HSP90 Molecular Chaperones, Metabolic Rewiring, and Epigenetics: Impact on Tumor Progression and Perspective for Anticancer Therapy

 , , , , and
, , , , and 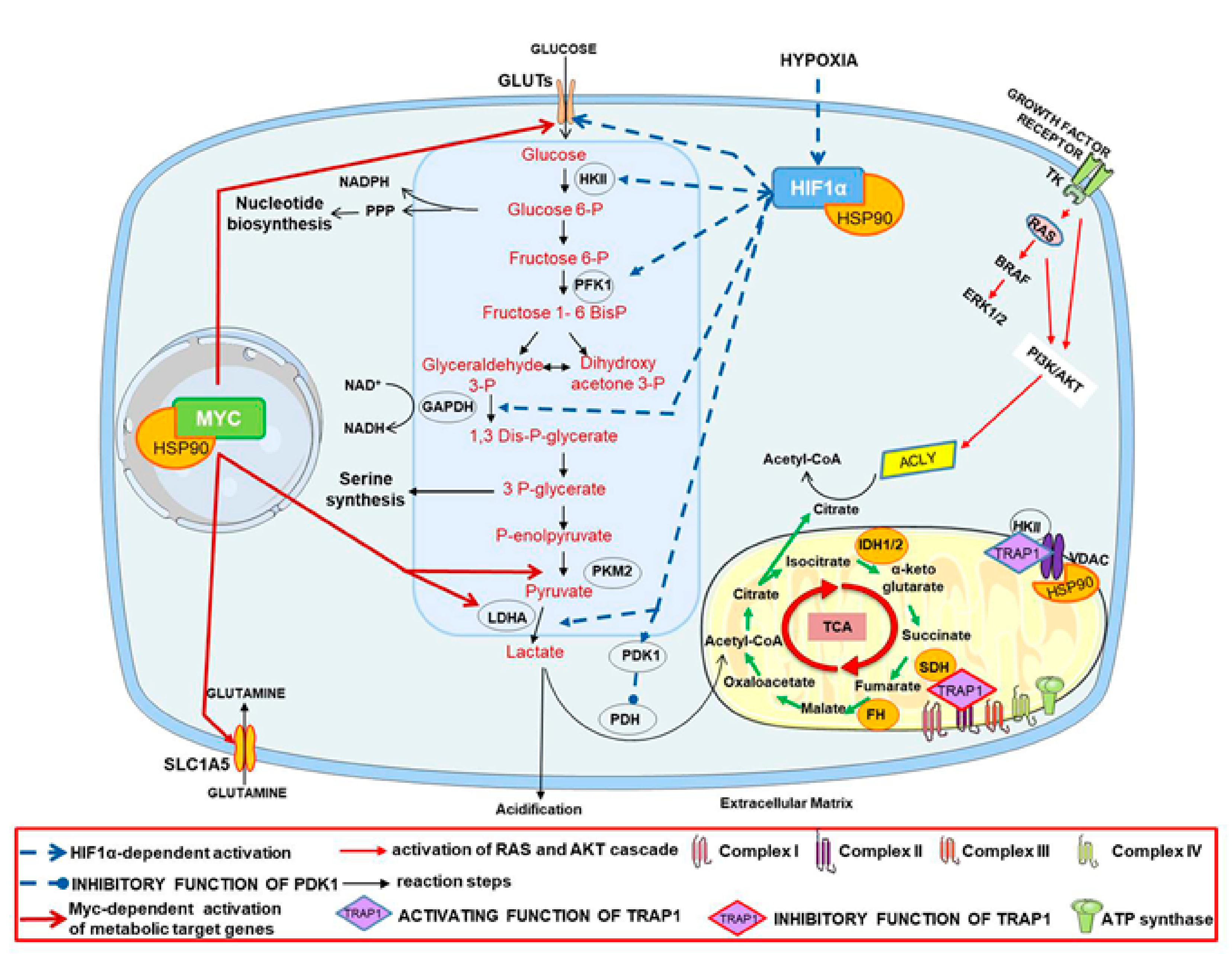{kind=link}
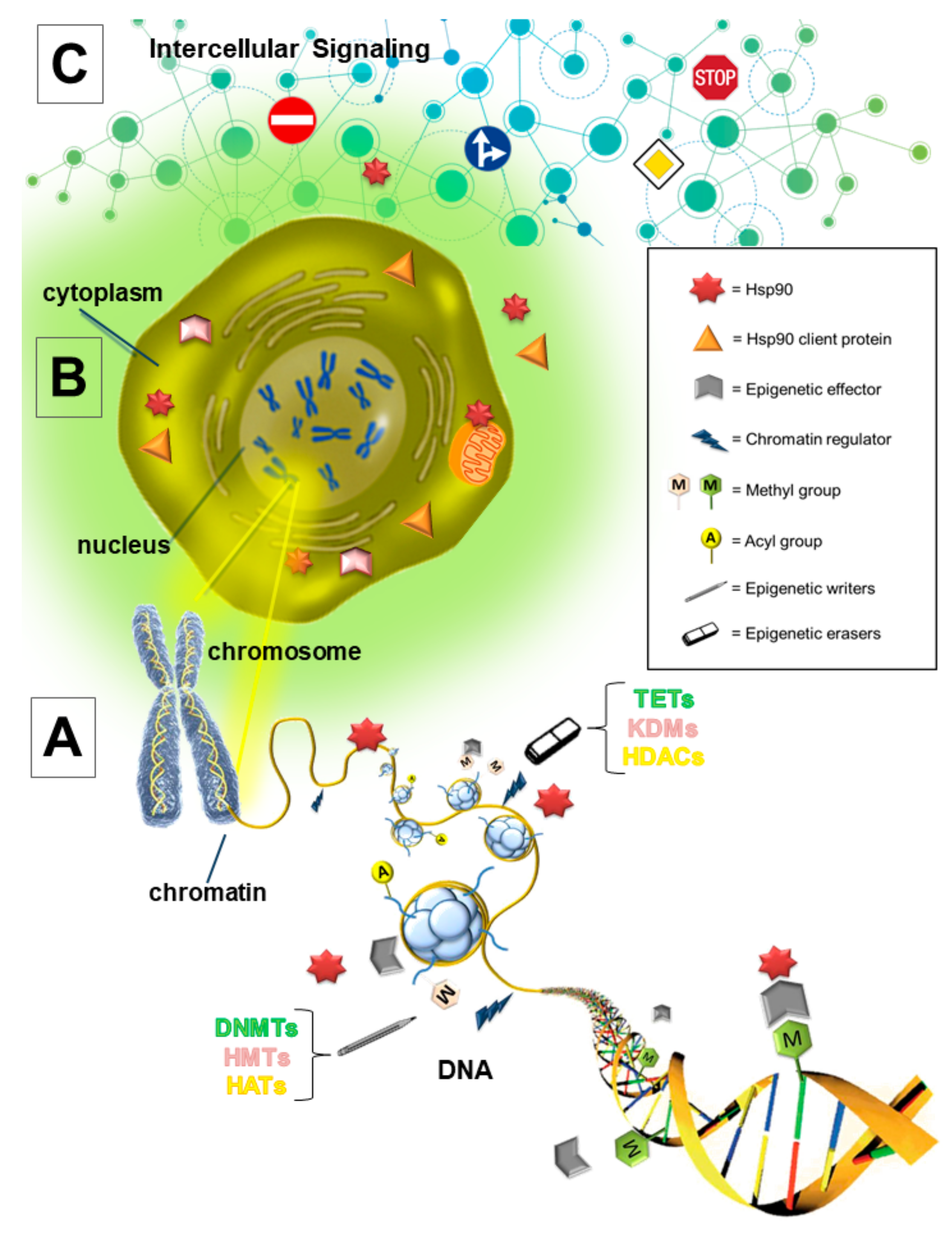{kind=link}
Abstract
:1. Introduction
2. Heat Shock Proteins
HSP90 Family
3. Role of HSP90 Family Members in Cancer Hallmarks
Role of HSP90 Family Members in Cancer Metabolic Reprogramming
4. HSP90 as an ‘Epigenetic Capacitor’ for Phenotypic Variation
4.1. Epigenetic Regulation of Gene Expression in Cancer Cells
4.2. HSP90 Epigenetic Mechanism of Action
4.3. HSP90 Role in Metabolism–Epigenetics Crosstalk
5. Conclusions: The Metabolism/Epigenetic Bidirectional Crosstalk as Novel Molecular Target
Author Contributions
Funding
Conflicts of Interest
References
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Joven, J.; Cufí, S.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyàs, E.; Martin-Castillo, B.; Lopez-Bonet, E.; Alarcón, T.; Vazquez-Martin, A. The Warburg effect version 2.0. Metabolic reprogramming of cancer stem cells. Cell Cycle 2013, 12, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetic tools (the Writers, the Readers and the Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Jerónimo, C. Metabolism and epigenetic interplay in cancer: Regulation and putative therapeutic targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Zabinsky, R.A.; Mason, G.A.; Queitsch, C.; Jarosz, D.F. It’s not magic—Hsp90 and its effects on genetic and epigenetic variation. Semin. Cell Dev. Biol. 2019, 88, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Pirkkala, L.; Sistonen, L. Heath Shock Proteins (HSPs): Structure, function and genetics. Encycl. Life Sci. 2006. [Google Scholar] [CrossRef]
- Lindquist, S.; Craig, E.A. The heat-shock proteins. Annu. Rev. Genet. 1988, 22, 631–677. [Google Scholar] [CrossRef]
- Glansdorff, N. About the last common ancestor, the universal life-tree and lateral gene transfer: A reappraisal. Mol. Microbiol. 2000, 38, 177–185. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J. Protein misassembly: Macromolecular crowding and molecular chaperones. Adv. Exp. Med. Biol. 2007, 594, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Whitley, D.; Goldberg, S.P.; Jordan, W.D. Heat shock proteins: A review of the molecular chaperones. J. Vasc. Surg. 1999, 29, 748–751. [Google Scholar] [CrossRef] [Green Version]
- Mathew, A.; Morimoto, R.I. Role of the heat-shock response in the life and death of proteins. Ann. N. Y. Acad. Sci. 1998, 851, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Gehrmann, M.; Brunet, M.; Multhoff, G.; Garrido, C. Intracellular and extracellular functions of heat shock proteins: Repercussions in cancer therapy. J. Leukoc. Biol. 2007, 81, 15–27. [Google Scholar] [CrossRef]
- Izawa, S.; Kita, T.; Ikeda, K.; Inoue, Y. Heat shock and ethanol stress provoke distinctly different responses in 3’-processing and nuclear export of HSP mRNA in Saccharomyces cerevisiae. Biochem. J. 2008, 414, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef]
- Rehana, K. Heat shock proteins in neurodegenerative disorders and aging. J. Cell Commun. Signal. 2014, 8, 293–310. [Google Scholar] [CrossRef]
- Yufu, Y.; Nishimura, J.; Nawata, H. High constitutive expression of heat shock protein 90 alpha in human acute leukemia cells. Leuk. Res. 1992, 16, 597–605. [Google Scholar] [CrossRef]
- Jego, G.; Hazoumé, A.; Seigneuric, R.; Garrido, C. Targeting heat shock proteins in cancer. Cancer Lett. 2013, 332, 275–285. [Google Scholar] [CrossRef]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef] [PubMed]
