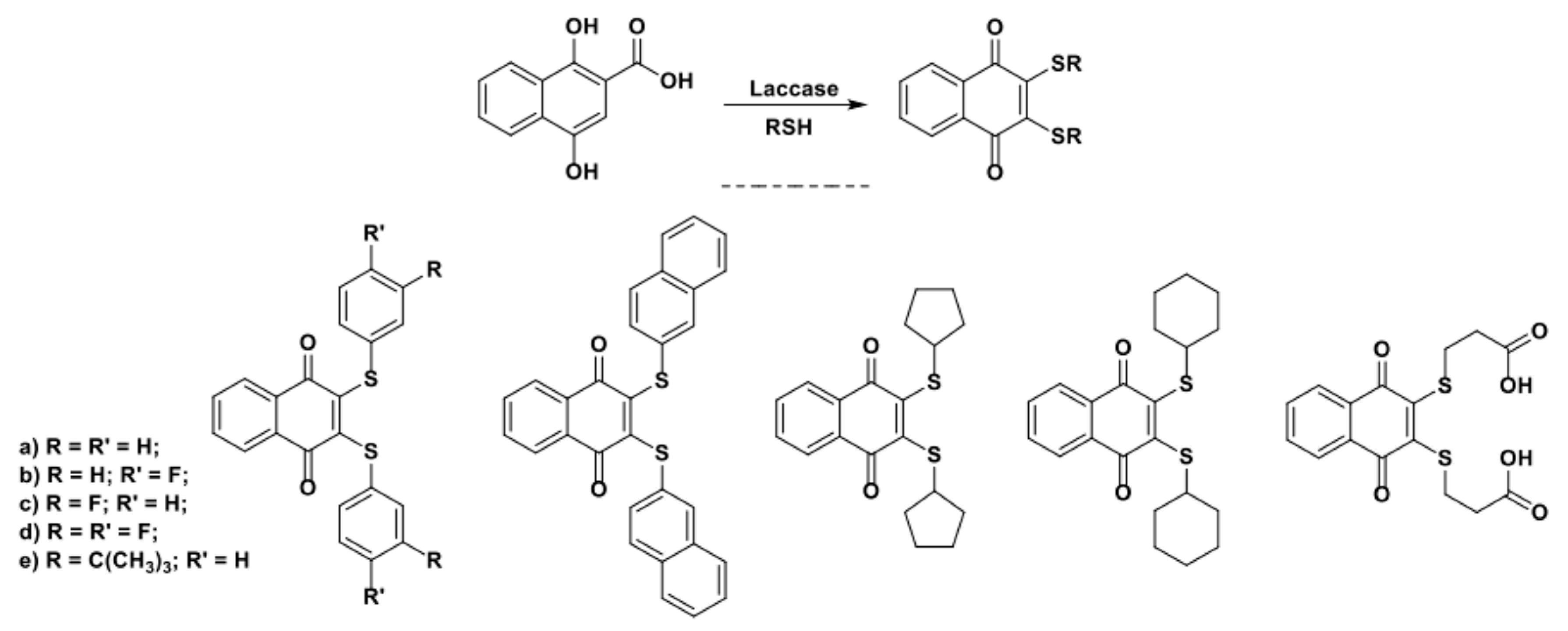{kind=link}
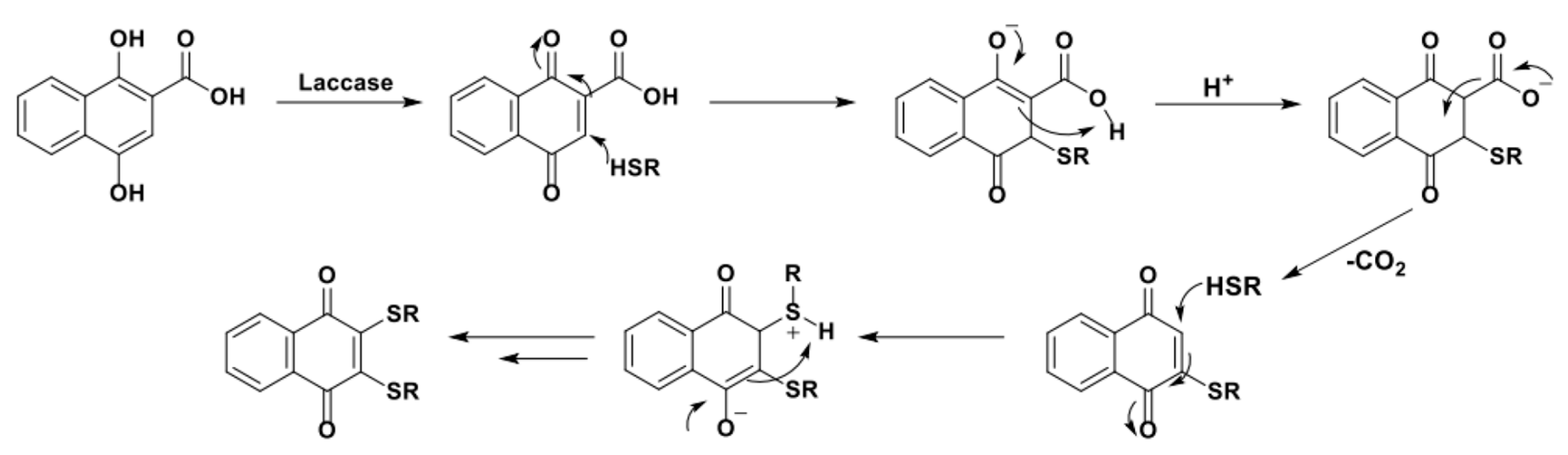{kind=link}
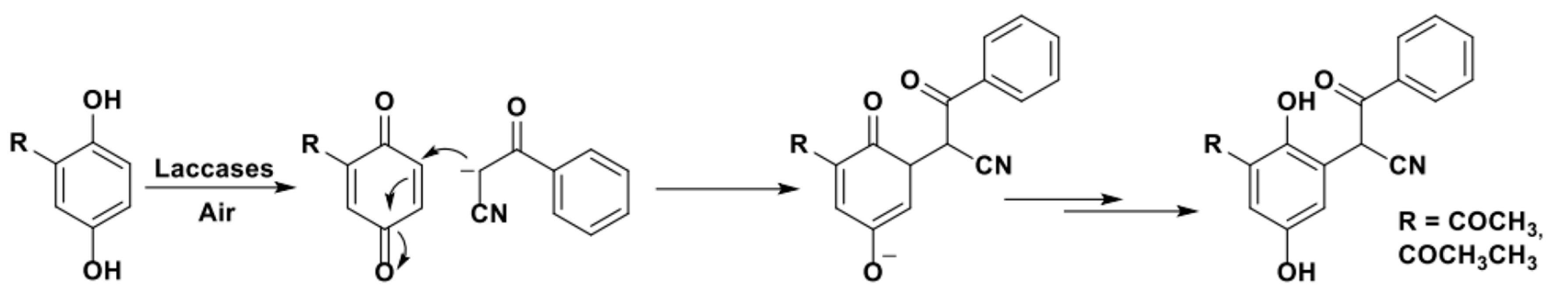{kind=link}
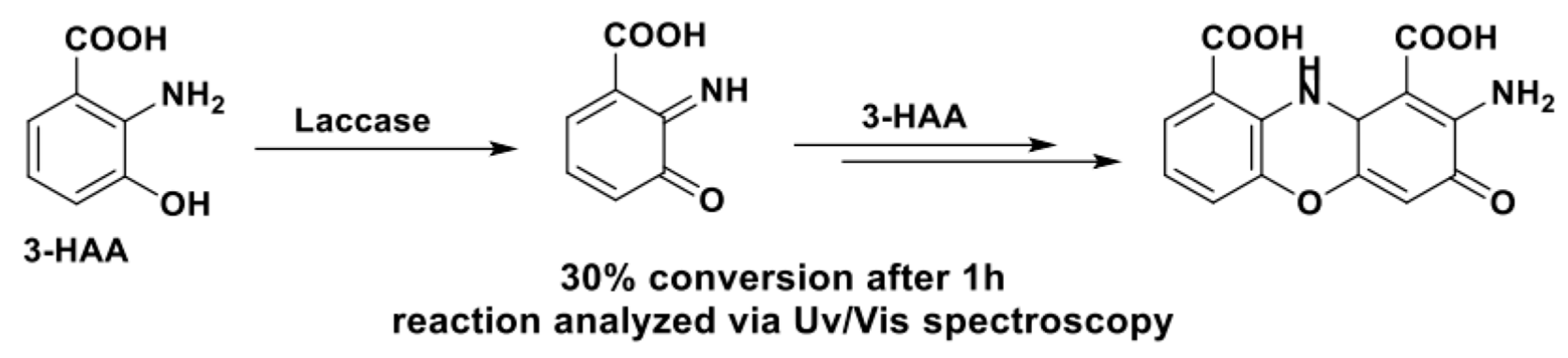{kind=link}
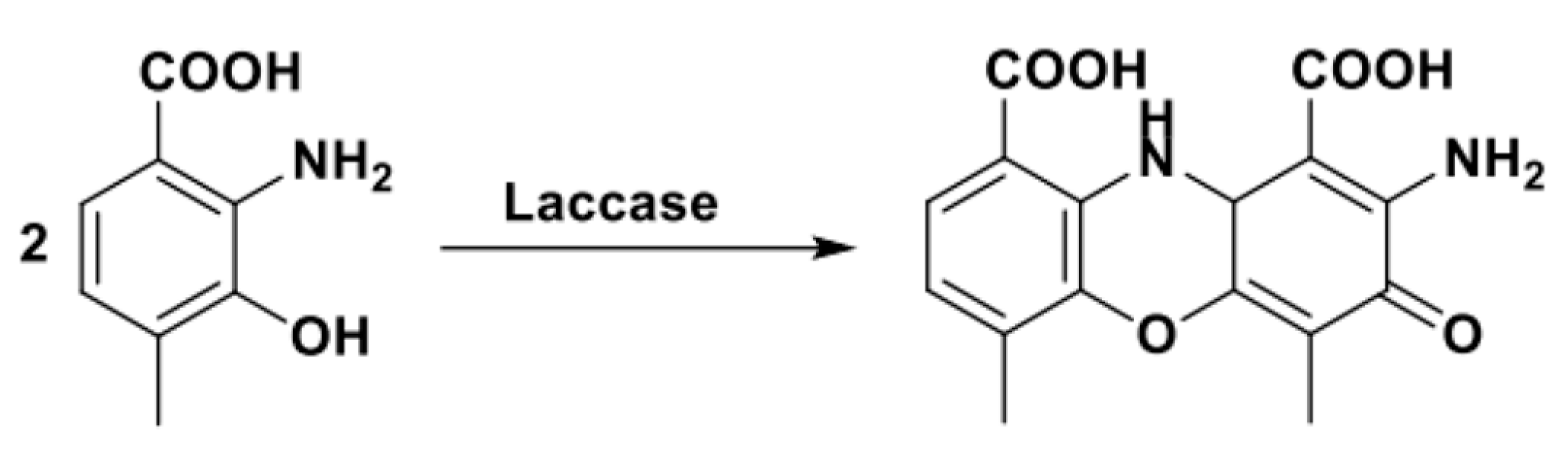{kind=link}
