
The Role of Cytokines and Chemokines in Shaping the Immune Microenvironment of Glioblastoma: Implications for Immunotherapy


Abstract
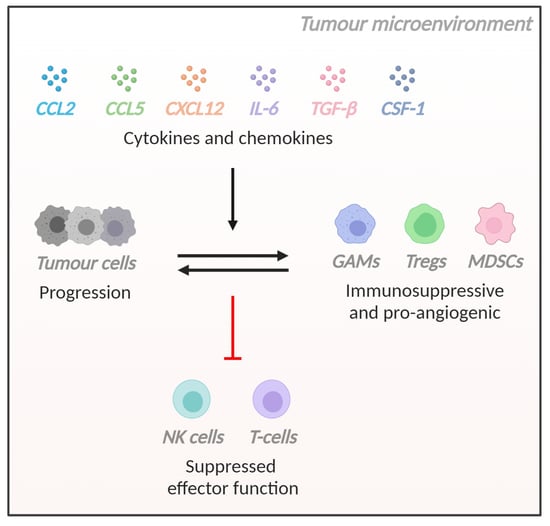
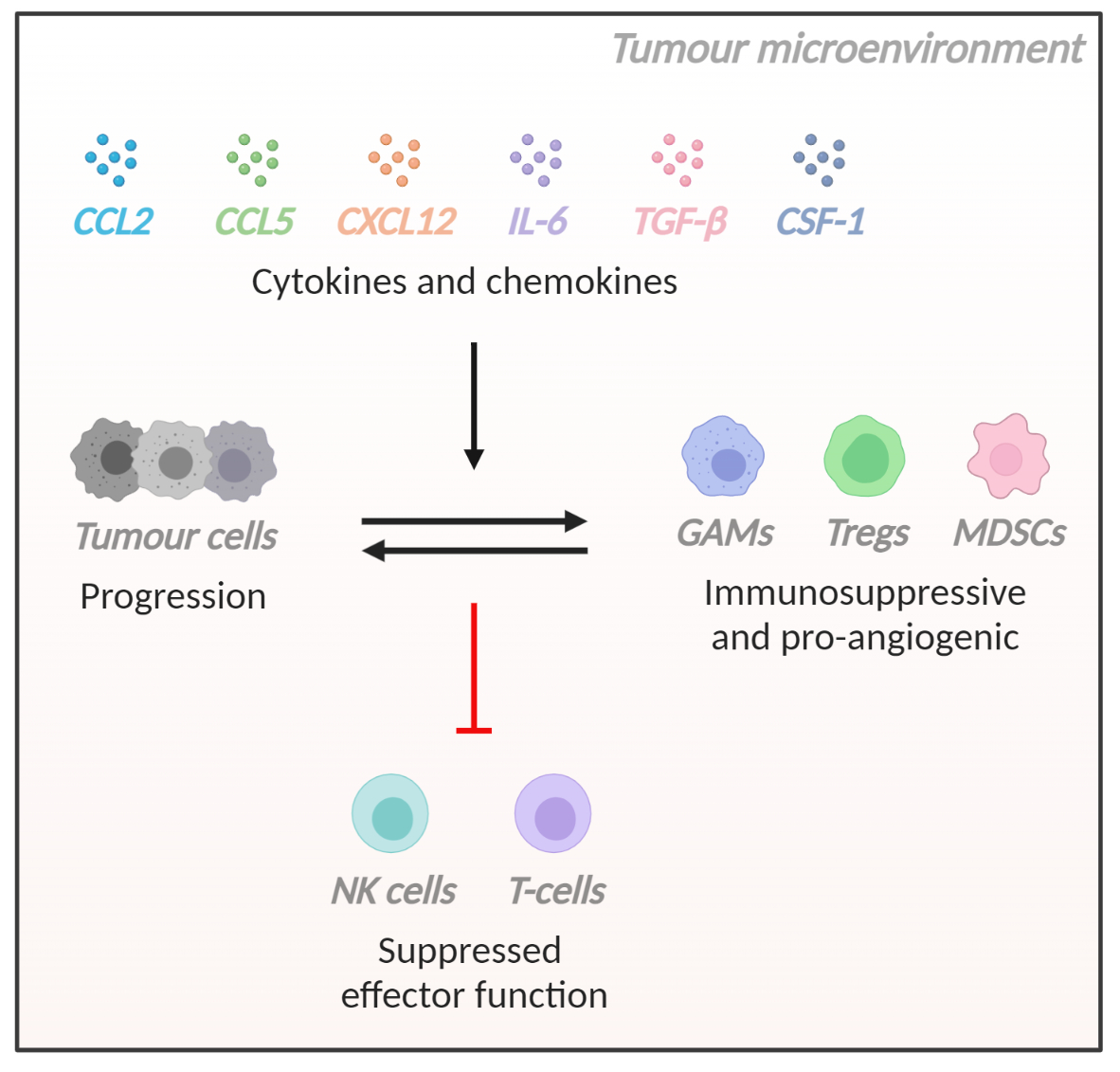
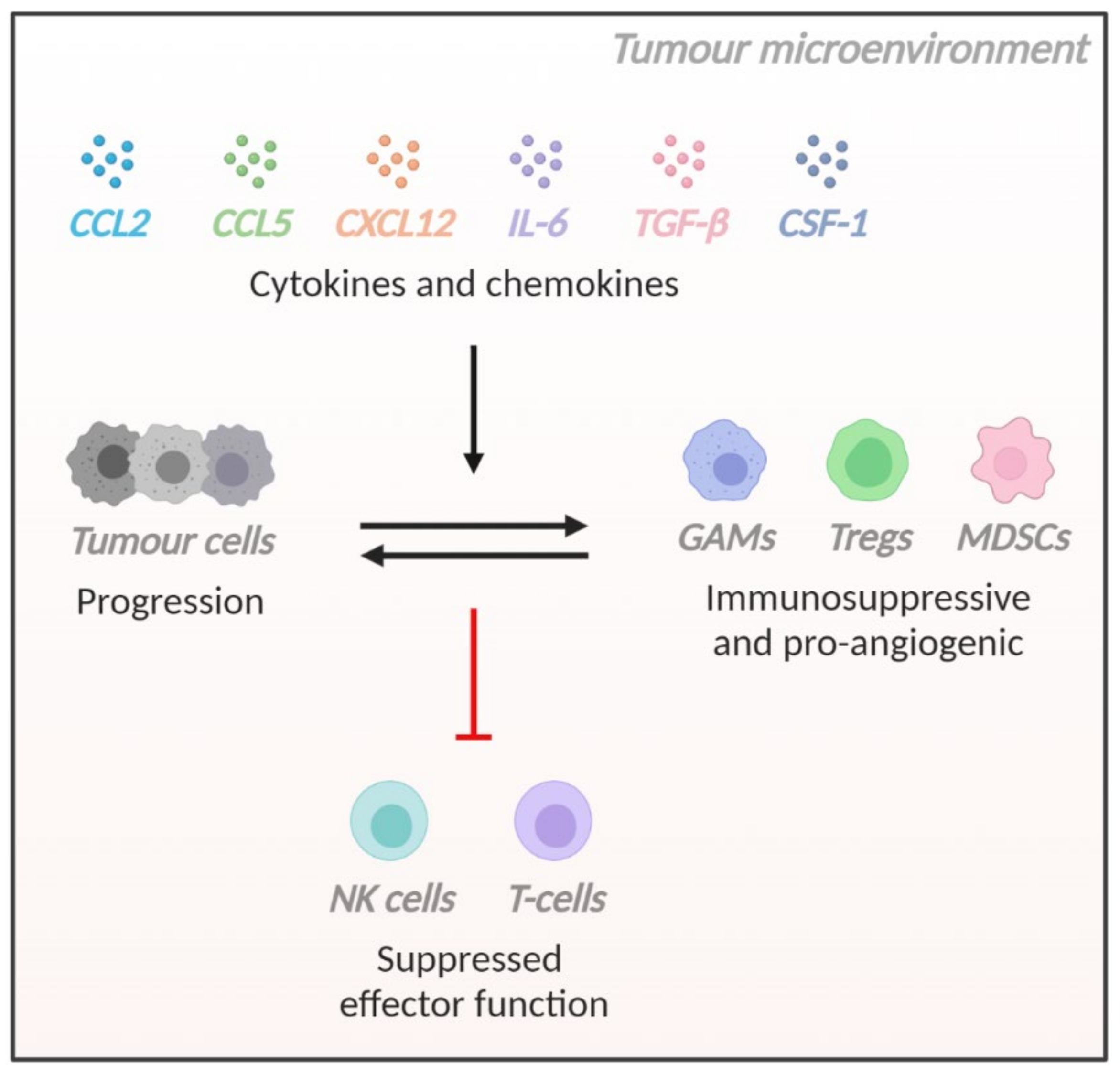
:
1. Introduction
2. The Glioblastoma TME
3. Cytokines and Chemokines That Regulate the Immune Microenvironment of Glioblastoma
3.1. Introduction
3.2. CCL2
3.3. CCL5
3.4. CXCL12
3.5. Interleukin-6 (IL-6)
3.6. Transforming Growth Factor-Beta (TGF-β)
3.7. Colony Stimulating Factor-1 (CSF-1)
3.8. Additional Soluble Factors with Immunomodulatory Roles in Glioblastoma
4. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar]
- Xu, H.; Chen, J.; Xu, H.; Qin, Z. Geographic variations in the incidence of glioblastoma and prognostic factors predictive of overall survival in us adults from 2004–2013. Front. Aging Neurosci. 2017, 9, 352. [Google Scholar] [CrossRef]
- Australian Institute of Health Welfare. Brain and other central nervous system cancers; AIHW: Canberra, Australia, 2017.