- De Maio, A.; Vazquez, D. Extracellular heat shock proteins: A new location, a new function. Shock 2013, 40, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Gong, J.; Murshid, A. Extracellular HSPs: The Complicated Roles of Extracellular HSPs in Immunity. Front. Immunol. 2016, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, E2560. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell. Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Sreedhar, A.S.; Kalmár, E.; Csermely, P.; Shen, Y.F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Li, J.; Soroka, J.; Buchner, J. The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 2012, 1823, 624–635. [Google Scholar] [CrossRef]
- Marzec, M.; Eletto, D.; Argona, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the Endoplasmic Reticulum. Biochim. Biophys. Acta 2012, 1823, 774–787. [Google Scholar] [CrossRef]
- Amoroso, M.R.; Matassa, D.S.; Sisinni, L.; Lettini, G.; Landriscina, M.; Esposito, F. TRAP1 revisited: Novel localizations and functions of a ‘next-generation’ biomarker. Int. J. Oncol. 2014, 45, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.E. Hsp90: Structure and function. Top Curr. Chem. 2013, 328, 155–240. [Google Scholar] [CrossRef]
- Zuehlke, A.; Johnson, J.L. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 2010, 93, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Gholami, A.M.; Kuster, B. Systematic identification of the HSP90 regulated proteome. Mol. Cell. Proteom. 2012, 11, M111.016675. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.A.; Forsberg, L.K.; Blagg, B.S.J. Alternative approaches to Hsp90 modulation for the treatment of cancer. Future Med. Chem. 2014, 6, 1587–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlejman, A.G.; Lagadari, M.; Toneatto, J.; Piwien-Pilipuk, G.; Galigniana, M.D. Regulatory role of the 90-kDa-heat-shock protein (Hsp90) and associated factors on gene expression. Biochim. Biophys. Acta 2014, 1839, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Csermely, P.; Sőti, C. Hsp90 chaperones PPARγ and regulates differentiation and survival of 3T3-L1 adipocytes. Cell Death Differ. 2013, 20, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sahu, D.; Tsen, F. Secreted heat shock protein-90 (Hsp90) in wound healing and cancer. Biochim. Biophys. Acta 2012, 1823, 730–741. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Bossy-Wetzel, E.; Burns, K.; Fadel, M.P.; Lozyk, M.; Goping, I.S.; Opas, M.; Bleackley, R.C.; Green, D.R.; Michalak, M. Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J. Cell Biol. 2000, 150, 731–740. [Google Scholar] [CrossRef]
- Biswas, C.; Ostrovsky, O.; Makarewich, C.A.; Wanderling, S.; Gidalevitz, T.; Argon, Y. The peptide-binding activity of GRP94 is regulated by calcium. Biochem. J. 2007, 405, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Song, H.Y.; Dunbar, J.D.; Zhang, Y.X.; Guo, D.; Donner, D.B. Identification of a protein with homology to hsp90 that binds the type 1 tumor necrosis factor receptor. J. Biol. Chem. 1995, 270, 3574–3581. [Google Scholar] [CrossRef]
- Chen, C.F.; Chen, Y.; Dai, K.; Chen, P.L.; Riley, D.J.; Lee, W.H. A new member of the hsp90 family of molecular chaperones interacts with the retinoblastoma protein during mitosis and after heat shock. Mol. Cell Biol. 1996, 16, 4691–4699. [Google Scholar] [CrossRef] [Green Version]
- Felts, S.J.; Owen, B.A.; Nguyen, P.; Trepel, J.; Donner, D.B.; Toft, D.O. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J. Biol. Chem. 2000, 275, 3305–3312. [Google Scholar] [CrossRef]
- Amoroso, M.R.; Matassa, D.S.; Laudiero, G.; Egorova, A.V.; Polishchuk, R.S.; Maddalena, F.; Piscazzi, A.; Paladino, S.; Sarnataro, D.; Garbi, C.; et al. TRAP1 and the proteasome regulatory particle TBP7/Rpt3 interact in the endoplasmic reticulum and control cellular ubiquitination of specific mitochondrial proteins. Cell Death Differ. 2012, 19, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Matassa, D.S.; Amoroso, M.R.; Agliarulo, I.; Maddalena, F.; Sisinni, L.; Paladino, S.; Romano, S.; Romano, M.F.; Sagar, V.; Loreni, F.; et al. Translational control in the stress adaptive response of cancer cells: A novel role for the heat shock protein TRAP1. Cell Death Dis. 2013, 4, e851. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Hsp90 Regulation of mitochondrial protein folding: From organelle integrity to cellular homeostasis. Cell Mol. Life Sci. 2013, 70, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Landriscina, M.; Laudiero, G.; Maddalena, F.; Amoroso, M.R.; Piscazzi, A.; Cozzolino, F.; Monti, M.; Garbi, C.; Fersini, A.; Pucci, P. Mitochondrial chaperone Trap1 and the calcium binding protein Sorcin interact and protect cells against apoptosis induced by antiblastic agents. Cancer Res. 2010, 70, 6577–6586. [Google Scholar] [CrossRef] [PubMed]
