An Autocrine Wnt5a Loop Promotes NF-κB Pathway Activation and Cytokine/Chemokine Secretion in Melanoma

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Constructs
2.4. Western Blotting
2.5. Luciferase Assays
2.6. Cell Fractioning
2.7. Cytokine Array
2.8. ELISA
2.9. Immunofluorescence
2.10. Bioinformatics Analysis
2.11. Statistics
3. Results
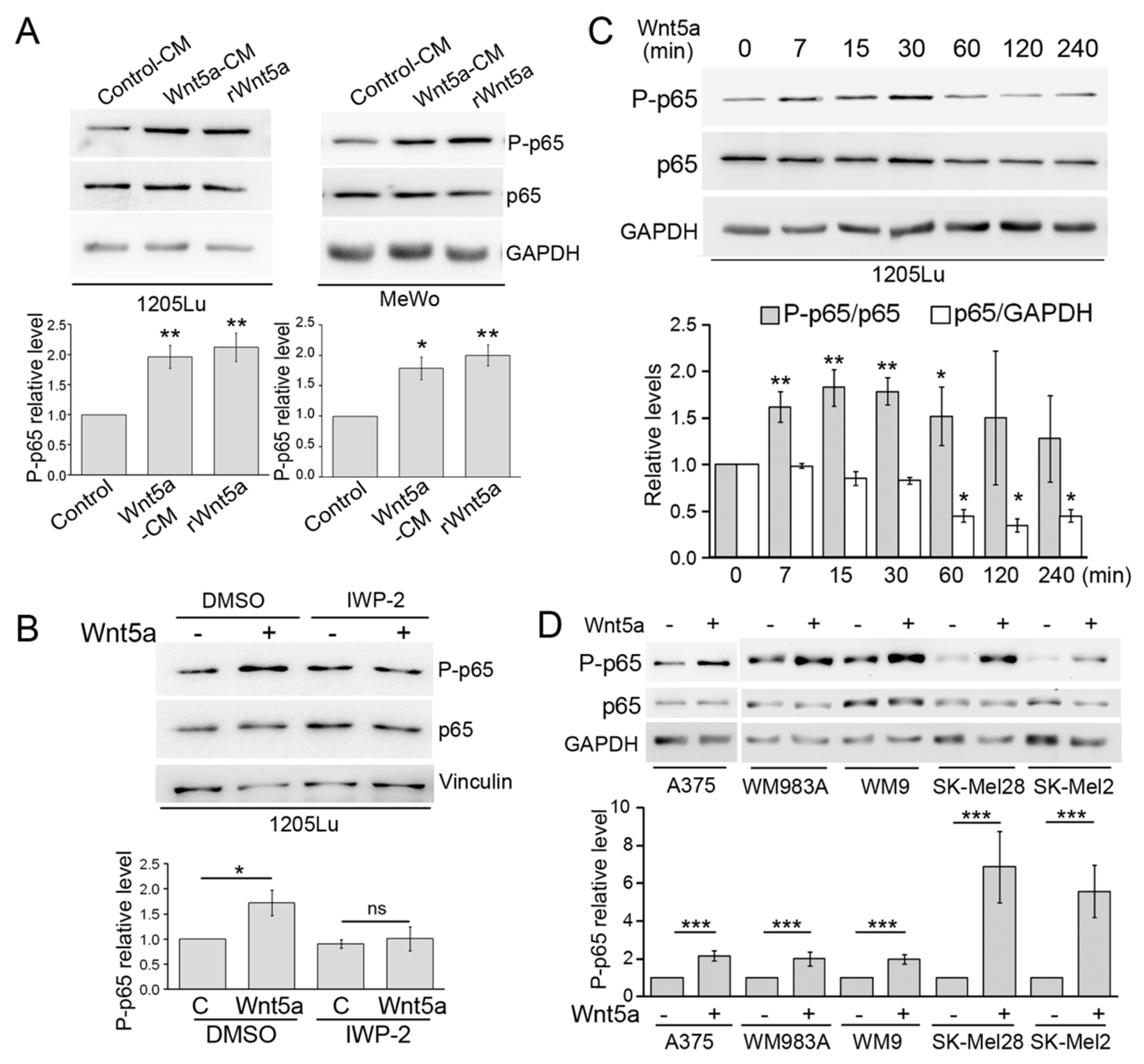
3.1. Wnt5a Induces p65 Phosphorylation in Melanoma
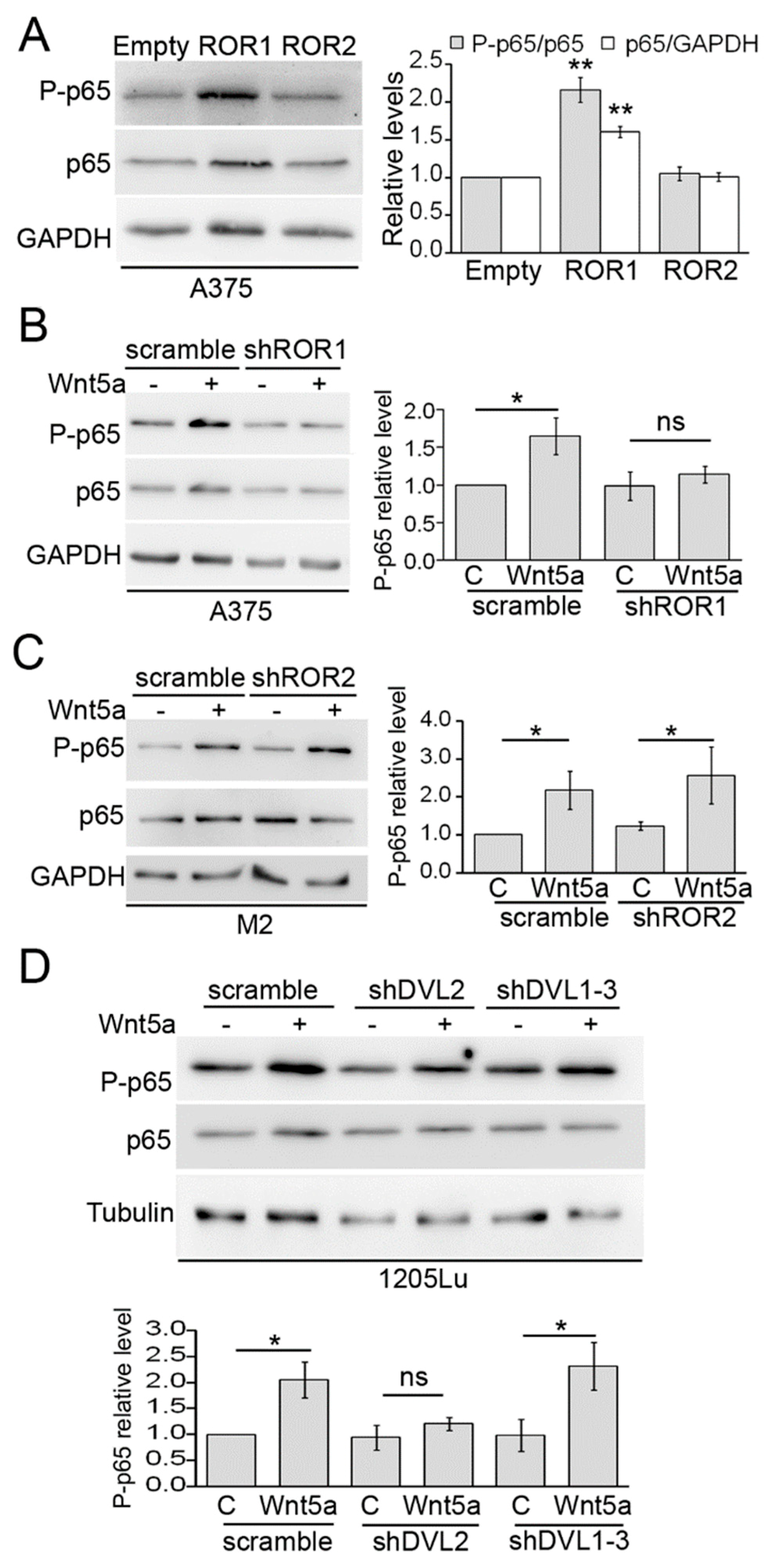
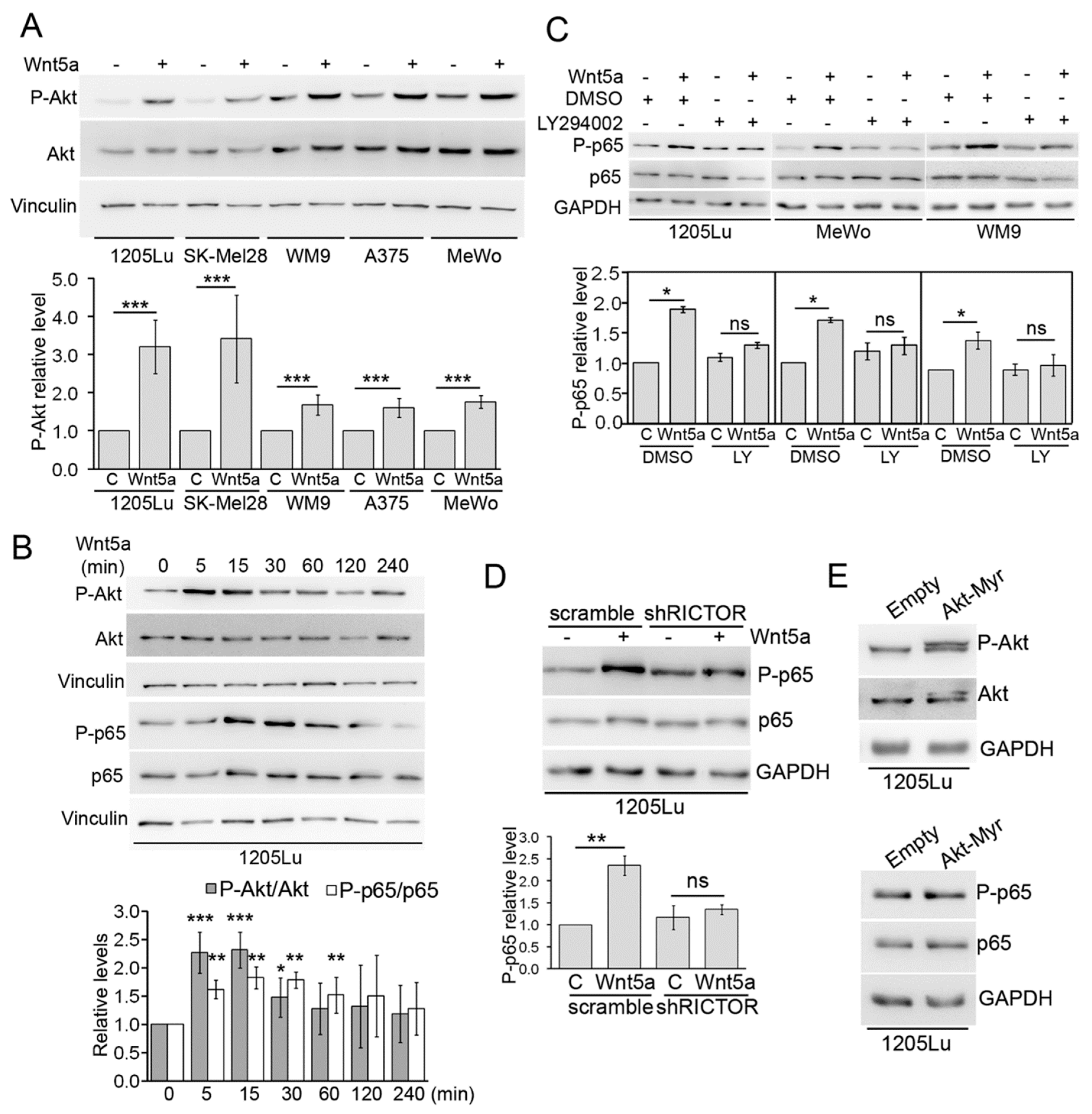
3.2. ROR1, Dvl2, and Akt are Required for Wnt5a-Dependent Activation of NF-κB
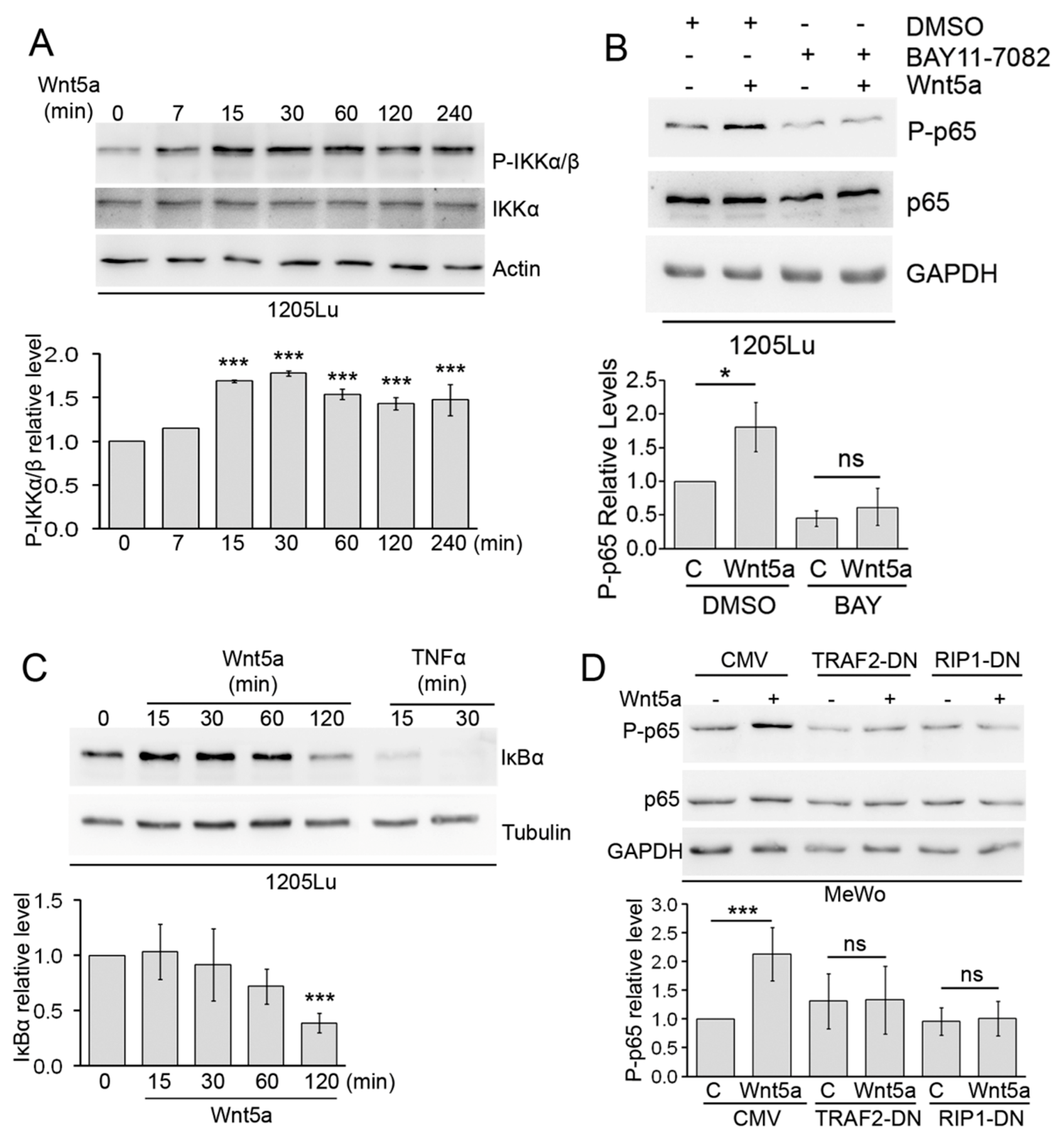
3.3. Wnt5a Activates Canonical Components of the NF-κB Pathway
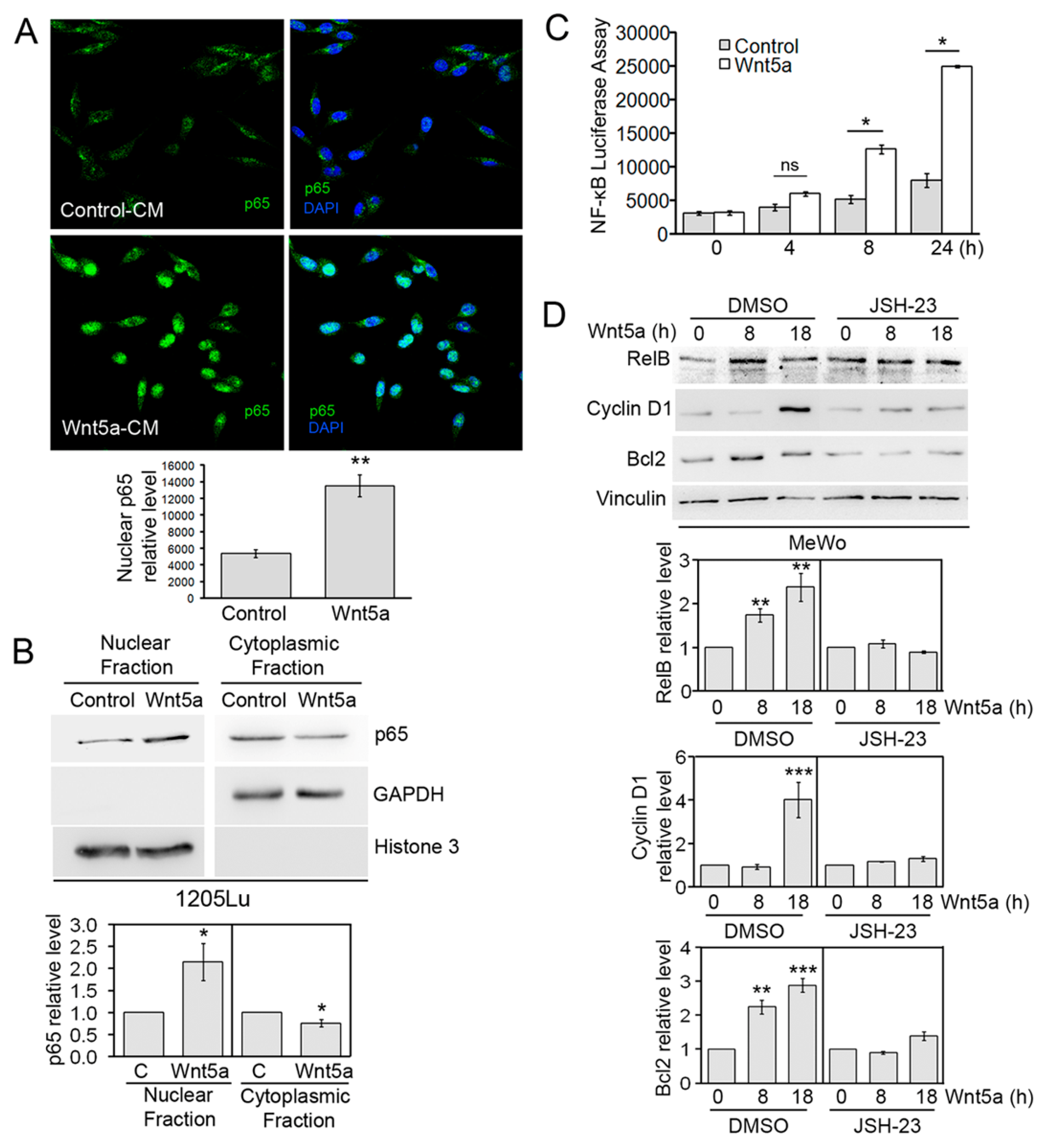
3.4. Wnt5a-Induced NF-κB Transcriptional Activity
3.5. Wnt5a Induced Secretion of Cytokines and Chemokines by Melanoma Cells
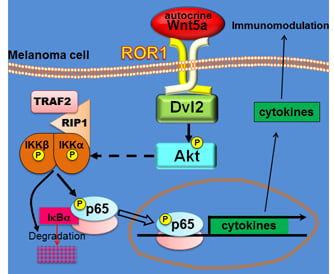
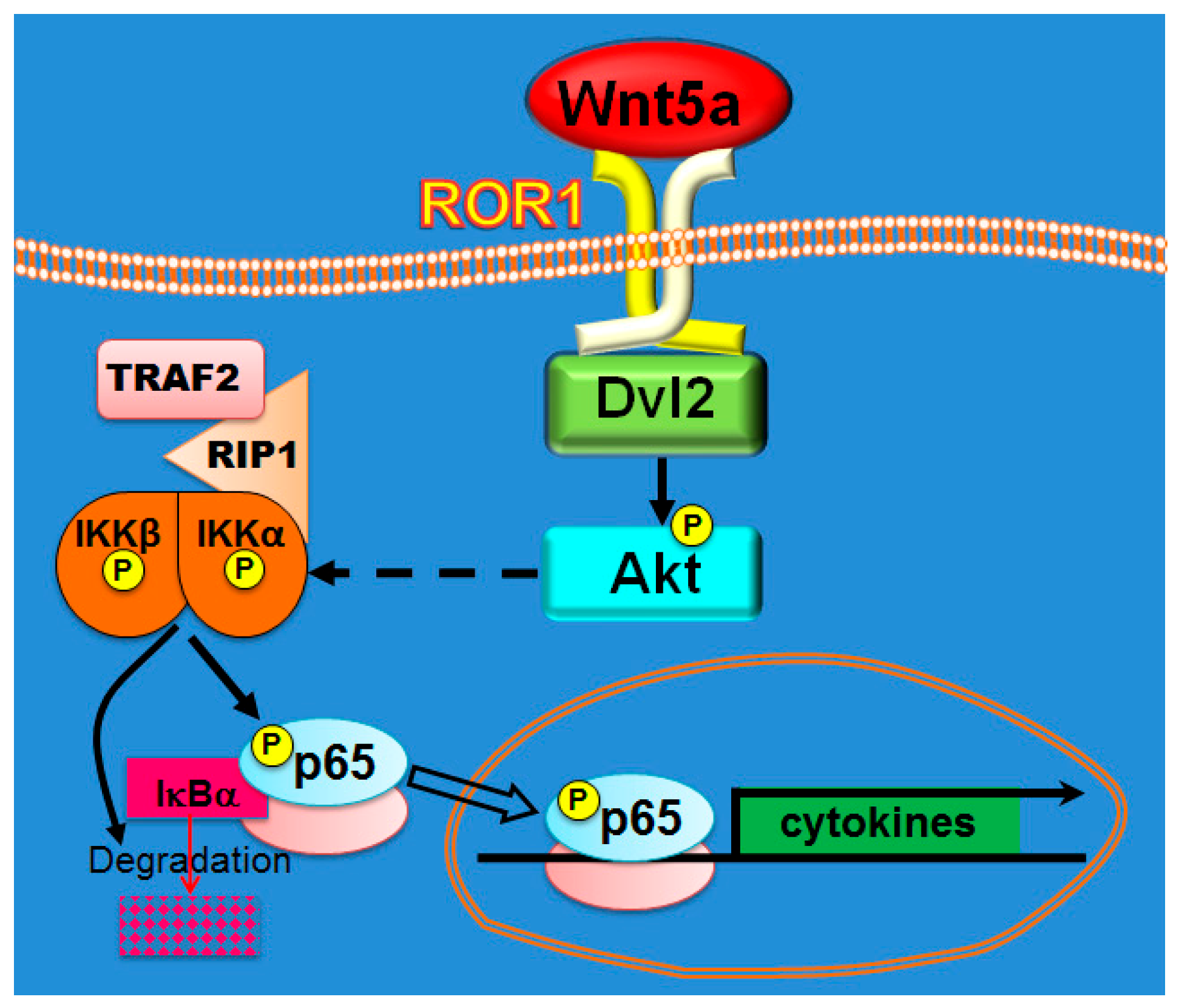
3.6. Melanoma Cells Present a Wnt5a Autocrine Circuit to Activate NF-κB
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures 2018; American Cancer Society: Atlanta, GA, USA, 2018. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.A.