- Youlden, D.R.; Baade, P.D.; Valery, P.C.; Ward, L.J.; Green, A.C.; Aitken, J.F. Childhood cancer mortality in australia. Cancer Epidemiol. 2012, 36, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Field, K.M.; Simes, J.; Nowak, A.K.; Cher, L.; Wheeler, H.; Hovey, E.J.; Brown, C.S.; Barnes, E.H.; Sawkins, K.; Livingstone, A.; et al. Randomized phase 2 study of carboplatin and bevacizumab in recurrent glioblastoma. Neuro Oncol. 2015, 17, 1504–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, egfrviii-expressing glioblastoma (act iv): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Schuessler, A.; Smith, C.; Beagley, L.; Boyle, G.M.; Rehan, S.; Matthews, K.; Jones, L.; Crough, T.; Dasari, V.; Klein, K.; et al. Autologous t-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 2014, 74, 3466–3476. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and ccl3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro Oncol. 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: The checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase i cohorts of checkmate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Brown, M.P.; Ebert, L.M.; Gargett, T. Clinical chimeric antigen receptor-t cell therapy: A new and promising treatment modality for glioblastoma. Clin. Transl. Immunol. 2019, 8, e1050. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of glioblastoma after chimeric antigen receptor t-cell therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma heterogeneity and the tumour microenvironment: Implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Invest. 2017, 97, 498–518. [Google Scholar] [CrossRef] [Green Version]
- Ebert, L.M.; Yu, W.; Gargett, T.; Toubia, J.; Kollis, P.M.; Tea, M.N.; Ebert, B.W.; Bardy, C.; van den Hurk, M.; Bonder, C.S.; et al. Endothelial, pericyte and tumor cell expression in glioblastoma identifies fibroblast activation protein (fap) as an excellent target for immunotherapy. Clin. Transl. Immunol. 2020, 9, e1191. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desland, F.A.; Hormigo, A. The cns and the brain tumor microenvironment: Implications for glioblastoma immunotherapy. Int. J. Mol. Sci. 2020, 21, 7358. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Catalano, M.; D’Alessandro, G.; Trettel, F.; Limatola, C. Role of infiltrating microglia/macrophages in glioma. Adv. Exp. Med. Biol. 2020, 1202, 281–298. [Google Scholar] [PubMed]
- Gutmann, D.H.; Kettenmann, H. Microglia/brain macrophages as central drivers of brain tumor pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble m0 macrophage phenotype. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Zeiner, P.S.; Preusse, C.; Golebiewska, A.; Zinke, J.; Iriondo, A.; Muller, A.; Kaoma, T.; Filipski, K.; Muller-Eschner, M.; Bernatz, S.; et al. Distribution and prognostic impact of microglia/macrophage subpopulations in gliomas. Brain Pathol. 2019, 29, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.C.; Showers, C.R.; Anderson, D.E.; Anderson, L.; Canoll, P.; Bruce, J.N.; Anderson, R.C. Tumor-associated macrophages in glioma: Friend or foe? J. Oncol. 2013, 2013, 486912. [Google Scholar] [CrossRef] [PubMed]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial mt1-mmp expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the m2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, H.; Villeneuve, J.; Gowing, G.; Julien, J.P.; Vallières, L. Increased glioma growth in mice depleted of macrophages. Cancer Res. 2007, 67, 8874–8881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladomersky, E.; Zhai, L.; Lauing, K.L.; Bell, A.; Xu, J.; Kocherginsky, M.; Zhang, B.; Wu, J.D.; Podojil, J.R.; Platanias, L.C.; et al. Advanced age increases immunosuppression in the brain and decreases immunotherapeutic efficacy in subjects with glioblastoma. Clin. Cancer Res. 2020, 26, 5232–5245. [Google Scholar] [CrossRef]
- Zhai, L.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Sosman, J.A.; Zhang, B.; Wu, J.D.; Miller, S.D.; Meeks, J.J.; et al. Immunosuppressive ido in cancer: Mechanisms of action, animal models, and targeting strategies. Front. Immunol 2020, 11, 1185. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Huo, R.; Yan, Z.; Wang, J.; Wang, S.; Wang, J.; Chen, D.; et al. Single-cell atlas reveals complexity of the immunosuppressive microenvironment of initial and recurrent glioblastoma. Front. Immunol. 2020, 11, 835. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.C.; et al. Effector t-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived tgf-beta. Clin. Cancer Res. 2011, 17, 4296–4308. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [Green Version]
- Gulati, K.; Guhathakurta, S.; Joshi, J.; Rai, N.; Ray, A.J.M.I. Cytokines and their role in health and disease: A brief overview. MOJ Immunol. 2016, 4, 1–9. [Google Scholar]
- Stenken, J.A.; Poschenrieder, A.J. Bioanalytical chemistry of cytokines--a review. Analytica chimica acta 2015, 853, 95–115. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Constantin, G.; Majeed, M.; Giagulli, C.; Piccio, L.; Kim, J.Y.; Butcher, E.C.; Laudanna, C. Chemokines trigger immediate β2 integrin affinity and mobility changes: Differential regulation and roles in lymphocyte arrest under flow. Immunity 2000, 13, 759–769. [Google Scholar] [CrossRef] [Green Version]
- Zlotnik, A.; Yoshie, O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lupardus, P.; Laporte, S.L.; Garcia, K.C. Structural biology of shared cytokine receptors. Annu. Rev. Immunol. 2009, 27, 29–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nibbs, R.J.B.; Graham, G.J. Immune regulation by atypical chemokine receptors. Nature Reviews Immunology 2013, 13, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A. The chemokine system: Redundancy for robust outputs. Immunology Today 1999, 20, 254–257. [Google Scholar] [CrossRef]
- Yoshimura, T.; Robinson, E.A.; Tanaka, S.; Appella, E.; Kuratsu, J.; Leonard, E.J. Purification and amino acid analysis of two human glioma-derived monocyte chemoattractants. J. Exp. Med. 1989, 169, 1449–1459. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (mcp-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More than just attractive: How ccl2 influences myeloid cell behavior beyond chemotaxis. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Akhter, N.; Hasan, A.; Shenouda, S.; Wilson, A.; Kochumon, S.; Ali, S.; Tuomilehto, J.; Sindhu, S.; Ahmad, R. Tlr4/myd88 -mediated ccl2 production by lipopolysaccharide (endotoxin): Implications for metabolic inflammation. J. Diabetes Metab Disord 2018, 17, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Barna, B.P.; Pettay, J.; Barnett, G.H.; Zhou, P.; Iwasaki, K.; Estes, M.L. Regulation of monocyte chemoattractant protein-1 expression in adult human non-neoplastic astrocytes is sensitive to tumor necrosis factor (tnf) or antibody to the 55-kda tnf receptor. J. Neuroimmunol. 1994, 50, 101–107. [Google Scholar] [CrossRef]