- Sisinni, L.; Maddalena, F.; Condelli, V.; Pannone, G.; Simeon, V.; Li Bergolis, V.; Lopes, E.; Piscazzi, A.; Matassa, D.S.; Mazzoccoli, C.; et al. TRAP1 controls cell cycle G2-M transition through the regulation of CDK1 and MAD2 expression/ubiquitination. J. Pathol. 2017, 243, 123–134. [Google Scholar] [CrossRef]
- Lettini, G.; Sisinni, L.; Condelli, V.; Matassa, D.S.; Simeon, V.; Maddalena, F.; Gemei, M.; Lopes, E.; Vita, G.; Del Vecchio, L.; et al. TRAP1 regulates stemness through Wnt/β-catenin pathway in human colorectal carcinoma. Cell Death Differ. 2016, 23, 1792–1803. [Google Scholar] [CrossRef]
- Condelli, V.; Piscazzi, A.; Sisinni, L.; Matassa, D.S.; Maddalena, F.; Lettini, G.; Simeon, V.; Palladino, G.; Amoroso, M.R.; Trino, S.; et al. TRAP1 is involved in BRAF regulation and downstream attenuation of ERK phosphorylation and cell-cycle progression: A novel target for BRAF-mutated colorectal tumors. Cancer Res. 2014, 74, 6693–6704. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013, 17, 988–999. [Google Scholar] [CrossRef]
- Choi, J.D.; Lee, J.S. Interplay between epigenetics and genetics in cancer. Genomics Inform. 2013, 11, 164–173. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Nakamoto, H.; Neckers, L. The therapeutic target Hsp90 and cancer hallmarks. Curr. Pharm. Des. 2013, 19, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W. HSP90 and immune modulation in cancer. Adv. Cancer Res. 2016, 129, 191–224. [Google Scholar] [CrossRef]
- Sevin, M.; Girodon, F.; Garrido, C.; de Thonel, A. HSP90 and HSP70: Implication in inflammation processes and therapeutic approaches for myeloproliferative neoplasms. Mediators Inflamm. 2015, 2015, 970242. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Amino acids are made from intermediates of the citric acid cycle and other major pathways. In Biochemistry, 5th ed.; WH Freeman: New York, NY, USA, 2002. [Google Scholar]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Warburg, O. The metabolism of carcinoma cells. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Moreira, J.D.; Hamraz, M.; Abolhassani, M.; Bigan, E.; Pérès, S.; Paulevé, L.; Nogueira, M.L.; Steyaert, J.M.; Schwartz, L. The redox status of cancer cells supports mechanisms behind the Warburg effect. Metabolites 2016, 6, E33. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Lehuédé, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res. 2016, 76, 5201–5208. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Tsutsumi, S.; Muhlebach, G.; Sourbier, C.; Lee, M.J.; Lee, S.; Vartholomaiou, E.; Tatokoro, M.; Beebe, K.; Miyajima, N.; et al. Molecular chaperone TRAP1 regulates a metabolic switch between mitochondrial respiration and aerobic glycolysis. Proc. Natl. Acad. Sci. USA 2013, 110, 1604–1612. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Tu, J.; Dou, C.; Zhang, J.; Yang, L.; Liu, X.; Lei, K.; Liu, Z.; Wang, Y.; Li, L.; et al. HSP90 promotes cell glycolysis, proliferation and inhibits apoptosis by regulating PKM2 abundance via Thr-328 phosphorylation in hepatocellular carcinoma. Mol. Cancer 2017, 16, 178. [Google Scholar] [CrossRef] [PubMed]
- Matassa, D.S.; Agliarulo, I.; Avolio, R.; Landriscina, M.; Esposito, F. TRAP1 regulation of cancer metabolism: Dual role as oncogene or tumor suppressor. Genes 2018, 9, E195. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhang, L.L.; Wu, W.; Guo, H.; Li, Y.; Sukhanova, M.; Venkataraman, G.; Zhang, H.; Alikhan, M.; Lu, P. Activation of myc, a bona fide client of HSP90, contributes to intrinsic ibrutinib resistance in mantle cell lymphoma. Blood Adv. 2018, 2, 2039–2051. [Google Scholar] [CrossRef]