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies-update 2017. Eur. J. Cancer 2017, 83, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Eldabaje, R.; Yang, L. Current Status of Biological Therapies for the Treatment of Metastatic Melanoma. Anticancer Res. 2016, 36, 3229–3241. [Google Scholar] [PubMed]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a Signaling in Cancer. Cancers 2016, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, R.; Mikels, A.; Nusse, R. Alternative wnt signaling is initiated by distinct receptors. Sci. Signal. 2009. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. WNT/PCP signaling pathway and human cancer (review). Oncol. Rep. 2005, 14, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Nishita, M.; Fujii, M.; Minami, Y. Insight into the role of Wnt5a-induced signaling in normal and cancer cells. Int. Rev. Cell Mol. Biol. 2015, 314, 117–148. [Google Scholar] [PubMed]
- Kikuchi, A.; Yamamoto, H.; Sato, A.; Matsumoto, S. Wnt5a: Its signalling, functions and implication in diseases. Acta Physiol. (Oxf.) 2012, 204, 17–33. [Google Scholar] [CrossRef]
- McDonald, S.L.; Silver, A. The opposing roles of Wnt-5a in cancer. Br. J. Cancer 2009, 101, 209–214. [Google Scholar] [CrossRef]
- Blanc, E.; Roux, G.L.; Benard, J.; Raguene, G. Low expression of Wnt-5a gene is associated with high-risk neuroblastoma. Oncogene 2005, 24, 1277–1283. [Google Scholar] [CrossRef]
- Liang, H.; Chen, Q.; Coles, A.H.; Anderson, S.J.; Pihan, G.; Bradley, A.; Gerstein, R.; Jurecic, R.; Jones, S.N. Wnt5a inhibits B cell proliferation and functions as a tumor suppressor in hematopoietic tissue. Cancer Cell 2003, 4, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, M.; Dejmek, J.; Bendahl, P.O.; Andersson, T. Loss of Wnt-5a protein is associated with early relapse in invasive ductal breast carcinomas. Cancer Res. 2002, 62, 409–416. [Google Scholar] [PubMed]
- Kobayashi, Y.; Kadoya, T.; Amioka, A.; Hanaki, H.; Sasada, S.; Masumoto, N.; Yamamoto, H.; Arihiro, K.; Kikuchi, A.; Okada, M. Wnt5a-induced cell migration is associated with the aggressiveness of estrogen receptor-positive breast cancer. Oncotarget 2018, 9, 20979–20992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.; Sun, B.; Liu, Z.; Zhao, X.; Qi, L.; Li, Y.; Gu, Q. Wnt5a suppresses colon cancer by inhibiting cell proliferation and epithelial-mesenchymal transition. J. Cell. Physiol. 2014, 229, 1908–1917. [Google Scholar] [CrossRef] [PubMed]