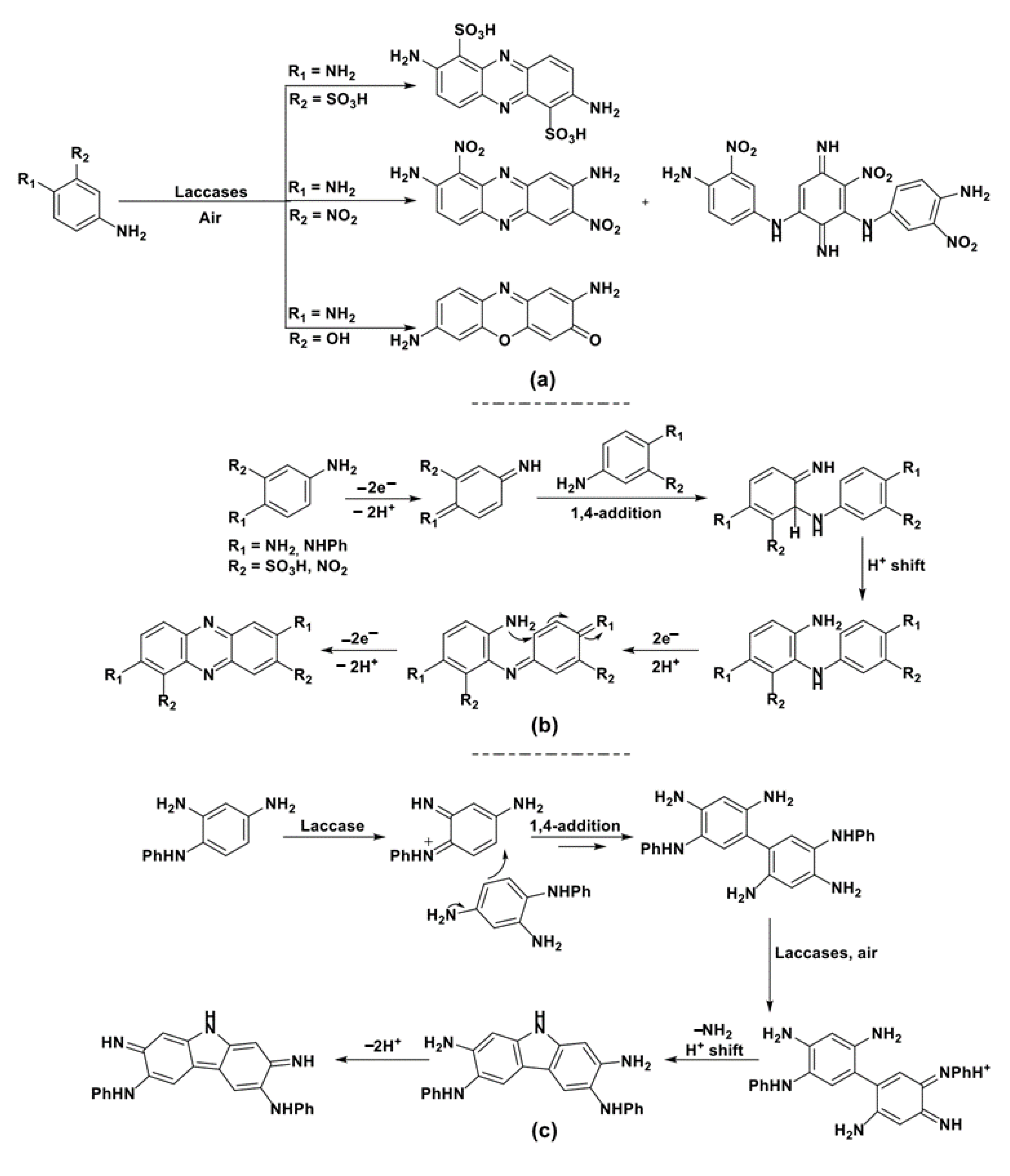{kind=link}
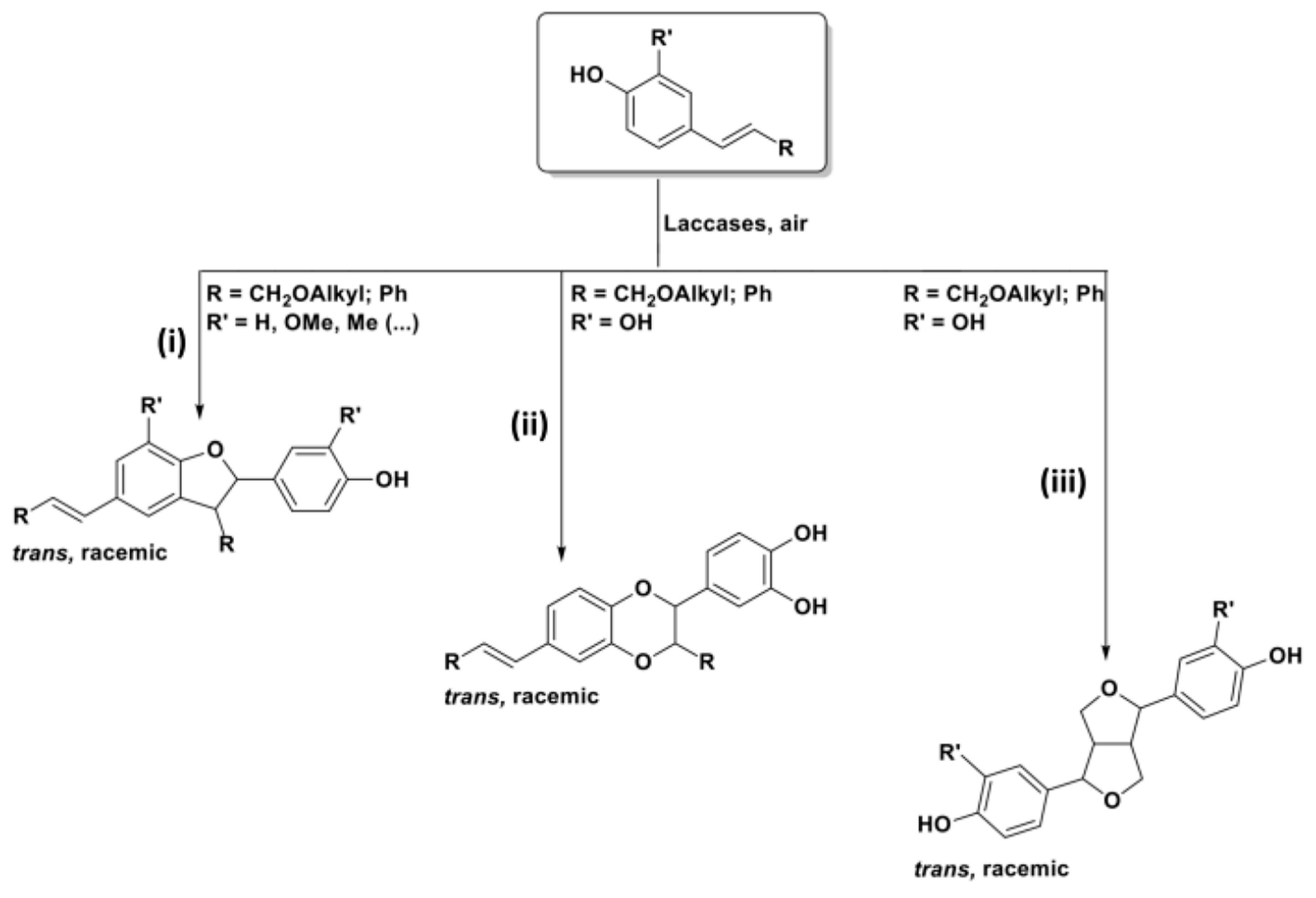{kind=link}
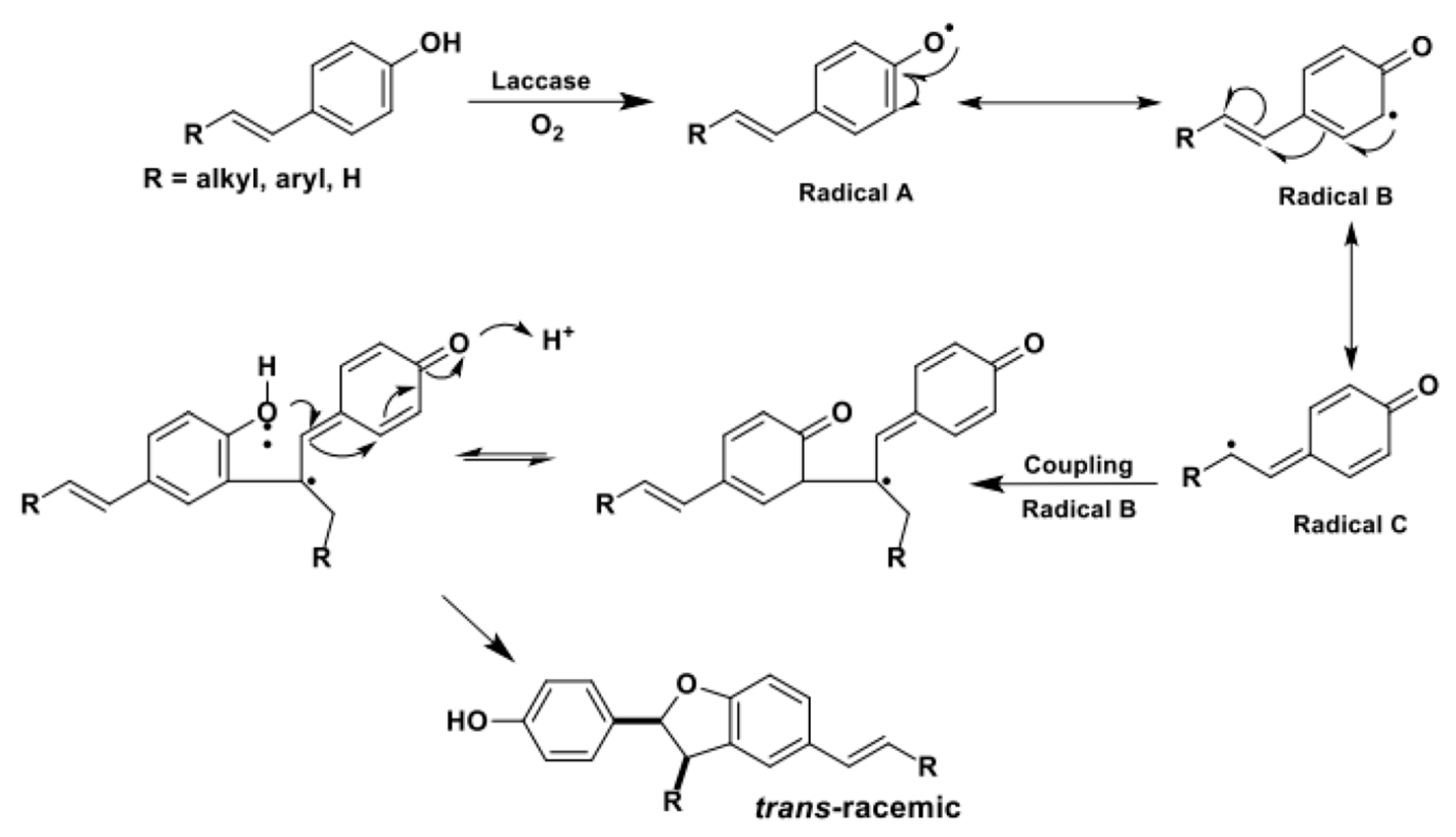{kind=link}
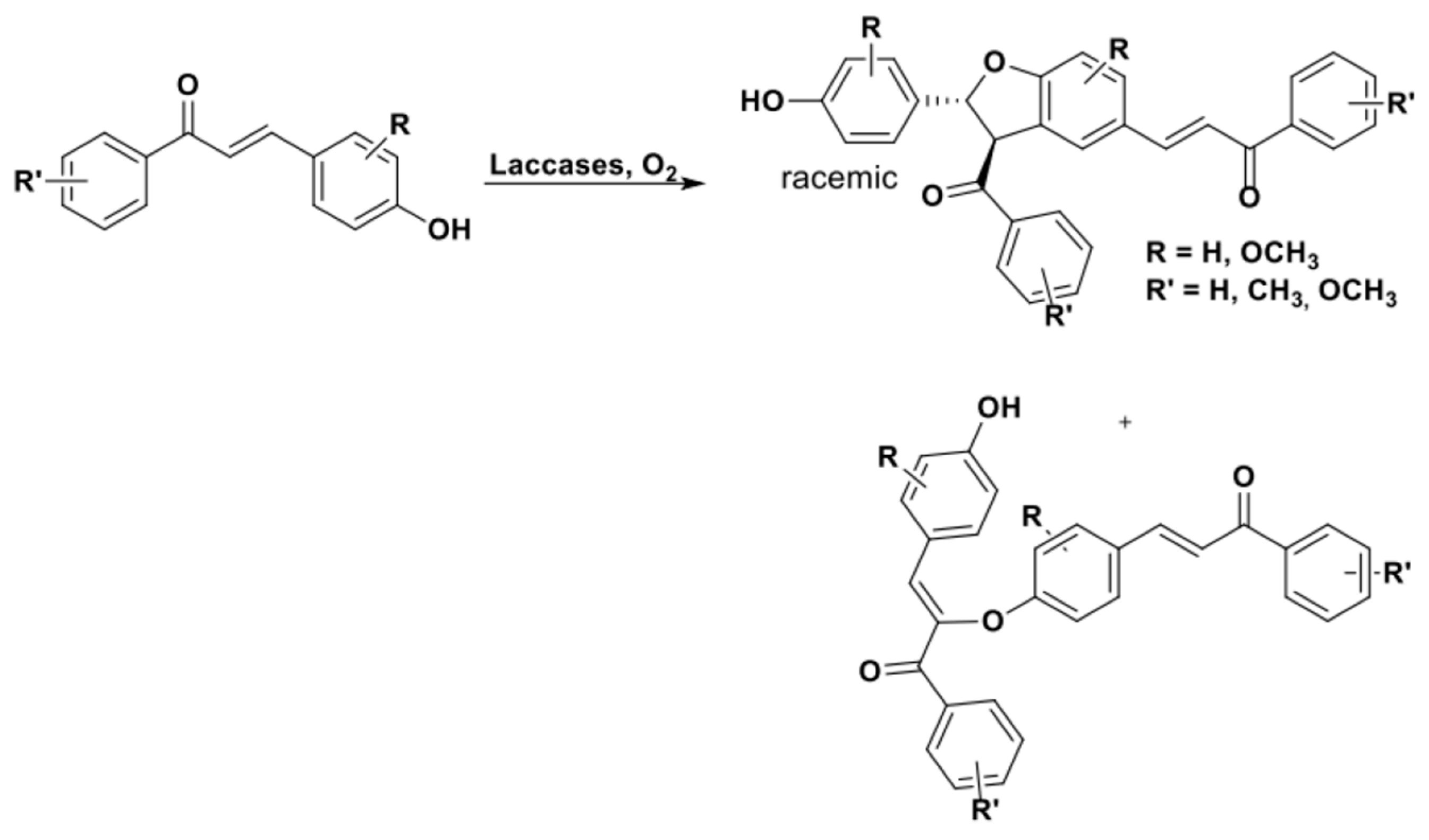{kind=link}
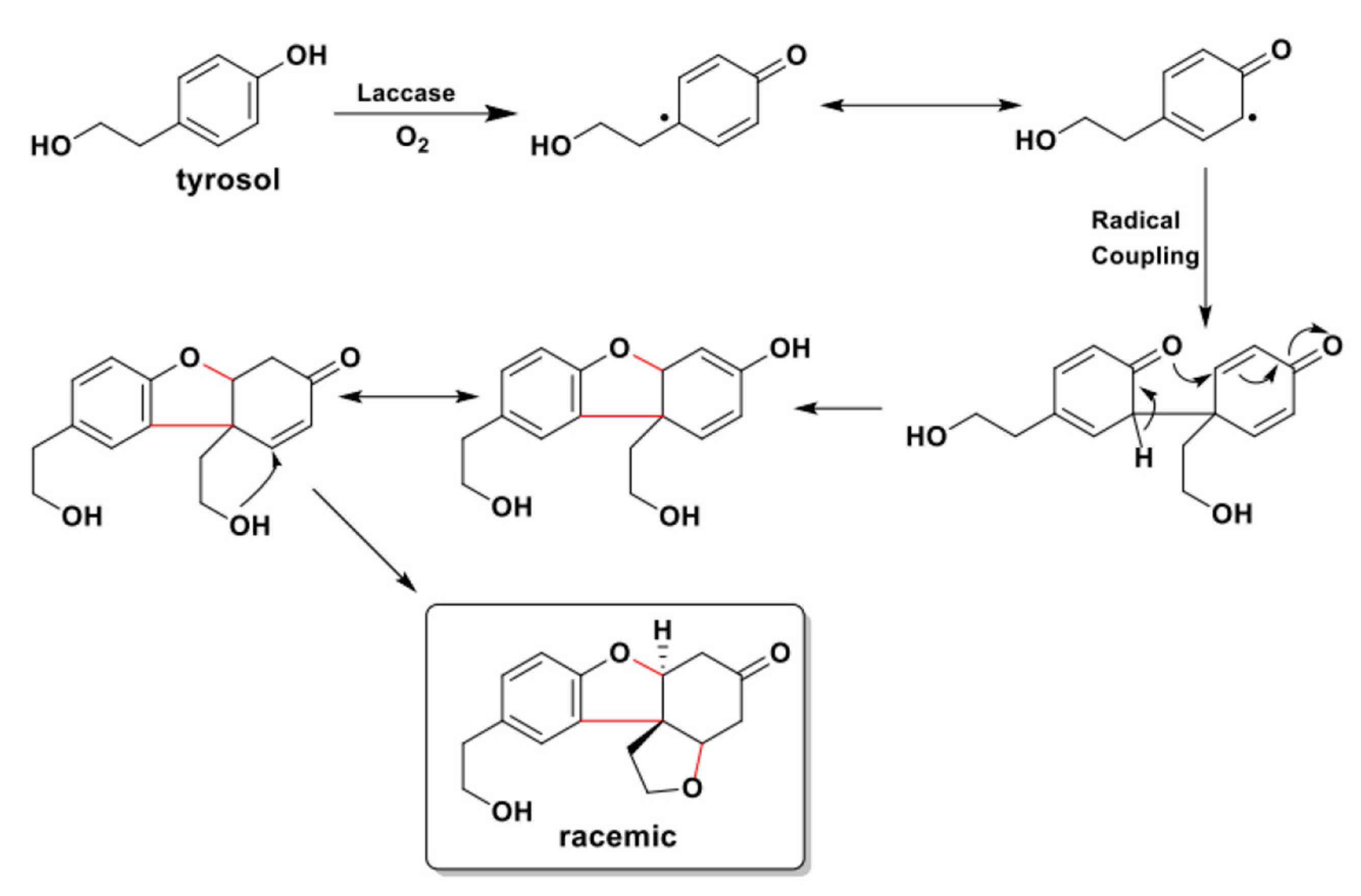{kind=link}