- Robey, I.F.; Lien, A.D.; Welsh, S.J.; Baggett, B.K.; Gillies, R.J. Hypoxia-inducible factor-1α and the glycolytic phenotype in tumors. Neoplasia 2005, 7, 324–330. [Google Scholar] [CrossRef]
- Corcoran, S.E.; O’Neill, L.A.J. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832–10837. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Gan, J.; Mosesson, Y.; Vereb, G.; Szollosi, J.; Yarden, Y. Hsp90 restrains ErbB-2/HER2 signalling by limiting heterodimer formation. EMBO Rep. 2004, 5, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Liu, Z.; Desai, S.; Zhao, Y.; Liu, H.; Pannell, L.K.; Yi, H.; Wright, E.R.; Owen, L.B.; Dean-Colomb, W.; et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat. Commun. 2012, 3, 1271. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Singer, M.A.; Lindquist, S. Maturation of the tyrosine kinase c-src as a kinase and as a substrate depends on the molecular chaperone Hsp90. Proc. Natl. Acad. Sci. USA 1999, 96, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Hinagata, J.; Tanaka, T.; Imanishi, T.; Wada, Y.; Kodama, T.; Doi, T. HSP90, HSP70, and GAPDH directly interact with the cytoplasmic domain of macrophage scavenger receptors. Biochem. Biophys. Res. Commun. 2002, 290, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Smith-Jones, P.M.; Solit, D.; Afroze, F.; Rosen, N.; Larson, S.M. Early tumor response to Hsp90 therapy using HER2 PET: Comparison with 18F-FDG PET. J. Nucl. Med. 2006, 47, 793–796. [Google Scholar] [PubMed]
- Mycielska, M.E.; Wachsmuth, C.J.; Dettmer, K.; Wagner, C.; Schlitt, H.J.; Oefner, P.J.; Geissler, E.K.; Lang, S.A. Hsp90 inhibition affects cell metabolism by disrupting mitochondrial protein insertion. J. Cell Biol. Cell Metab. 2014, 1, 2. [Google Scholar] [CrossRef]
- Lettini, G.; Maddalena, F.; Sisinni, L.; Condelli, V.; Matassa, D.S.; Costi, M.P.; Simoni, D.; Esposito, F.; Landriscina, M. TRAP1: A viable therapeutic target for future cancer treatments? Expert Opin. Ther. Targets 2017, 21, 805–815. [Google Scholar] [CrossRef]
- Maddalena, F.; Simeon, V.; Vita, G.; Bochicchio, A.; Possidente, L.; Sisinni, L.; Lettini, G.; Condelli, V.; Matassa, D.S.; Li Bergolis, V.; et al. TRAP1 protein signature predicts outcome in human metastatic colorectal carcinoma. Oncotarget 2017, 8, 21229–21240. [Google Scholar] [CrossRef]
- Chae, Y.C.; Caino, M.C.; Lisanti, S.; Ghosh, J.C.; Dohi, T.; Danial, N.N.; Villanueva, J.; Ferrero, S.; Vaira, V.; Santambrogio, L.; et al. Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer Cell 2012, 22, 331–344. [Google Scholar] [CrossRef]
- Matassa, D.S.; Amoroso, M.R.; Lu, H.; Avolio, R.; Arzeni, D.; Procaccini, C.; Faicchia, D.; Maddalena, F.; Simeon, V.; Agliarulo, I.; et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. 2016, 23, 1542–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegelin, M.D. Inhibition of the mitochondrial Hsp90 chaperone network: A novel, efficient treatment strategy for cancer? Cancer Lett. 2013, 333, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. Canalization of development and the inheritance of acquired characters. Nature 1942, 150, 563–565. [Google Scholar] [CrossRef]
- Waddington, C.H. Genetic assimilation of an acquired character. Evolution 1953, 7, 118–126. [Google Scholar] [CrossRef]
- Pigliucci, M. Epigenetics is back! Cell Cycle 2003, 2, 34–35. [Google Scholar] [CrossRef] [PubMed]
- Ruden, D.M.; Xiao, L.; Garfinkel, M.D.; Lu, X. Hsp90 and environmental impacts on epigenetic states: A model for the trans-generational effects of diethylstibesterol on uterine development and cancer. Hum. Mol. Genet. 2005, 14, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Queitsch, C.; Sanster, T.A.; Lindquist, S. Hsp90 as a capacitor of phenotypic variation. Nature 2002, 417, 618–624. [Google Scholar] [CrossRef]