- Kremenevskaja, N.; von Wasielewski, R.; Rao, A.S.; Schofl, C.; Andersson, T.; Brabant, G. Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene 2005, 24, 2144–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Tu, Z.; Xiong, M.; Tembo, K.; Zhou, L.; Liu, P.; Pan, S.; Xiong, J.; Yang, X.; Leng, J.; et al. Wnt5a and CCL25 promote adult T-cell acute lymphoblastic leukemia cell migration, invasion and metastasis. Oncotarget 2017, 8, 39033–39047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Forno, P.D.; Pringle, J.H.; Hutchinson, P.; Osborn, J.; Huang, Q.; Potter, L.; Hancox, R.A.; Fletcher, A.; Saldanha, G.S. WNT5A expression increases during melanoma progression and correlates with outcome. Clin. Cancer Res. 2008, 14, 5825–5832. [Google Scholar] [CrossRef]
- Kurayoshi, M.; Oue, N.; Yamamoto, H.; Kishida, M.; Inoue, A.; Asahara, T.; Yasui, W.; Kikuchi, A. Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res. 2006, 66, 10439–10448. [Google Scholar] [CrossRef]
- Huang, C.L.; Liu, D.; Nakano, J.; Ishikawa, S.; Kontani, K.; Yokomise, H.; Ueno, M. Wnt5a expression is associated with the tumor proliferation and the stromal vascular endothelial growth factor—An expression in non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 8765–8773. [Google Scholar] [CrossRef]