- Cushing, S.D.; Berliner, J.A.; Valente, A.J.; Territo, M.C.; Navab, M.; Parhami, F.; Gerrity, R.; Schwartz, C.J.; Fogelman, A.M. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. USA 1990, 87, 5134–5138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, J.L.; Torroella-Kouri, M.; Handel-Fernandez, M.E.; Iragavarapu-Charyulu, V. Gm-csf up-regulates the expression of ccl2 by t lymphocytes in mammary tumor-bearing mice. Int J. Mol. Med. 2007, 20, 129–136. [Google Scholar] [CrossRef]
- Standiford, T.J.; Kunkel, S.L.; Phan, S.H.; Rollins, B.J.; Strieter, R.M. Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type ii-like epithelial cells. J. Biol. Chem. 1991, 266, 9912–9918. [Google Scholar] [CrossRef]
- Strieter, R.M.; Wiggins, R.; Phan, S.H.; Wharram, B.L.; Showell, H.J.; Remick, D.G.; Chensue, S.W.; Kunkel, S.L. Monocyte chemotactic protein gene expression by cytokine-treated human fibroblasts and endothelial cells. Biochem. Biophys. Res. Commun. 1989, 162, 694–700. [Google Scholar] [CrossRef] [Green Version]
- White, F.A.; Sun, J.; Waters, S.M.; Ma, C.; Ren, D.; Ripsch, M.; Steflik, J.; Cortright, D.N.; Lamotte, R.H.; Miller, R.J. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc. Natl. Acad. Sci. USA 2005, 102, 14092–14097. [Google Scholar] [CrossRef] [Green Version]
- Rollins, B.J.; Pober, J.S. Interleukin-4 induces the synthesis and secretion of mcp-1/je by human endothelial cells. Am. J. Pathol. 1991, 138, 1315–1319. [Google Scholar]
- Schmouder, R.L.; Strieter, R.M.; Kunkel, S.L. Interferon-γ regulation of human renal cortical epithelial cell-derived monocyte chemotactic peptide-1. Kidney Int. 1993, 44, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.F.; Valente, A.J.; Lorenzo, J.A.; Carnes, D.; Graves, D.T. Expression of monocyte chemoattractant protein 1 in human osteoblastic cells stimulated by proinflammatory mediators. J. Bone Miner. Res. 1994, 9, 1123–1130. [Google Scholar] [CrossRef]
- Allavena, P.; Bianchi, G.; Zhou, D.; van Damme, J.; Jílek, P.; Sozzani, S.; Mantovani, A. Induction of natural killer cell migration by monocyte chemotactic protein-1, -2 and -3. Eur. J. Immunol. 1994, 24, 3233–3236. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.W.; Roth, S.J.; Luther, E.; Rose, S.S.; Springer, T.A. Monocyte chemoattractant protein 1 acts as a t-lymphocyte chemoattractant. Proceedings of the National Academy of Sciences of the United States of America 1994, 91, 3652–3656. [Google Scholar] [CrossRef] [Green Version]
- Frade, J.M.; Mellado, M.; del Real, G.; Gutierrez-Ramos, J.C.; Lind, P.; Martinez, A.C. Characterization of the ccr2 chemokine receptor: Functional ccr2 receptor expression in b cells. J. Immunol. 1997, 159, 5576–5584. [Google Scholar] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory t cells in tumor immunity. Int J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef]
- Yoshie, O.; Matsushima, K. Ccr4 and its ligands: From bench to bedside. Int. Immunol. 2015, 27, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. Ccl2 produced by the glioma microenvironment is essential for the recruitment of regulatory t cells and myeloid-derived suppressor cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Fujita, M.; Snyder, L.A.; Okada, H. Systemic delivery of neutralizing antibody targeting ccl2 for glioma therapy. J. Neurooncol. 2011, 104, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.; Ahn, S.H.; Park, H.; Park, S.H.; Choi, K.; Choi, C.; Kang, J.L.; Choi, Y.H. Mcp-1 and mip-3α secreted from necrotic cell-treated glioblastoma cells promote migration/infiltration of microglia. Cell Physiol. Biochem. 2018, 48, 1332–1346. [Google Scholar] [CrossRef]
- Jordan, J.T.; Sun, W.; Hussain, S.F.; DeAngulo, G.; Prabhu, S.S.; Heimberger, A.B. Preferential migration of regulatory t cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol. Immunother. 2008, 57, 123–131. [Google Scholar] [CrossRef]
- Platten, M.; Kretz, A.; Naumann, U.; Aulwurm, S.; Egashira, K.; Isenmann, S.; Weller, M. Monocyte chemoattractant protein–1 increases microglial infiltration and aggressiveness of gliomas. Ann. Neurol. 2003, 54, 388–392. [Google Scholar] [CrossRef]
- Felsenstein, M.; Blank, A.; Bungert, A.D.; Mueller, A.; Ghori, A.; Kremenetskaia, I.; Rung, O.; Broggini, T.; Turkowski, K.; Scherschinski, L.; et al. Ccr2 of tumor microenvironmental cells is a relevant modulator of glioma biology. Cancers 2020, 12, 1882. [Google Scholar] [CrossRef]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. Ccr2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef]
- Aldinucci, D.; Colombatti, A. The inflammatory chemokine ccl5 and cancer progression. Mediators Inflamm. 2014, 2014, 292376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kameyoshi, Y.; Dörschner, A.; Mallet, A.I.; Christophers, E.; Schröder, J.M. Cytokine rantes released by thrombin-stimulated platelets is a potent attractant for human eosinophils. J. Exp. Med. 1992, 176, 587–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Deskin, R.W.; Casola, A.; Haeberle, H.; Olszewska, B.; Ernst, P.B.; Alam, R.; Ogra, P.L.; Garofalo, R. Respiratory syncytial virus induces selective production of the chemokine rantes by upper airway epithelial cells. J. Infect. Dis 1997, 175, 497–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, W.; Shimizu, K.; Kojo, S.; Okeke, A.; Kohwi-Shigematsu, T.; Fujii, S.-i.; Taniuchi, I. Runx-mediated regulation of ccl5 via antagonizing two enhancers influences immune cell function and anti-tumor immunity. Nat. Commun. 2020, 11, 1562. [Google Scholar] [CrossRef] [PubMed]
- Dieu, M.C.; Vanbervliet, B.; Vicari, A.; Bridon, J.M.; Oldham, E.; Aït-Yahia, S.; Brière, F.; Zlotnik, A.; Lebecque, S.; Caux, C. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 1998, 188, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Rot, A.; Krieger, M.; Brunner, T.; Bischoff, S.C.; Schall, T.J.; Dahinden, C.A. Rantes and macrophage inflammatory protein 1 alpha induce the migration and activation of normal human eosinophil granulocytes. J. Exp. Med. 1992, 176, 1489–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schall, T.J.; Bacon, K.; Toy, K.J.; Goeddel, D.V. Selective attraction of monocytes and t lymphocytes of the memory phenotype by cytokine rantes. Nature 1990, 347, 669–671. [Google Scholar] [CrossRef]
- Appay, V.; Rowland-Jones, S.L. Rantes: A versatile and controversial chemokine. Trends Immunol. 2001, 22, 83–87. [Google Scholar] [CrossRef]
- Roscic-Mrkic, B.; Fischer, M.; Leemann, C.; Manrique, A.; Gordon, C.J.; Moore, J.P.; Proudfoot, A.E.I.; Trkola, A. Rantes (ccl5) uses the proteoglycan cd44 as an auxiliary receptor to mediate cellular activation signals and hiv-1 enhancement. Blood 2003, 102, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, L.; Dang, W.-q.; Cao, M.-f.; Xiao, J.-f.; Lv, S.-q.; Jiang, W.-j.; Yao, X.-h.; Lu, H.-m.; Miao, J.-y.; et al. Ccl8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via erk1/2 signaling. Lab. Invest. 2020, 100, 619–629. [Google Scholar] [CrossRef]
- Dragic, T.; Litwin, V.; Allaway, G.P.; Martin, S.R.; Huang, Y.; Nagashima, K.A.; Cayanan, C.; Maddon, P.J.; Koup, R.A.; Moore, J.P.; et al. Hiv-1 entry into cd4+ cells is mediated by the chemokine receptor cc-ckr-5. Nature 1996, 381, 667–673. [Google Scholar] [CrossRef]