- Isaacs, J.S. Hsp90 as a “chaperone” of the epigenome: Insights and opportunities for cancer therapy. Adv. Cancer Res. 2016, 129, 107–140. [Google Scholar] [CrossRef]
- Sollars, V.; Lu, X.; Xiao, L.; Wang, X.; Garfinkel, M.D.; Ruden, D.M. Evidence for an epigenetic mechanism by which Hsp90 acts as a capacitor for morphological evolution. Nat. Genet. 2003, 33, 70–74. [Google Scholar] [CrossRef]
- Horowitz, M. Epigenetics and cytoprotection with heat acclimation. J. Appl. Physiol. 2016, 120, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Ruden, D.M.; Lu, X. Hsp90 affecting chromatin remodeling might explain transgenerational epigenetic inheritance in Drosophila. Curr. Genomics 2008, 9, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.L.; Lindquist, S. Quantitative analysis of Hsp90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Tucker, G.; Peng, J.; Krykbaeva, I.; Lin, Z.Y.; Larsen, B.; Choi, H.; Berger, B.; Gingras, AC.; Lindquist, S. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 2014, 158, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Sawarkar, R.; Paro, R. [email protected]: An emerging hub of a networker. Trends Cell Biol. 2013, 23, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, D. Hsp90: A global regulator of the genotype-to-phenotype map in cancers. Adv. Cancer Res. 2016, 129, 225–247. [Google Scholar] [CrossRef] [PubMed]
- Claderwood, S.K.; Neckers, L. Hsp90 in cancer: Transcriptional roles in the nucleus. Adv. Cancer Res. 2016, 129, 89–106. [Google Scholar] [CrossRef]
- Sawarkar, R.; Sievers, C.; Paro, R. Hsp90 globally targets paused RNA polymerase to regulate gene expression in response to environmental stimuli. Cell 2012, 149, 807–818. [Google Scholar] [CrossRef]
- Morcillo, G.; Diez, J.L.; Carbajal, M.E.; Tanguay, R.M. HSP90 associates with specific heat shock puffs (hsp omega) in polytene chromosomes of Drosophila and Chironomus. Chromosoma 1993, 102, 648–659. [Google Scholar] [CrossRef]
- Zhao, R.; Davey, M.; Hsu, Y.C.; Kaplanek, P.; Tong, A.; Parsons, A.B.; Krogan, N.; Cagney, G.; Mai, D.; Greenblatt, J.; et al. Navigating the chaperone network: An integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell 2005, 120, 715–727. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.A. Throwing the cancer switch: Reciprocal roles of polycomb and trithorax proteins. Nat. Rev. Cancer 2010, 10, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Kingston, R.E.; Tamkun, J.W. Transcriptional regulation by trithorax-group proteins. Cold Spring Harb. Perspect. Biol. 2014, 6, a019349. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Herz, H.M.; Takahashi, Y.H.; Lin, C.; Lai, K.C.; Zhang, Y.; Washburn, M.P.; Florens, L.; Shilatifard, A. Linking H3K79trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev. 2010, 24, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; Ho, J.; Coutinho, F.J.; Vanner, R.; Lee, L.; Head, R.; Ling, E.K.; Clarke, I.D.; Dirks, P.B. A tumorigenic MLL-homeobox network in human glioblastoma stem cells. Cancer Res. 2013, 73, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Liu, H.; Sasagawa, S.; Dong, Y.; Trainor, P.A.; Cheng, E.H.; Hsieh, J.J. HGF-MET signals via the MLL-ETS2 complex in hepatocellular carcinoma. J. Clin. Investig. 2013, 123, 3154–3165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Farha, M.; Lambert, J.P.; Al-Madhoun, A.S.; Elisma, F.; Skerjanc, I.S.; Figeys, D. The tale of two domains: Proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol. Cell. Proteomics 2008, 7, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Sparmann, A.; van Lohuizan, M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006, 6, 846–856. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, DF.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Rhee, I.; Backman, K.E.; Park, B.H.; Jair, K.W.; Yen, R.W.; Schuebel, K.E.; Cui, H.; Feinberg, A.P.; Lengauer, C.; Kinzler, K.W.; et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002, 416, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Uzvolgyi, E.; Lianng, G.; Talmadge, C.; Sumegi, J.; Gonzales, F.A.; Jones, P.A. The human DNA methyltransferases (DNMTs) 1,3a and 3b: Coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999, 27, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Simon, M.C. Molecular pathways: Targeting MYC-induced metabolic reprogramming and oncogenic stress in cancer. Clin. Cancer Res. 2013, 19, 5835–5841. [Google Scholar] [CrossRef] [PubMed]