- Ripka, S.; Konig, A.; Buchholz, M.; Wagner, M.; Sipos, B.; Kloppel, G.; Downward, J.; Gress, T.; Michl, P. WNT5A—Target of CUTL1 and potent modulator of tumor cell migration and invasion in pancreatic cancer. Carcinogenesis 2007, 28, 1178–1187. [Google Scholar] [CrossRef]
- Yamamoto, H.; Oue, N.; Sato, A.; Hasegawa, Y.; Yamamoto, H.; Matsubara, A.; Yasui, W.; Kikuchi, A. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene 2010, 29, 2036–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, C.P.; Mohapatra, P.; Andersson, T. Therapy for BRAFi-Resistant Melanomas: Is WNT5A the Answer? Cancers (Basel) 2015, 7, 1900–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pashirzad, M.; Shafiee, M.; Rahmani, F.; Behnam-Rassouli, R.; Hoseinkhani, F.; Ryzhikov, M.; Moradi Binabaj, M.; Parizadeh, M.R.; Avan, A.; Hassanian, S.M. Role of Wnt5a in the Pathogenesis of Inflammatory Diseases. J. Cell. Physiol. 2017, 232, 1611–1616. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Zheng, Q.; Wang, W.; Xin, N.; Song, X.; Zhao, C. Biological functions of macrophage-derived Wnt5a, and its roles in human diseases. Oncotarget 2016, 7, 