- Pan, Y.; Smithson, L.J.; Ma, Y.; Hambardzumyan, D.; Gutmann, D.H. Ccl5 establishes an autocrine high-grade glioma growth regulatory circuit critical for mesenchymal glioblastoma survival. Oncotarget 2017, 8, 32977–32989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu-Ju Wu, C.; Chen, C.H.; Lin, C.Y.; Feng, L.Y.; Lin, Y.C.; Wei, K.C.; Huang, C.Y.; Fang, J.Y.; Chen, P.Y. Ccl5 of glioma-associated microglia/macrophages regulates glioma migration and invasion via calcium-dependent matrix metalloproteinase 2. Neuro Oncol. 2020, 22, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.; Koprivnikar Krajnc, M.; Hrastar, B.; Breznik, B.; Majc, B.; Mlinar, M.; Rotter, A.; Porčnik, A.; Mlakar, J.; Stare, K.; et al. Ccr5-mediated signaling is involved in invasion of glioblastoma cells in its microenvironment. Int J. Mol. Sci 2020, 21, 4199. [Google Scholar] [CrossRef] [PubMed]
- Pham, K.; Luo, D.; Liu, C.; Harrison, J.K. Ccl5, ccr1 and ccr5 in murine glioblastoma: Immune cell infiltration and survival rates are not dependent on individual expression of either ccr1 or ccr5. J. Neuroimmunol. 2012, 246, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Yi, L.; Xiao, H.; Xu, M.; Ye, X.; Hu, J.; Li, F.; Li, M.; Luo, C.; Yu, S.; Bian, X.; et al. Glioma-initiating cells: A predominant role in microglia/macrophages tropism to glioma. J. Neuroimmunol. 2011, 232, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Y.; Xue, Y.; Lv, W.; Zhang, Y.; He, S. Critical roles of chemokine receptor ccr5 in regulating glioblastoma proliferation and invasion. Acta Biochim. Biophys. Sin. 2015, 47, 890–898. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, T.; Yang, N.; Xu, S.; Li, X.; Wang, D. Hypoxia and macrophages promote glioblastoma invasion by the ccl4-ccr5 axis. Oncol. Rep. 2016, 36, 3522–3528. [Google Scholar] [CrossRef]
- Laudati, E.; Currò, D.; Navarra, P.; Lisi, L. Blockade of ccr5 receptor prevents m2 microglia phenotype in a microglia-glioma paradigm. Neurochem. Int. 2017, 108, 100–108. [Google Scholar] [CrossRef]
- Koul, D.; Fu, J.; Shen, R.; LaFortune, T.A.; Wang, S.; Tiao, N.; Kim, Y.-W.; Liu, J.-L.; Ramnarian, D.; Yuan, Y.; et al. Antitumor activity of nvp-bkm120—a selective pan class i pi3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin. Cancer Res. 2012, 18, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revach, O.-Y.; Geiger, B. The interplay between the proteolytic, invasive, and adhesive domains of invadopodia and their roles in cancer invasion. Cell Adh. Migr. 2014, 8, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Kahn, J.; Hayman, T.J.; Jamal, M.; Rath, B.H.; Kramp, T.; Camphausen, K.; Tofilon, P.J. The mtorc1/mtorc2 inhibitor azd2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro Oncol. 2014, 16, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecca, C.; Giambanco, I.; Bruscoli, S.; Bereshchenko, O.; Fioretti, B.; Riccardi, C.; Donato, R.; Arcuri, C. Pp242 counteracts glioblastoma cell proliferation, migration, invasiveness and stemness properties by inhibiting mtorc2/akt. Front. Cell Neurosci. 2018, 12, 99. [Google Scholar] [CrossRef]
- Nagasawa, T.; Hirota, S.; Tachibana, K.; Takakura, N.; Nishikawa, S.-i.; Kitamura, Y.; Yoshida, N.; Kikutani, H.; Kishimoto, T. Defects of b-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the cxc chemokine pbsf/sdf-1. Nature 1996, 382, 635–638. [Google Scholar] [CrossRef]
- Bai, Z.; Hayasaka, H.; Kobayashi, M.; Li, W.; Guo, Z.; Jang, M.H.; Kondo, A.; Choi, B.-i.; Iwakura, Y.; Miyasaka, M. Cxc chemokine ligand 12 promotes ccr7-dependent naive t cell trafficking to lymph nodes and peyer’s patches. J. Immunol. 2009, 182, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.; Hao, D.; Chai, J. Processing of cxcl12 impedes the recruitment of endothelial progenitor cells in diabetic wound healing. FEBS J. 2014, 281, 5054–5062. [Google Scholar] [CrossRef] [PubMed]
- García-Cuesta, E.M.; Santiago, C.A.; Vallejo-Díaz, J.; Juarranz, Y.; Rodríguez-Frade, J.M.; Mellado, M. The role of the cxcl12/cxcr4/ackr3 axis in autoimmune diseases. Front. Endocrinol. (Lausanne) 2019, 10, 585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtani, Y.; Minami, M.; Kawaguchi, N.; Nishiyori, A.; Yamamoto, J.; Takami, S.; Satoh, M. Expression of stromal cell-derived factor-1 and cxcr4 chemokine receptor mrnas in cultured rat glial and neuronal cells. Neurosci. Lett. 1998, 249, 163–166. [Google Scholar] [CrossRef]
- Santiago, B.; Calonge, E.; Rey, M.J.D.; Gutierrez-Cañas, I.; Izquierdo, E.; Usategui, A.; Galindo, M.; Alcamí, J.; Pablos, J.L. Cxcl12 gene expression is upregulated by hypoxia and growth arrest but not by inflammatory cytokines in rheumatoid synovial fibroblasts. Cytokine 2011, 53, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Infantino, S.; Moepps, B.; Thelen, M. Expression and regulation of the orphan receptor rdc1 and its putative ligand in human dendritic and b cells. J. Immunol. 2006, 176, 2197–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schutyser, E.; Su, Y.; Yu, Y.; Gouwy, M.; Zaja-Milatovic, S.; Van Damme, J.; Richmond, A. Hypoxia enhances cxcr4 expression in human microvascular endothelial cells and human melanoma cells. Eur. Cytokine Netw. 2007, 18, 59–70. [Google Scholar] [PubMed]
- Rizzo, P.; Perico, N.; Gagliardini, E.; Novelli, R.; Alison, M.R.; Remuzzi, G.; Benigni, A. Nature and mediators of parietal epithelial cell activation in glomerulonephritides of human and rat. Am. J. Pathol. 2013, 183, 1769–1778. [Google Scholar] [CrossRef]
- Janssens, R.; Struyf, S.; Proost, P. The unique structural and functional features of cxcl12. Cell Mol. Immunol. 2018, 15, 299–311. [Google Scholar] [CrossRef]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Β-arrestin- but not g protein-mediated signaling by the “decoy” receptor cxcr7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Naumann, U.; Cameroni, E.; Pruenster, M.; Mahabaleshwar, H.; Raz, E.; Zerwes, H.G.; Rot, A.; Thelen, M. Cxcr7 functions as a scavenger for cxcl12 and cxcl11. PLoS One 2010, 5, e9175. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. Pillars article: Hiv-1 entry cofactor: Functional cdna cloning of a seven-transmembrane, g protein-coupled receptor. J. Immunol. 2011, 186, 6076–6081. [Google Scholar]
- D’Huys, T.; Claes, S.; Van Loy, T.; Schols, D. Cxcr7/ackr3-targeting ligands interfere with x7 hiv-1 and hiv-2 entry and replication in human host cells. Heliyon 2018, 4, e00557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattermann, K.; Mentlein, R.; Held-Feindt, J. Cxcl12 mediates apoptosis resistance in rat c6 glioma cells. Oncol. Rep. 2012, 27, 1348–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; VandenBerg, S.; Johnson, R.S.; Werb, Z.; et al. Hif1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Walters, M.J.; Ebsworth, K.; Berahovich, R.D.; Penfold, M.E.T.; Liu, S.C.; Al Omran, R.; Kioi, M.; Chernikova, S.B.; Tseng, D.; Mulkearns-Hubert, E.E.; et al. Inhibition of cxcr7 extends survival following irradiation of brain tumours in mice and rats. Br. J. Cancer 2014, 110, 1179–1188. [Google Scholar] [CrossRef]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Invest. 2010, 120, 694–705. [Google Scholar] [CrossRef]