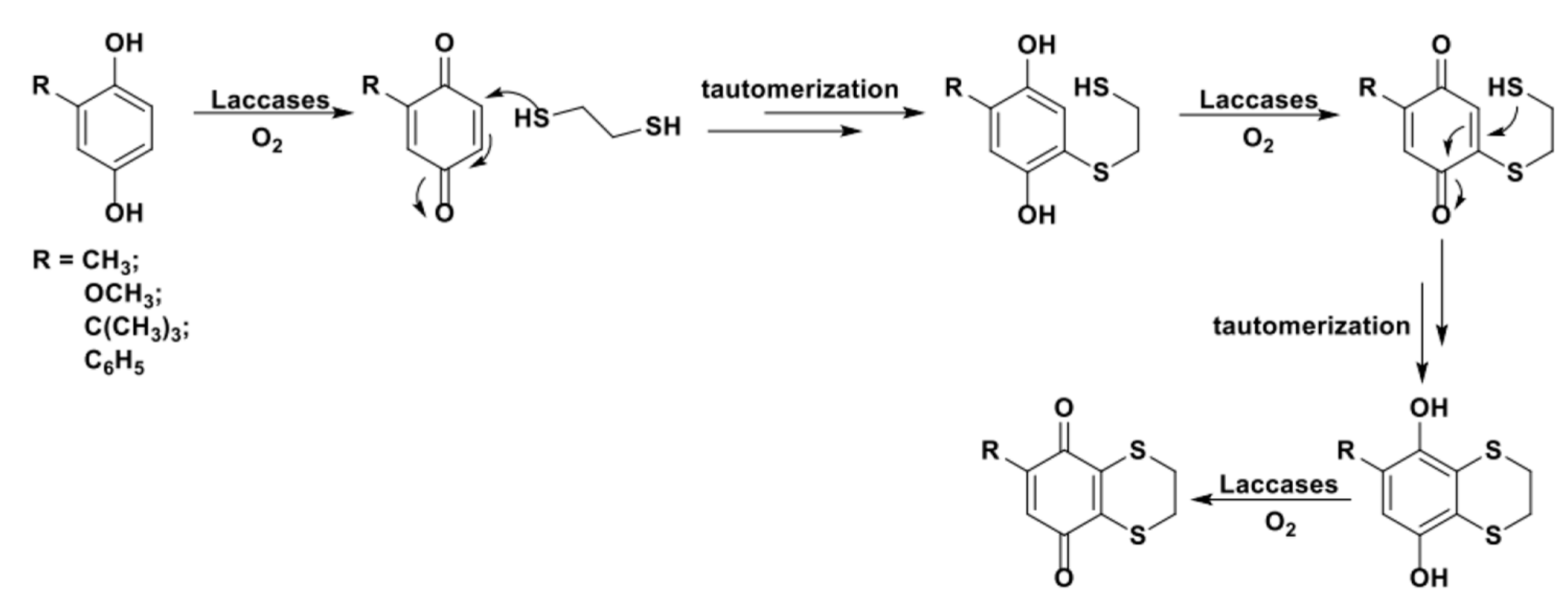{kind=link}
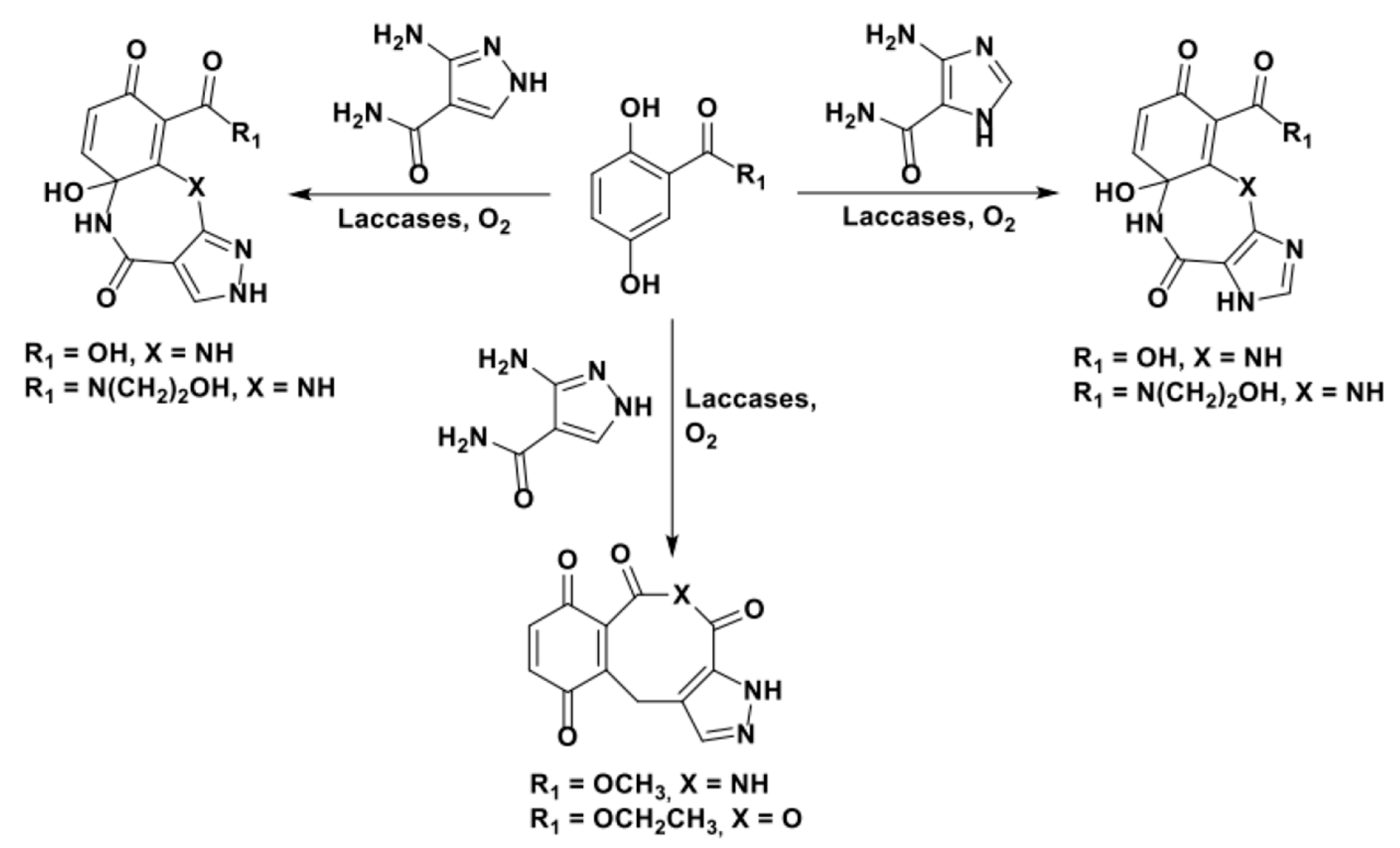{kind=link}
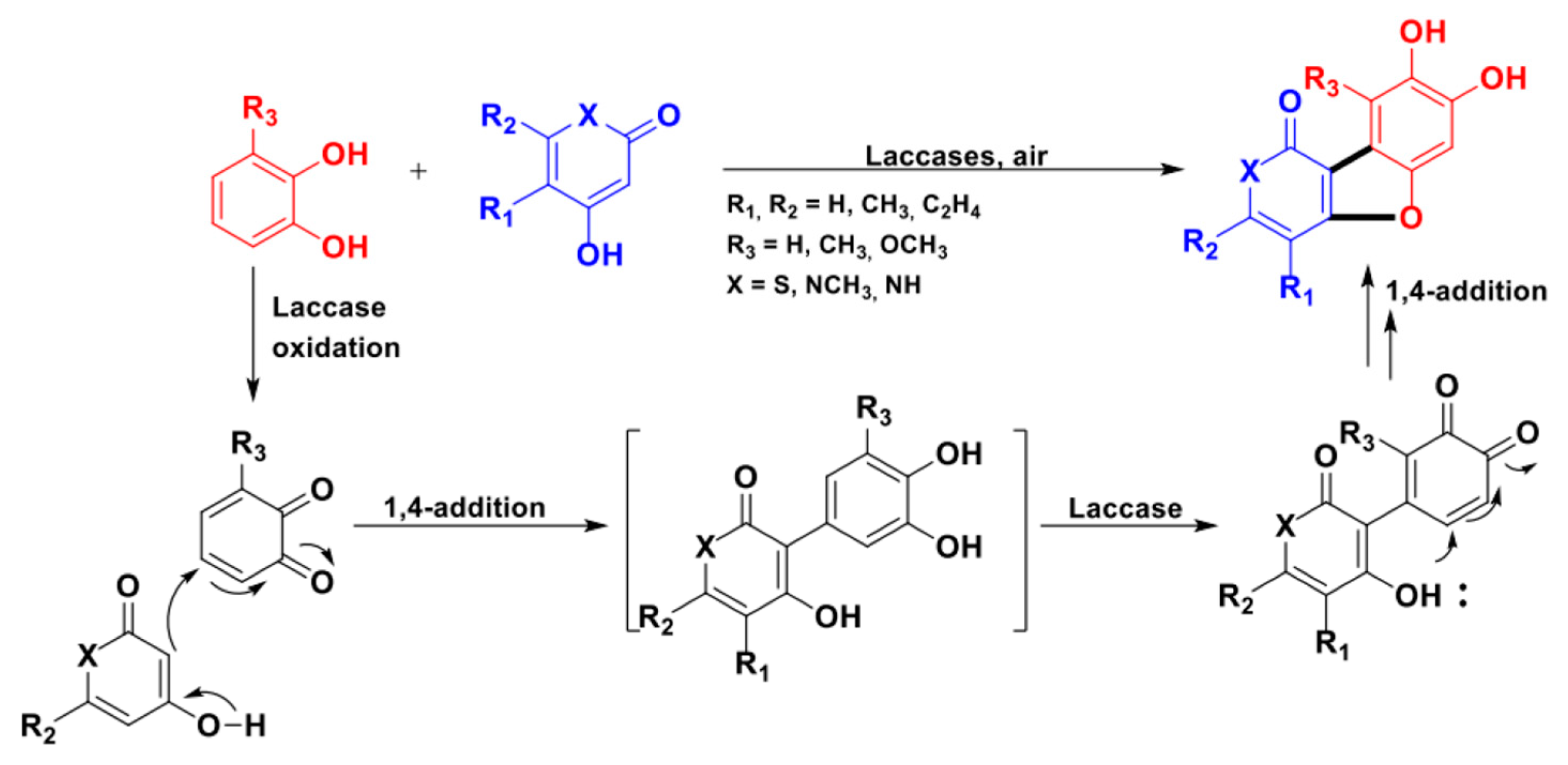{kind=link}
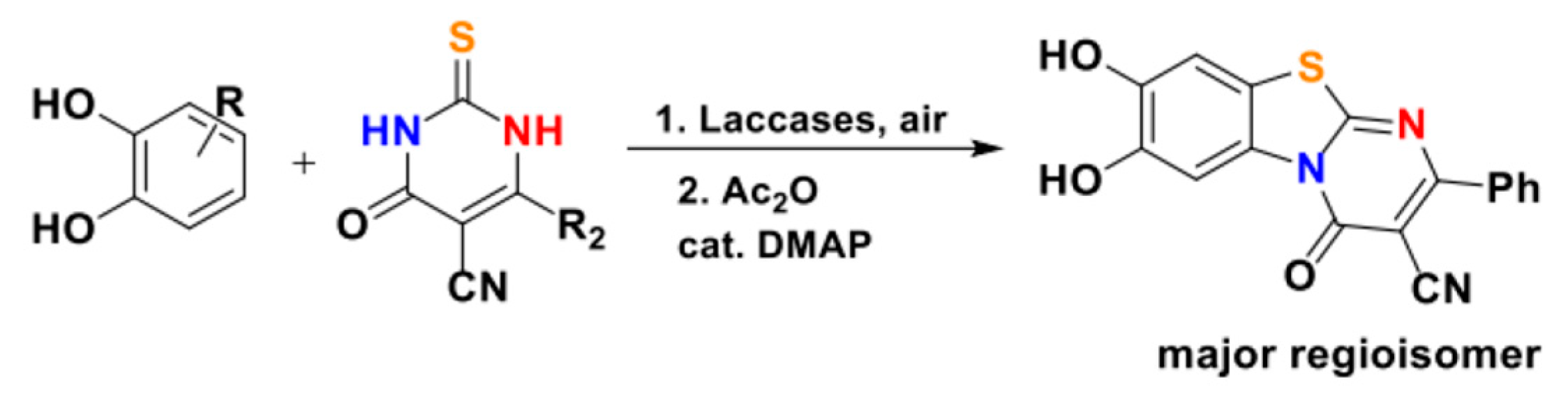{kind=link}
{kind=link}
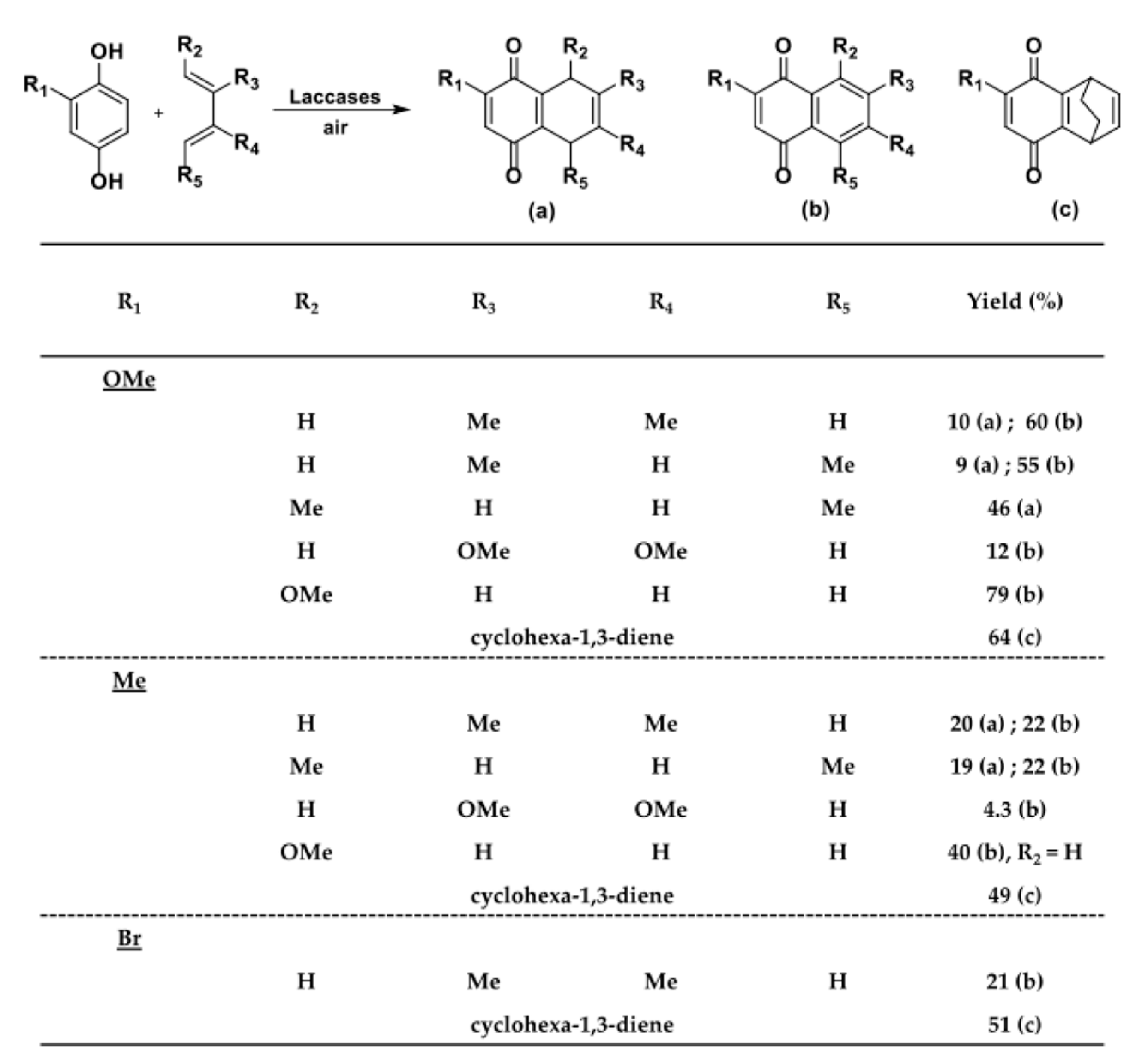{kind=link}