- Poole, C.J.; Zheng, W.; Lee, H.; Young, D.; Lodh, A.; Chadli, A.; van Riggelen, J. Targeting the MYC oncogene in Burkitt lymphoma through Hsp90 inhibition. Cancers 2018, 10, E448. [Google Scholar] [CrossRef] [PubMed]
- Paul, I.; Ahmed, S.F.; Bhowmik, A.; Deb, S.; Ghosh, M.K. The ubiquitin ligase CHIP regulates c-Myc stability and transcriptional activity. Oncogene 2013, 32, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.D.; Franco, O.E.; Hance, M.W.; Hayward, S.W.; Isaacs, J.S. Tumor-secreted Hsp90 subverts polycomb function to drive prostate tumor growth and invasion. J. Biol. Chem. 2015, 290, 8271–8282. [Google Scholar] [CrossRef] [PubMed]
- Sidera, K.; Gaitanou, M.; Stellas, D.; Matsas, R.; Patsavoudi, E. A critical role for HSP90 in cancer cell invasion involves interaction with the extracellular domain of HER-2. J. Biol. Chem. 2008, 283, 2031–2041. [Google Scholar] [CrossRef]
- Chen, J.S.; Hsu, Y.M.; Chen, C.C.; Chen, L.L.; Lee, C.C.; Huang, T.S. Secreted heat shock protein 90alpha induces colorectal cancer cell invasion through CD91/LRP-1 and NF-kappaB-mediated integrin alphaV expression. J. Biol. Chem. 2010, 285, 25458–25466. [Google Scholar] [CrossRef]
- Bohonowych, J.E.; Hance, M.V.; Nolan, K.D.; Defee, M.; Parsons, C.H.; Isaacs, J.S. Extracellular Hsp90 mediates an NF-kappaB dependent inflammatory stromal program: Implications for the prostate tumor microenvironment. Prostate 2014, 74, 395–407. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Martín-Martín, N.; Carracedo, A.; Torrano, V. Metabolism and transcription in cancer: Merging two classic tales. Front. Cell Dev. Biol. 2018, 5, 119. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.P.; Mostoslavsky, R. Interplay between metabolism and epigenetics: A nuclear adaptation to environmental changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.L.; Wellen, K.E. Metabolic signaling to the nucleus in cancer. Mol. Cell 2018, 71, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Yeom, Y. Metabolic signaling to epigenetic alterations in cancer. Biomol. Ther. 2018, 26, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef]
- Xu, W.; Wang, F.; Yu, Z.; Xin, F. Epigenetics and cellular metabolism. Genet. Epigenet. 2016, 8, 43–51. [Google Scholar] [CrossRef]
- Dong, Z.; Cui, H. Epigenetic modulation of metabolism in glioblastoma. Semin. Cancer Biol. 2018, in press, ISSN 1044-579X. [Google Scholar] [CrossRef]
- Agarwal., E.; Altman, B.J.; Seo, J.H.; Ghosh, J.C.; Kossenkov, A.V.; Tang, H.Y.; Krishn, S.R.; Languino., LR.; Gabrilovich, D.I.; Speicher, D.W.; et al. Myc-mediated transcriptional regulation of the mitochondrial chaperone TRAP1 controls primary and metastatic tumor growth. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Locasale, J.W. Serine, glycine and the one-carbon cycle: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef]
- Luo, J.; Li, Y.N.; Wang, F.; Zhang, W.M.; Geng, X. S-adenosylmethionine inhibits the growth of cancer cells by reversing the hypomethylation status of c-myc and H-ras in human gastric cancer and colon cancer. Int. J. Biol. Sci. 2010, 6, 784–795. [Google Scholar] [CrossRef]
- Shukeir, N.; Stefanska, B.; Parashar, S.; Chik, F.; Arakelian, A.; Szyf, M.; Rabbani, S.A. Pharmacological methyl group donors block skeletal metastasis in vitro and in vivo. Br. J. Pharmacol. 2015, 172, 2769–2781. [Google Scholar] [CrossRef]
- Ilisso, C.P.; Sapio, L.; Cave, D.D.; Illiano, M.; Spina, A.; Cacciapuoti, G.; Naviglio, S.; Porcelli, M. S-adenosylmethionine affects ERK1/2 and Stat3 pathways and induces apotosis in osteosarcoma cells. J. Cell. Physiol. 2016, 231, 428–435. [Google Scholar] [CrossRef]