- Hattermann, K.; Held-Feindt, J.; Lucius, R.; Müerköster, S.S.; Penfold, M.E.T.; Schall, T.J.; Mentlein, R. The chemokine receptor cxcr7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res. 2010, 70, 3299–3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.; Sengupta, R.; Choe, E.J.; Woerner, B.M.; Jackson, E.; Sun, T.; Leonard, J.; Piwnica-Worms, D.; Rubin, J.B. Cxcl12 mediates trophic interactions between endothelial and tumor cells in glioblastoma. PLoS ONE 2012, 7, e33005. [Google Scholar] [CrossRef] [Green Version]
- Ehtesham, M.; Mapara, K.Y.; Stevenson, C.B.; Thompson, R.C. Cxcr4 mediates the proliferation of glioblastoma progenitor cells. Cancer Lett. 2009, 274, 305–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatti, M.; Pattarozzi, A.; Bajetto, A.; Würth, R.; Daga, A.; Fiaschi, P.; Zona, G.; Florio, T.; Barbieri, F. Inhibition of cxcl12/cxcr4 autocrine/paracrine loop reduces viability of human glioblastoma stem-like cells affecting self-renewal activity. Toxicology 2013, 314, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Verbovšek, U.; Breznik, B.; Srdič, M.; Novinec, M.; Kakar, H.; Wormer, J.; der Swaan, B.V.; Lenarčič, B.; Juliano, L.; et al. Cathepsin k cleavage of sdf-1α inhibits its chemotactic activity towards glioblastoma stem-like cells. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 594–603. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Vitale, F.; Vetuschi, A.; Pompili, S.; Rossi, G.; Marampon, F.; Richardson, P.J.; Patient, L.; et al. The novel cxcr4 antagonist, prx177561, reduces tumor cell proliferation and accelerates cancer stem cell differentiation in glioblastoma preclinical models. Tumour Biol. 2017, 39, 1010428317695528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, Y.-f.; Yao, X.-h.; Jiang, J.-y.; Zhao, L.-t.; Yu, S.-c.; Jiang, T.; Lin, M.C.; Chen, J.-h.; Wang, B.; Zhang, R.; et al. The chemokine cxcl12 and its receptor cxcr4 promote glioma stem cell-mediated vegf production and tumour angiogenesis via pi3k/akt signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through hif-1 induction of sdf-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef]
- Liu, S.C.; Alomran, R.; Chernikova, S.B.; Lartey, F.; Stafford, J.; Jang, T.; Merchant, M.; Zboralski, D.; Zöllner, S.; Kruschinski, A.; et al. Blockade of sdf-1 after irradiation inhibits tumor recurrences of autochthonous brain tumors in rats. Neuro Oncol. 2014, 16, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, L.; Ajmone-Cat, M.A.; Cecchetti, S.; Ricci, A.; Bozzuto, G.; Molinari, A.; Manni, I.; Pollo, B.; Scala, S.; Carpinelli, G.; et al. Targeting cxcr4 by a selective peptide antagonist modulates tumor microenvironment and microglia reactivity in a human glioblastoma model. J. Exp. Clin. Cancer Res. 2016, 35. [Google Scholar] [CrossRef]
- Barbero, S.; Bonavia, R.; Bajetto, A.; Porcile, C.; Pirani, P.; Ravetti, J.L.; Zona, G.L.; Spaziante, R.; Florio, T.; Schettini, G. Stromal cell-derived factor 1α stimulates human glioblastoma cell growth through the activation of both extracellular signal-regulated kinases 1/2 and akt. Cancer Res. 2003, 63, 1969–1974. [Google Scholar] [PubMed]
- Zhang, J.; Sarkar, S.; Yong, V.W. The chemokine stromal cell derived factor-1 (cxcl12) promotes glioma invasiveness through mt2-matrix metalloproteinase. Carcinogenesis 2005, 26, 2069–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Taga, T.; Kishimoto, T. Interleukin-6 in biology and medicine. In Advances in Immunology; Dixon, F.J., Ed.; Academic Press: Cambridge, MA, USA, 1993; Volume 54, pp. 1–78. [Google Scholar]
- Weissenbach, J.; Chernajovsky, Y.; Zeevi, M.; Shulman, L.; Soreq, H.; Nir, U.; Wallach, D.; Perricaudet, M.; Tiollais, P.; Revel, M. Two interferon mrnas in human fibroblasts: In vitro translation and escherichia coli cloning studies. Proc. Natl. Acad. Sci. USA 1980, 77, 7152–7156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, V.; Montero-Julian, F.A.; Grès, S.; Boulay, V.; Bongrand, P.; Farnarier, C.; Kaplanski, G. The il-6-soluble il-6rα autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: An experimental model involving thrombin. J. Immunol. 2001, 167, 3435–3442. [Google Scholar] [CrossRef] [Green Version]
- Sancéau, J.; Falcoff, R.; Zilberstein, A.; Béranger, F.; Lebeau, J.; Revel, M.; Vaquero, C. Interferon-beta 2 (bsf-2) mrna is expressed in human monocytes. J. Interferon Res. 1988, 8, 473–481. [Google Scholar] [CrossRef]
- Zimmermann, M.; Arruda-Silva, F.; Bianchetto-Aguilera, F.; Finotti, G.; Calzetti, F.; Scapini, P.; Lunardi, C.; Cassatella, M.A.; Tamassia, N. Ifnα enhances the production of il-6 by human neutrophils activated via tlr8. Sci Rep. 2016, 6, 19674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Snick, J.; Cayphas, S.; Vink, A.; Uyttenhove, C.; Coulie, P.G.; Rubira, M.R.; Simpson, R.J. Purification and nh2-terminal amino acid sequence of a t-cell-derived lymphokine with growth factor activity for b-cell hybridomas. Proc. Natl. Acad. Sci. USA 1986, 83, 9679–9683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeland, E.B.; Blomhoff, H.K.; Funderud, S.; Shalaby, M.R.; Espevik, T. Interleukin 4 induces selective production of interleukin 6 from normal human b lymphocytes. J. Exp. Med. 1989, 170, 1463–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopf, M.; Baumann, H.; Freer, G.; Freudenberg, M.; Lamers, M.; Kishimoto, T.; Zinkernagel, R.; Bluethmann, H.; Köhler, G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 1994, 368, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Hösel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis b virus infection. Hepatology 2009, 50, 1773–1782. [Google Scholar]
- Smith, K.A.; Maizels, R.M. Il-6 controls susceptibility to helminth infection by impeding th2 responsiveness and altering the treg phenotype in vivo. Eur. J. Immunol. 2014, 44, 150–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romani, L.; Mencacci, A.; Cenci, E.; Spaccapelo, R.; Toniatti, C.; Puccetti, P.; Bistoni, F.; Poli, V. Impaired neutrophil response and cd4+ t helper cell 1 development in interleukin 6-deficient mice infected with candida albicans. J. Exp. Med. 1996, 183, 1345–1355. [Google Scholar] [CrossRef]
- Hurst, S.M.; Wilkinson, T.S.; McLoughlin, R.M.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Mitani, H.; Katayama, N.; Araki, H.; Ohishi, K.; Kobayashi, K.; Suzuki, H.; Nishii, K.; Masuya, M.; Yasukawa, K.; Minami, N.; et al. Activity of interleukin 6 in the differentiation of monocytes to macrophages and dendritic cells. Br. J. Haematol. 2000, 109, 288–295. [Google Scholar] [CrossRef]
- Yang, R.; Masters, A.R.; Fortner, K.A.; Champagne, D.P.; Yanguas-Casás, N.; Silberger, D.J.; Weaver, C.T.; Haynes, L.; Rincon, M. Il-6 promotes the differentiation of a subset of naive cd8+ t cells into il-21-producing b helper cd8+ t cells. J. Exp. Med. 2016, 213, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, M.; Hayakawa, N.; Suzuki, M.; Mihara, M. Il-6/sil-6r trans-signalling, but not tnf-alpha induced angiogenesis in a huvec and synovial cell co-culture system. Rheumatol. Int. 2009, 29, 1449–1454. [Google Scholar] [CrossRef]