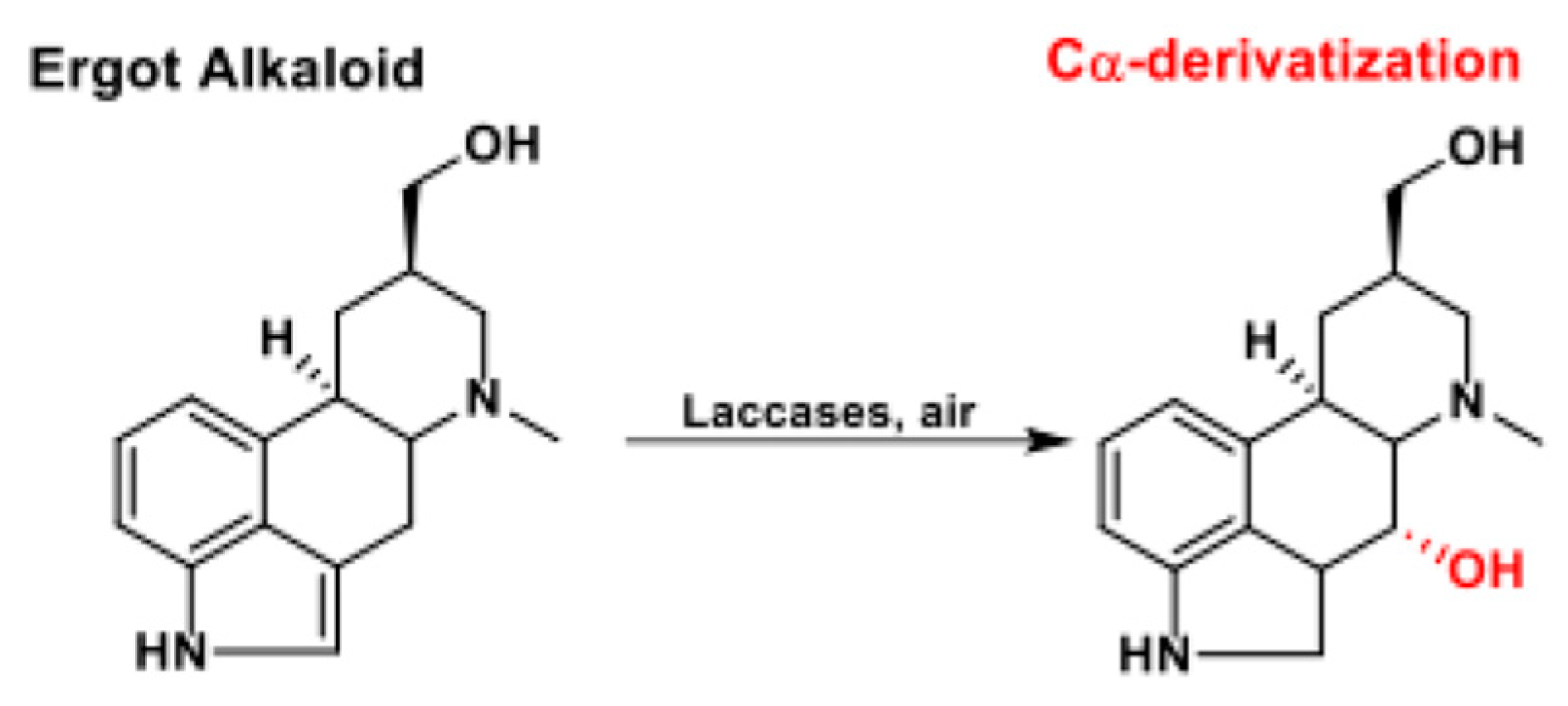{kind=link}
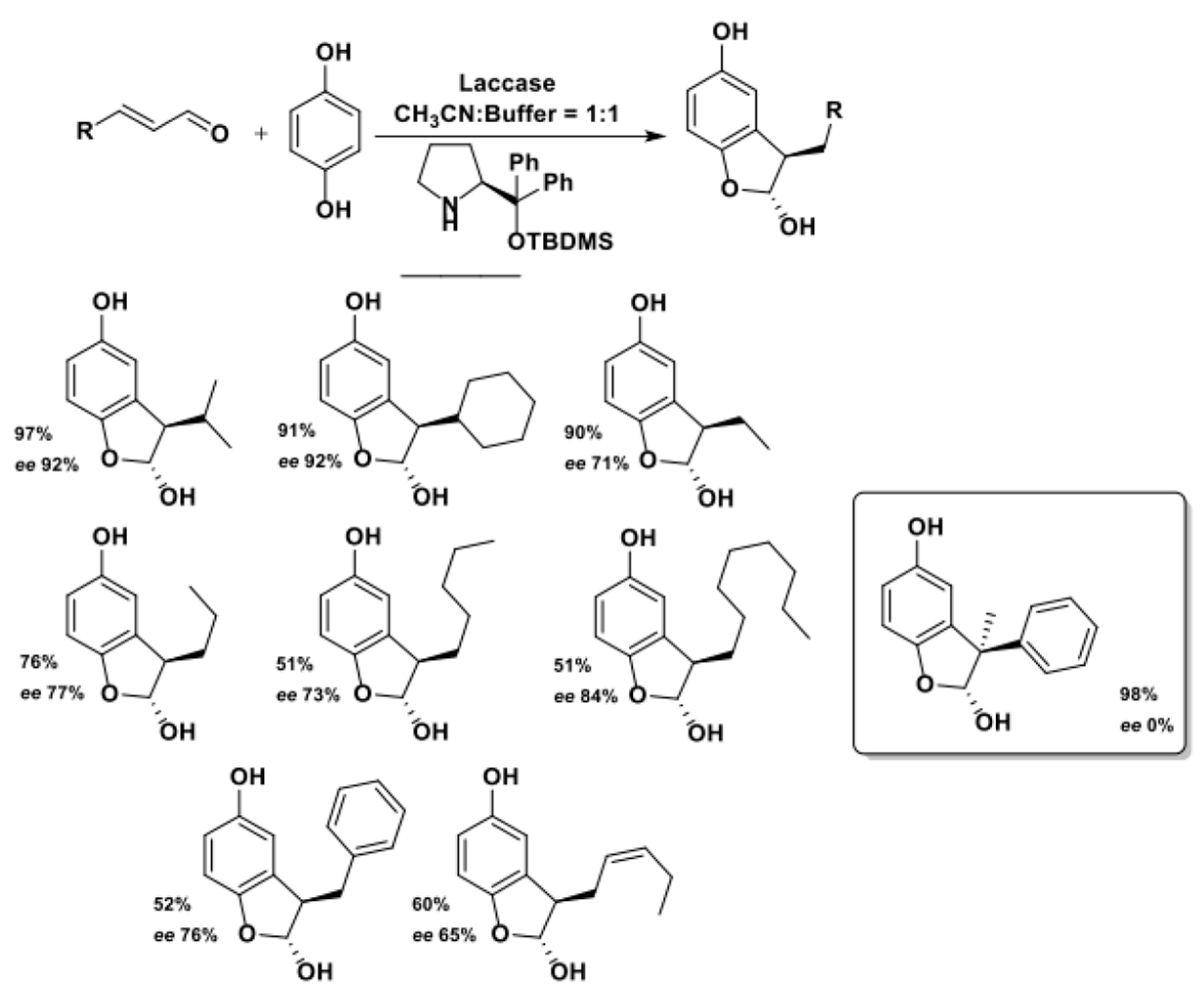{kind=link}
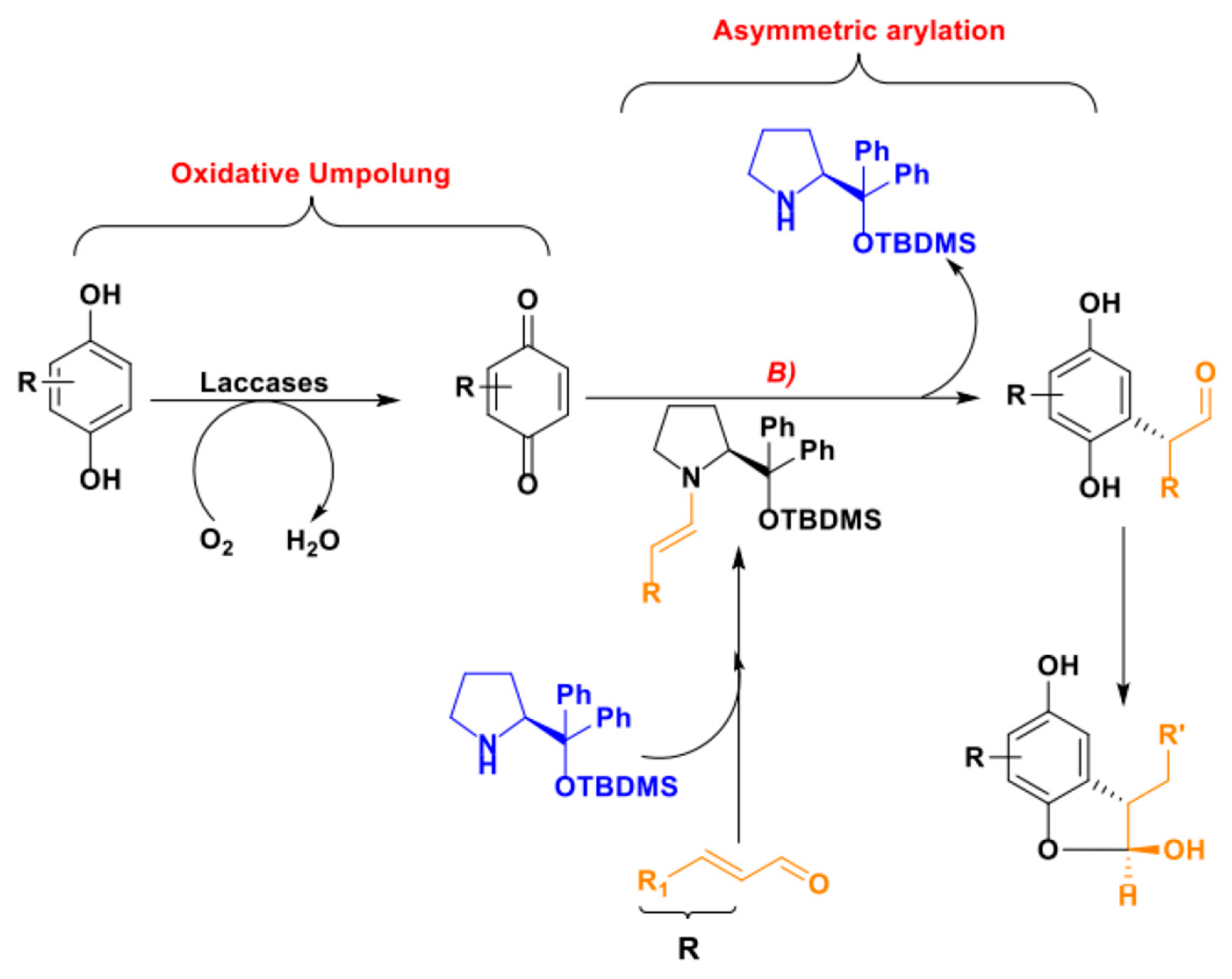{kind=link}
Table 1.
Representative examples of the oxidation of benzyl alcohols to the corresponding benzaldehydes via laccase/ABTS catalysis.
Table 1.
Representative examples of the oxidation of benzyl alcohols to the corresponding benzaldehydes via laccase/ABTS catalysis.
| Substrate | Product | Yield (%) |
|---|---|---|
| toluene | benzaldehyde | 92 |
| p-nitrotoluene | p-nitrobenzaldehyde | 98 |
| m-chlorotoluene | m-chlorobenzaldehyde | 89 |
| 3,4-dimethoxytoluene | 3,4-dimethoxybenzaldehyde | 90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bassanini, I.; Ferrandi, E.E.; Riva, S.; Monti, D. Biocatalysis with Laccases: An Updated Overview. Catalysts 2021, 11, 26. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010026
AMA Style
Bassanini I, Ferrandi EE, Riva S, Monti D. Biocatalysis with Laccases: An Updated Overview. Catalysts. 2021; 11(1):26. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010026
Chicago/Turabian StyleBassanini, Ivan, Erica Elisa Ferrandi, Sergio Riva, and Daniela Monti. 2021. "Biocatalysis with Laccases: An Updated Overview" Catalysts 11, no. 1: 26. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11010026
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.