- Cave, D.D.; Ilisso, C.P.; Mosca, L.; Pagano, M.; Martino, E.; Porcelli, M.; Cacciapuoti, G. The anticancer effects of S-adenosylmethionine on breast cancer cells. JSM Chem. 2017, 5, 1049. [Google Scholar]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.F.; Lim, H.W.; Liu, S.E.; et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef]
- Collins, R.R.J.; Patel, K.; Putnam, W.C.; Kapur, P.; Rakheja, D. Oncometabolites: A new paradigm for oncology, metabolism, and the clinical laboratory. Clin. Chem. 2017, 63, 1812–1820. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signal. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.S.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef]
- Katada, S.; Imhof, A.; Sassone-Corsi, P. Connecting threads: Epigenetics and metabolism. Cell 2012, 148, 24–28. [Google Scholar] [CrossRef]
- Nieborak, A.; Schneider, R. Metabolic intermediates–cellular messangers talking to chromatin modifiers. Mol. Metab. 2018, 14, 39–52. [Google Scholar] [CrossRef]
- Yang, W.; Lu, Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle 2013, 12, 3154–3158. [Google Scholar] [CrossRef]
- Desai, S.; Ding, M.; Wang, B.; Lu, Z.; Zhao, Q.; Shaw, K.; Yung, W.K.A.; Weinstein, J.N.; Tan, M.; Yao, J. Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers. Oncotarget 2014, 5, 8202–8210. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef]
- Wang, H.J.; Hsieh, Y.J.; Cheng, W.C.; Lin, C.P.; Lin, Y.S.; Yang, S.F.; Chen, C.C.; Izumiya, Y.; Yu, J.S.; Kung, H.J.; et al. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1α–mediated glucose metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 279–284. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Yung, W.K.A.; Lu, Z. PKM2 Phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 17, 685–696. [Google Scholar] [CrossRef]
- Matsuda, S.; Adachi, J.; Ihara, M.; Tanuma, N.; Shima, H.; Kakizuka, A.; Ikura, M.; Ikura, T.; Matsuda, T. Nuclear pyruvate kinase M2 complex serves as transcriptional coactivator of aryl hydrocarbon receptor. Nucleic Acids Res. 2016, 44, 636–647. [Google Scholar] [CrossRef]
- Li, S.; Gogol, S.K.; Florens, L.; Washburn, M.P.; Jerry, L. Serine and SAM responsive complex SESAME regulates histone modification crosstalk by sensing cellular metabolism. Mol. Cell 2015, 60, 408–421. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Zhang, F.; Hong, C.Q.; Giuliano, A.E.; Cui, X.J.; Zhou, G.J.; Zhang, G.J.; Cui, Y.K. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol. Med. 2015, 12, 10–22. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell. 2005, 18, 601–607. [Google Scholar] [CrossRef]
- Mollapour, M.; Neckers, L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 2012, 1823, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Nimmanapalli, R.; Fuino, L.; Bali, P.; Gasparetto, M.; Glozak, M.; Tao, J.; Moscinski, L.; Smith, C.; Wu, J.; Jove, R.; et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003, 63, 5126–5135. [Google Scholar]
- Fuino, L.; Bali, P.; Wittmann, S.; Donapaty, S.; Guo, F.; Yamaguchi, H.; Wang, H.G.; Atadja, P.; Bhalla, K. Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol. Cancer. Ther. 2003, 2, 971–984. [Google Scholar]
- Fiskus, W.; Rao, R.; Fernandez, P.; Herger, B.; Yang, Y.; Chen, J.; Kolhe, R.; Mandawat, A.; Wang, Y.; Joshi, R.; et al. Molecular and biologic characterization and drug sensitivity of pan-histone deacetylase inhibitor-resistant acute myeloid leukemia cells. Blood 2008, 112, 2896–2905. [Google Scholar] [CrossRef]
- Zhou, Q.; Agoston, A.T.; Atadja, P.; Nelson, W.G.; Davidson, N.E. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol. Cancer Res. 2008, 6, 873–883. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, S.H.; Choi, M.C.; Lee, J.; Oh, D.Y.; Im, S.A.; Bang, Y.J.; Kim, T.Y. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem. Biophys. Res. Commun. 2008, 368, 318–322. [Google Scholar] [CrossRef]