- Geisterfer, M.; Richards, C.; Baumann, M.; Fey, G.; Gywnne, D.; Gauldie, J. Regulation of il-6 and the hepatic il-6 receptor in acute inflammation in vivo. Cytokine 1993, 5, 1–7. [Google Scholar] [CrossRef]
- Farahi, N.; Paige, E.; Balla, J.; Prudence, E.; Ferreira, R.C.; Southwood, M.; Appleby, S.L.; Bakke, P.; Gulsvik, A.; Litonjua, A.A.; et al. Neutrophil-mediated il-6 receptor trans-signaling and the risk of chronic obstructive pulmonary disease and asthma. Hum. Mol. Genet. 2017, 26, 1584–1596. [Google Scholar] [CrossRef]
- Oberg, H.-H.; Wesch, D.; Grüssel, S.; Rose-John, S.; Kabelitz, D. Differential expression of cd126 and cd130 mediates different stat-3 phosphorylation in cd4+cd25− and cd25high regulatory t cells. Int. Immunol. 2006, 18, 555–563. [Google Scholar] [CrossRef]
- Rose-John, S.; Winthrop, K.; Calabrese, L. The role of il-6 in host defence against infections: Immunobiology and clinical implications. Nat. Rev. Rheumatol. 2017, 13, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Barkhausen, T.; Tschernig, T.; Rosenstiel, P.; van Griensven, M.; Vonberg, R.-P.; Dorsch, M.; Mueller-Heine, A.; Chalaris, A.; Scheller, J.; Rose-John, S.; et al. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Crit. Care Med. 2011, 39. [Google Scholar] [CrossRef]
- Lissilaa, R.; Buatois, V.; Magistrelli, G.; Williams, A.S.; Jones, G.W.; Herren, S.; Shang, L.; Malinge, P.; Guilhot, F.; Chatel, L.; et al. Although il-6 trans-signaling is sufficient to drive local immune responses, classical il-6 signaling is obligate for the induction of t cell-mediated autoimmunity. J. Immunol. 2010, 1002015. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; He, Z.; Huang, M.; Liu, T.; Wang, Y.; Xu, H.; Duan, H.; Ma, P.; Zhang, L.; Zamvil, S.S.; et al. Vascular niche il-6 induces alternative macrophage activation in glioblastoma through hif-2α. Nat. Commun. 2018, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Xia, T.; Wang, D.; Huang, B.; Zhao, P.; Wang, J.; Qu, X.; Li, X. Human astrocytes secrete il-6 to promote glioma migration and invasion through upregulation of cytomembrane mmp14. Oncotarget 2016, 7, 62425–62438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, T.; Sasayama, T.; Tanaka, K.; Koma, Y.I.; Nishihara, M.; Tanaka, H.; Nakamizo, S.; Nagashima, H.; Maeyama, M.; Fujita, Y.; et al. Tumor-associated macrophage related interleukin-6 in cerebrospinal fluid as a prognostic marker for glioblastoma. J. Clin. Neurosci. 2019, 68, 281–289. [Google Scholar] [CrossRef]
- Lamano, J.B.; Lamano, J.B.; Li, Y.D.; DiDomenico, J.D.; Choy, W.; Veliceasa, D.; Oyon, D.E.; Fakurnejad, S.; Ampie, L.; Kesavabhotla, K.; et al. Glioblastoma-derived il6 induces immunosuppressive peripheral myeloid cell pd-l1 and promotes tumor growth. Clin. Cancer Res. 2019, 25, 3643–3657. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-Y.; Li, M.-C.; Liao, S.-L.; Huang, Y.-L.; Shen, C.-C.; Pan, H.-C. Prognostic and clinical implication of il-6 expression in glioblastoma multiforme. J. Clin. Neurosci. 2005, 12, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Lin, L.-T.; Wang, M.-L.; Lee, S.-H.; Tsai, M.-L.; Tsai, C.-C.; Liu, W.-H.; Chen, T.-C.; Yang, Y.-P.; Lee, Y.-Y.; et al. Musashi-1 regulates akt-derived il-6 autocrinal/paracrinal malignancy and chemoresistance in glioblastoma. Oncotarget 2016, 7, 42485–42501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasi, F.; Facoetti, A.; Nano, R. Il-8 and il-6 bystander signalling in human glioblastoma cells exposed to gamma radiation. Anticancer Res. 2010, 30, 2769–2772. [Google Scholar]
- Xue, H.; Yuan, G.; Guo, X.; Liu, Q.; Zhang, J.; Gao, X.; Guo, X.; Xu, S.; Li, T.; Shao, Q.; et al. A novel tumor-promoting mechanism of il6 and the therapeutic efficacy of tocilizumab: Hypoxia-induced il6 is a potent autophagy initiator in glioblastoma via the p-stat3-mir155-3p-crebrf pathway. Autophagy 2016, 12, 1129–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; Eyler, C.E.; Elderbroom, J.; Gallagher, J.; Schuschu, J.; et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Ni, Z.; Liu, X.; Feng, S.; Dong, X.; Shi, X.; Zhai, J.; Mai, S.; Jiang, J.; Wang, Z.; et al. Beyond t cells: Understanding the role of pd-1/pd-l1 in tumor-associated macrophages. J. Immunol. Res. 2019, 2019, 1919082. [Google Scholar] [CrossRef] [Green Version]
- Sica, G.L.; Choi, I.-H.; Zhu, G.; Tamada, K.; Wang, S.-D.; Tamura, H.; Chapoval, A.I.; Flies, D.B.; Bajorath, J.; Chen, L. B7-h4, a molecule of the b7 family, negatively regulates t cell immunity. Immunity 2003, 18, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by cb-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [Green Version]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.-M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta 2009, 1793, 1524–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubost, J.-J.; Rolhion, C.; Tchirkov, A.; Bertrand, S.; Chassagne, J.; Dosgilbert, A.; Verrelle, P. Interleukin-6-producing cells in a human glioblastoma cell line are not affected by ionizing radiation. J. Neurooncol. 2002, 56, 29–34. [Google Scholar] [CrossRef]
- Tamari, Y.; Kashino, G.; Mori, H. Acquisition of radioresistance by il-6 treatment is caused by suppression of oxidative stress derived from mitochondria after γ-irradiation. J. Radiat. Res. 2017, 58, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Li, G.; Li, R.; Shen, J.; He, Q.; Deng, L.; Zhang, C.; Zhang, J. Il-6 promotion of glioblastoma cell invasion and angiogenesis in u251 and t98g cell lines. J. Neurooncol. 2010, 100, 165–176. [Google Scholar] [CrossRef]
- Li, R.; Li, G.; Deng, L.; Liu, Q.; Dai, J.; Shen, J.; Zhang, J. Il-6 augments the invasiveness of u87mg human glioblastoma multiforme cells via up-regulation of mmp-2 and fascin-1. Oncol. Rep. 2010, 23, 1553–1559. [Google Scholar] [CrossRef] [Green Version]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Tgf-β—an excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef] [Green Version]
- Branton, M.H.; Kopp, J.B. Tgf-beta and fibrosis. Microbes Infect. 1999, 1, 1349–1365. [Google Scholar] [CrossRef]
- Frei, K.; Gramatzki, D.; Tritschler, I.; Schroeder, J.J.; Espinoza, L.; Rushing, E.J.; Weller, M. Transforming growth factor-β pathway activity in glioblastoma. Oncotarget 2015, 6, 5963–5977. [Google Scholar] [CrossRef] [Green Version]
- Roy, L.-O.; Poirier, M.-B.; Fortin, D. Differential expression and clinical significance of transforming growth factor-beta isoforms in gbm tumors. Int J. Mol. Sci 2018, 19, 1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caja, L.; Bellomo, C.; Moustakas, A. Transforming growth factor β and bone morphogenetic protein actions in brain tumors. FEBS Lett 2015, 589, 1588–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Yi, L.; Wang, X.; Zhou, C.; Xu, L. Interleukin-17 facilitates the immune suppressor capacity of high-grade glioma-derived cd4 (+) cd25 (+) foxp3 (+) t cells via releasing transforming growth factor beta. Scand. J. Immunol. 2014, 80, 144–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhl, M.; Aulwurm, S.; Wischhusen, J.; Weiler, M.; Ma, J.Y.; Almirez, R.; Mangadu, R.; Liu, Y.-W.; Platten, M.; Herrlinger, U.; et al. Sd-208, a novel transforming growth factor beta receptor i kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004, 64, 7954–7961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. Nkg2d and its ligands: “One for all, all for one”. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Friese, M.A.; Wischhusen, J.; Wick, W.; Weiler, M.; Eisele, G.; Steinle, A.; Weller, M. Rna interference targeting transforming growth factor-beta enhances nkg2d-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004, 64, 7596–7603. [Google Scholar] [CrossRef] [Green Version]
- Alter, G.; Malenfant, J.M.; Altfeld, M. Cd107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef]
- Betts, M.R.; Brenchley, J.M.; Price, D.A.; De Rosa, S.C.; Douek, D.C.; Roederer, M.; Koup, R.A. Sensitive and viable identification of antigen-specific cd8+ t cells by a flow cytometric assay for degranulation. J. Immunol. Methods 2003, 281, 65–78. [Google Scholar] [CrossRef]
- Tran, T.-T.; Uhl, M.; Ma, J.Y.; Janssen, L.; Sriram, V.; Aulwurm, S.; Kerr, I.; Lam, A.; Webb, H.K.; Kapoun, A.M.; et al. Inhibiting tgf-β signaling restores immune surveillance in the sma-560 glioma model. Neuro Oncol. 2007, 9, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocaña, A.; Peñuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High tgfβ-smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the pdgf-b gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziembowska, M.; Danilkiewicz, M.; Wesolowska, A.; Zupanska, A.; Chouaib, S.; Kaminska, B. Cross-talk between smad and p38 mapk signalling in transforming growth factor β signal transduction in human glioblastoma cells. Biochem. Biophys. Res. Commun. 2007, 354, 1101–1106. [Google Scholar] [CrossRef]
- Hjelmeland, M.D.; Hjelmeland, A.B.; Sathornsumetee, S.; Reese, E.D.; Herbstreith, M.H.; Laping, N.J.; Friedman, H.S.; Bigner, D.D.; Wang, X.-F.; Rich, J.N. Sb-431542, a small molecule transforming growth factor-β-receptor antagonist, inhibits human glioma cell line proliferation and motility. Mol. Cancer Ther. 2004, 3, 737–745. [Google Scholar]
- Nickl-Jockschat, T.; Arslan, F.; Doerfelt, A.; Bogdahn, U.; Bosserhoff, A.; Hau, P. An imbalance between smad and mapk pathways is responsible for tgf-β tumor promoting effects in high-grade gliomas. Int J. Oncol. 2007, 30, 499–507. [Google Scholar]
- Arslan, F.; Bosserhoff, A.K.; Nickl-Jockschat, T.; Doerfelt, A.; Bogdahn, U.; Hau, P. The role of versican isoforms v0/v1 in glioma migration mediated by transforming growth factor-beta2. Br. J. Cancer 2007, 96, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Baumann, F.; Leukel, P.; Doerfelt, A.; Beier, C.P.; Dettmer, K.; Oefner, P.J.; Kastenberger, M.; Kreutz, M.; Nickl-Jockschat, T.; Bogdahn, U.; et al. Lactate promotes glioma migration by tgf-beta2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol. 2009, 11, 368–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.-z.; Xu, S.-l.; Xin, Y.-h.; Yu, S.-c.; Ping, Y.-f.; Chen, L.; Xiao, H.-l.; Wang, B.; Yi, L.; Wang, Q.-l.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via tgf-β1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Hardee, M.E.; Marciscano, A.E.; Medina-Ramirez, C.M.; Zagzag, D.; Narayana, A.; Lonning, S.M.; Barcellos-Hoff, M.H. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-β. Cancer Res. 2012, 72, 4119–4129. [Google Scholar] [CrossRef] [Green Version]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine tgf-beta signaling maintains tumorigenicity of glioma-initiating cells through sry-related hmg-box factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef] [Green Version]
- Peñuelas, S.; Anido, J.; Prieto-Sánchez, R.M.; Folch, G.; Barba, I.; Cuartas, I.; García-Dorado, D.; Poca, M.A.; Sahuquillo, J.; Baselga, J.; et al. Tgf-β increases glioma-initiating cell self-renewal through the induction of lif in human glioblastoma. Cancer Cell 2009, 15, 315–327. [Google Scholar]
- Anido, J.; Sáez-Borderías, A.; Gonzàlez-Juncà, A.; Rodón, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martínez-Sáez, E.; Prudkin, L.; et al. Tgf-β receptor inhibitors target the cd44high/id1high glioma-initiating cell population in human glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitu, V.; Stanley, E.R. Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol. 2006, 18, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Borrello, M.A.; Phipps, R.P. Fibroblast-secreted macrophage colony-stimulating factor is responsible for generation of biphenotypic b/macrophage cells from a subset of mouse b lymphocytes. J. Immunol. 1999, 163, 3605–3611. [Google Scholar] [PubMed]
- Elford, P.R.; Felix, R.; Cecchini, M.; Trechsel, U.; Fleisch, H. Murine osteoblastlike cells and the osteogenic cell mc3t3-e1 release a macrophage colony-stimulating activity in culture. Calcif Tissue Int. 1987, 41, 151–156. [Google Scholar] [CrossRef]
- Clinton, S.K.; Underwood, R.; Hayes, L.; Sherman, M.L.; Kufe, D.W.; Libby, P. Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am. J. Pathol. 1992, 140, 301–316. [Google Scholar]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, I.; Steffen, M.D.; Clark, P.A.; Patros, C.J.; Sokn, E.; Bishop, S.M.; Litscher, S.; Maklakova, V.I.; Kuo, J.S.; Rodriguez, F.J.; et al. Csf1 overexpression promotes high-grade glioma formation without impacting the polarization status of glioma-associated microglia and macrophages. Cancer Res. 2016, 76, 2552–2560. [Google Scholar] [CrossRef] [Green Version]
- Stafford, J.H.; Hirai, T.; Deng, L.; Chernikova, S.B.; Urata, K.; West, B.L.; Brown, J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. 2016, 18, 797–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.M.; Collier, L.S.; Rodriguez, F.J.; Tieu, C.; Larson, J.D.; Halder, C.; Mahlum, E.; Kollmeyer, T.M.; Akagi, K.; Sarkar, G.; et al. Sleeping beauty–mediated somatic mutagenesis implicates csf1 in the formation of high-grade astrocytomas. Cancer Res. 2010, 70, 3557–3565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. Csf-1r inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; Joyce, J.A. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef] [PubMed]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal csf-1r inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef] [PubMed]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Moughon, D.; Orpilla, J.R.; Shin, N.P.; Sedighim, S.; Treger, J.; Odesa, S.; Tucker, A.; et al. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a pd-1/pd-l1 mechanism in glioblastoma. Neuro Oncol. 2017, 19, 796–807. [Google Scholar] [CrossRef]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (egfr) and colony stimulating factor 1 receptor (csf-1r) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance mechanisms and barriers to successful immunotherapy for treating glioblastoma. Cells 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro Oncol. 2012, 14, 958–978. [Google Scholar] [CrossRef] [Green Version]