- Park, H.K.; Hong, J.H.; Oh, Y.T.; Kim, S.S.; Yin, J.; Lee, A.J.; Chae, Y.C.; Kim, J.H.; Park, S.H.; Park, C.K.; et al. Interplay between TRAP1 and Sirtuin-3 modulates mitochondrial respiration and oxidative stress to maintain stemness of glioma stem cells. Cancer Res. 2019, 79, 1369–1382. [Google Scholar] [CrossRef]
- Rondanin, R.; Lettini, G.; Oliva, P.; Baruchello, R.; Costantini, C.; Trapella, C.; Simoni, D.; Bernardi, T.; Sisinni, L.; Pietrafesa, M. New TRAP1 and Hsp90 chaperone inhibitors with cationic components: Preliminary studies on mitochondrial targeting. Bioorg. Med. Chem. Lett. 2018, 28, 2289–2293. [Google Scholar] [CrossRef]
- Moser, C.; Lang, S.A.; Hackl, C.; Wagner, C.; Scheiffert, E.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O. Targeting HSP90 by the novel inhibitor NVP-AUY922 reduces growth and angiogenesis of pancreatic cancer. Anticancer Res. 2012, 32, 2551–2561. [Google Scholar]
- Ganji, P.N.; Diaz, R.; El-Rayes, B. Antiangiogenic activity of the HSP90 inhibitor genetespib in pancreatic cancer models. FASEB J. 2013, 27, lb572. [Google Scholar] [CrossRef]
- Boroumand, N.; Saghi, H.; Avan, A.; Bahreyni, A.; Ryzhikov, M.; Khazaei, M.; Hassanian, S.M. Therapeutic potency of heat-shock protein-90 pharmacological inhibitors in the treatment of gastrointestinal cancer, current status and perspectives. J. Pharm. Pharmacol. 2018, 70, 151–158. [Google Scholar] [CrossRef]
- Woodford, M.R.; Dunn, D.; Miller, J.B.; Jamal, S.; Neckers, L.; Mollapour, M. Impact of posttranslational modifications on the anticancer activity of Hsp90 inhibitors. Adv. Cancer Res. 2016, 129, 31–50. [Google Scholar] [CrossRef]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors. Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef]
- Borodovsky, A.; Salmasi, V.; Turcan, S.; Fabius, A.W.; Baia, G.S.; Eberhart, C.G.; Gallia, G.L.; Baylin, S.B.; Chan, T.A.; Riggins, G.J.; et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget 2013, 4, 1737–1747. [Google Scholar] [CrossRef]
- Li, L.; Paz, A.C.; Wilky, B.A.; Johnson, B.; Galoian, K.; Rosenberg, A.; Hu, G.; Tinoco, G.; Bodamer, O.; Trent, J.C. Treatment with a small molecule mutant IDH1 inhibitor suppresses tumorigenic activity and decreases production of the oncometabolite 2-hydroxyglutarate in human chondrosarcoma cells. PLoS ONE 2015, 10, e0133813. [Google Scholar] [CrossRef]
- Rodrigues, M.F.; Carvalho, E.; Pezzuto, P.; Rumjanek, F.D.; Amoedo, N.D. Reciprocal modulation of histone deacetylase inhibitors sodium butyrate and trichostatin A on the energy metabolism of breast cancer cells. J. Cell. Biochem. 2015, 116, 797–808. [Google Scholar] [CrossRef]
- Lu, X.; Xiao, L.; Wang, L.; Ruden, D.M. Hsp90 inhibitors and drug resistance in cancer: The potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem. Pharmacol. 2012, 83, 995–1004. [Google Scholar] [CrossRef] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Condelli, V.; Crispo, F.; Pietrafesa, M.; Lettini, G.; Matassa, D.S.; Esposito, F.; Landriscina, M.; Maddalena, F. HSP90 Molecular Chaperones, Metabolic Rewiring, and Epigenetics: Impact on Tumor Progression and Perspective for Anticancer Therapy. Cells 2019, 8, 532. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8060532
Condelli V, Crispo F, Pietrafesa M, Lettini G, Matassa DS, Esposito F, Landriscina M, Maddalena F. HSP90 Molecular Chaperones, Metabolic Rewiring, and Epigenetics: Impact on Tumor Progression and Perspective for Anticancer Therapy. Cells. 2019; 8(6):532. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8060532
Chicago/Turabian StyleCondelli, Valentina, Fabiana Crispo, Michele Pietrafesa, Giacomo Lettini, Danilo Swann Matassa, Franca Esposito, Matteo Landriscina, and Francesca Maddalena. 2019. "HSP90 Molecular Chaperones, Metabolic Rewiring, and Epigenetics: Impact on Tumor Progression and Perspective for Anticancer Therapy" Cells 8, no. 6: 532. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8060532