- Prosniak, M.; Harshyne, L.A.; Andrews, D.W.; Kenyon, L.C.; Bedelbaeva, K.; Apanasovich, T.V.; Heber-Katz, E.; Curtis, M.T.; Cotzia, P.; Hooper, D.C. Glioma grade is associated with the accumulation and activity of cells bearing m2 monocyte markers. Clin. Cancer Res. 2013, 19, 3776–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, B.; Zhang, D.; Wang, C.; Tao, J.; Tie, X.; Qiao, Y.; Xu, K.; Wang, Y.; Wu, A. Il-10 and tgf-β2 are overexpressed in tumor spheres cultured from human gliomas. Mol. Biol. Rep. 2011, 38, 3585–3591. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of b7-h1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Huang, X.; Li, J.; Fan, H.; Yang, F.; Zhang, R.; Yang, Y.; Feng, S.; He, D.; Sun, W.; et al. Interleukin 10 promotes growth and invasion of glioma cells by up-regulating kpna 2 in vitro. J. Cancer Res. Ther. 2019, 15, 927–932. [Google Scholar]
- Mittelbronn, M.; Platten, M.; Zeiner, P.; Dombrowski, Y.; Frank, B.; Zachskorn, C.; Harter, P.N.; Weller, M.; Wischhusen, J. Macrophage migration inhibitory factor (mif) expression in human malignant gliomas contributes to immune escape and tumour progression. Acta Neuropathol. 2011, 122, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Otvos, B.; Silver, D.J.; Mulkearns-Hubert, E.E.; Alvarado, A.G.; Turaga, S.M.; Sorensen, M.D.; Rayman, P.; Flavahan, W.A.; Hale, J.S.; Stoltz, K.; et al. Cancer stem cell-secreted macrophage migration inhibitory factor stimulates myeloid derived suppressor cell function and facilitates glioblastoma immune evasion. Stem Cells 2016, 34, 2026–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Xu, S.; Gao, X.; Wang, J.; Xue, H.; Chen, Z.; Zhang, J.; Guo, X.; Qian, M.; Qiu, W.; et al. Macrophage migration inhibitory factor promotes vasculogenic mimicry formation induced by hypoxia via cxcr4/akt/emt pathway in human glioblastoma cells. Oncotarget 2017, 8, 80358–80372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alban, T.J.; Bayik, D.; Otvos, B.; Rabljenovic, A.; Leng, L.; Jia-Shiun, L.; Roversi, G.; Lauko, A.; Momin, A.A.; Mohammadi, A.M.; et al. Glioblastoma myeloid-derived suppressor cell subsets express differential macrophage migration inhibitory factor receptor profiles that can be targeted to reduce immune suppression. Front. Immunol. 2020, 11, 1191. [Google Scholar] [CrossRef] [PubMed]
- Lund, S.A.; Giachelli, C.M.; Scatena, M. The role of osteopontin in inflammatory processes. J. Cell Commun. Signal. 2009, 3, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Invest. 2019, 129, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Toy, H.; Yavas, O.; Eren, O.; Genc, M.; Yavas, C. Correlation between osteopontin protein expression and histological grade of astrocytomas. Pathol. Oncol. Res. 2009, 15, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Ellert-Miklaszewska, A.; Wisniewski, P.; Kijewska, M.; Gajdanowicz, P.; Pszczolkowska, D.; Przanowski, P.; Dabrowski, M.; Maleszewska, M.; Kaminska, B. Tumour-processed osteopontin and lactadherin drive the protumorigenic reprogramming of microglia and glioma progression. Oncogene 2016, 35, 6366–6377. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. Her2-specific chimeric antigen receptor-modified virus-specific t cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced t cells targeting egfrviii in patients with glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused egfrviii-directed car t cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and safety of il13rα2-redirected chimeric antigen receptor cd8+ t cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. Erbb2/her2-specific nk cells for targeted therapy of glioblastoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. Car-engineered nk cells targeting wild-type egfr and egfrviii enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in car t cell therapy. Nat. Rev. Clin. Oncol 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. Cxcr1- or cxcr2-modified car t cells co-opt il-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Lei, W.; Zhang, C.; Yang, C.; Wei, J.; Guo, Q.; Guo, X.; Chen, Z.; Lu, Y.; Young, K.H.; et al. Cd19-specific car t cells that express a pd-1/cd28 chimeric switch-receptor are effective in patients with pd-l1-positive b-cell lymphoma. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Hartley, J.; Abken, H. Chimeric antigen receptors designed to overcome transforming growth factor-β-mediated repression in the adoptive t-cell therapy of solid tumors. Clin. Transl. Immunol. 2019, 8, e1064. [Google Scholar] [CrossRef]
- Li, Y.; Wu, H.; Chen, G.; Wei, X.; Wang, C.; Zhou, S.; Huang, A.; Zhang, Z.; Zhan, C.; Wu, Y.; et al. Arming anti-egfrviii car-t with tgfβ trap improves antitumor efficacy in glioma mouse models. Front. Oncol. 2020, 10, 1117. [Google Scholar] [CrossRef]
- Repellin, C.E.; Patel, P.; Beviglia, L.; Javitz, H.; Sambucetti, L.; Bhatnagar, P. Modular antigen-specific t-cell biofactories for calibrated in vivo synthesis of engineered proteins. Adv. Biosyst. 2018, 2. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. Il-12 release by engineered t cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. Car-t cells secreting bites circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A phase ii randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016, 18, 1146–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M.; et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor i, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Invest. New Drugs 2020, 38, 1570–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor plx3397 in recurrent glioblastoma: An ivy foundation early phase clinical trials consortium phase ii study. Neuro Oncol. 2016, 18, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Ligand | Alternative Name | Receptor |
|---|---|---|
| CCL2 | MCP-1 | CCR2 and CCR4 |
| CCL5 | RANTES | CCR1, CCR5 and CD44 |
| CXCL12 | PBGF or SDF-1 | CXCR4 and ACKR3 |
| IL-6 | BSF-2, IFN-β2, HGF or HSF | IL-6 receptor |
| TGF-β | - | TGF-β receptor |
| CSF-1 | M-CSF | CSF-1R |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo, E.C.F.; Brown, M.P.; Gargett, T.; Ebert, L.M. The Role of Cytokines and Chemokines in Shaping the Immune Microenvironment of Glioblastoma: Implications for Immunotherapy. Cells 2021, 10, 607. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030607
Yeo ECF, Brown MP, Gargett T, Ebert LM. The Role of Cytokines and Chemokines in Shaping the Immune Microenvironment of Glioblastoma: Implications for Immunotherapy. Cells. 2021; 10(3):607. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030607
Chicago/Turabian StyleYeo, Erica C. F., Michael P. Brown, Tessa Gargett, and Lisa M. Ebert. 2021. "The Role of Cytokines and Chemokines in Shaping the Immune Microenvironment of Glioblastoma: Implications for Immunotherapy" Cells 10, no. 3: 607. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030607