67674–67684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanzawa, M.; Semba, S.; Hara, S.; Itoh, T.; Yokozaki, H. WNT5A is a key regulator of the epithelial-mesenchymal transition and cancer stem cell properties in human gastric carcinoma cells. Pathobiology 2013, 80, 235–244. [Google Scholar] [CrossRef]
- Arabzadeh, S.; Hossein, G.; Zarnani, A.H. Wnt5A exerts immunomodulatory activity in the human ovarian cancer cell line SKOV-3. Cell Biol. Int. 2016, 40, 177–187. [Google Scholar] [CrossRef]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef]
- Ueda, Y.; Richmond, A. NF-kappaB activation in melanoma. Pigment Cell Res. 2006, 19, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Werner, S.L.; Barken, D.; Hoffmann, A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science 2005, 309, 1857–1861. [Google Scholar] [CrossRef] [PubMed]
- Picco, M.E.; Castro, M.V.; Quezada, M.J.; Barbero, G.; Villanueva, M.B.; Fernández, N.B.; Kim, H.; Lopez-Bergami, P. STAT3 enhances the constitutive activity of AGC kinases in melanoma by transactivating PDK1. Cell Biosci. 2019, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Fernández, N.B.; Lorenzo, D.; Picco, M.E.; Barbero, G.; Dergan-Dylon, L.S.; Marks, M.P.; García-Rivello, H.; Gimenez, L.; Labovsky, V.; Grumolato, L.; et al. ROR1 contributes to melanoma cell growth and migration by regulating N-cadherin expression via the PI3K/Akt pathway. Mol. Carcinog. 2016, 55, 1772–1785. [Google Scholar] [CrossRef] [PubMed]
- DeMorrow, S.; Francis, H.; Gaudio, E.; Venter, J.; Franchitto, A.; Kopriva, S.; Onori, P.; Mancinelli, R.; Frampton, G.; Coufal, M.; et al. The endocannabinoid anandamide inhibits cholangiocarcinoma growth via activation of the noncanonical Wnt signaling pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G1150-8. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Mao, J.; Sun, L.; Liu, W.; Wu, D. Second cysteine-rich domain of Dickkopf-2 activates canonical Wnt signaling pathway via LRP-6 independently of dishevelled. J. Biol. Chem. 2002, 277, 5977–5981. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Bafico, A.; Aaronson, S.A. The mechanism of endogenous receptor activation functionally distinguishes prototype canonical and noncanonical Wnts. Mol. Cell. Biol. 2005, 25, 3475–3482. [Google Scholar] [CrossRef]
- Lopez-Bergami, P.; Huang, C.; Goydos, J.S.; Yip, D.; Bar-Eli, M.; Herlyn, M.; Smalley, K.S.; Mahale, A.; Eroshkin, A.; Aaronson, S.; et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell 2007, 11, 447–460. [Google Scholar] [CrossRef]
- Smale, S.T. Beta-galactosidase assay. Cold Spring Harb. Protoc. 2010. [Google Scholar] [CrossRef]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef]
- Chen, B.; Dodge, M.; Tang, M.; Lu, J.; Ma, Z.; Fan, C.; Wei, S.; Hao, W.; Kilgore, J.; Williams, N.S.; et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Bebien, M.; Liu, G.Y.; Nizet, V.; Karin, M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature 2005, 434, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.V.; Barbero, G.; Lopez Bergami, P. ROR2 promotes melanoma progression. Unpublished work. 2019. [Google Scholar]
- Whang, Y.M.; Jo, U.; Sung, J.S.; Ju, H.J.; Kim, H.K.; Park, K.H.; Lee, J.W.; Koh, I.S.; Kim, Y.H. Wnt5a is associated with cigarette smoke-related lung carcinogenesis via protein kinase C. PLoS ONE 2013, 8, e53012. [Google Scholar] [CrossRef] [PubMed]
- Freland, L.; Beaulieu, J.M. Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front. Mol. Neurosci. 2012, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habelhah, H.; Takahashi, S.; Cho, S.G.; Kadoya, T.; Watanabe, T.; Ronai, Z. Ubiquitination and translocation of TRAF2 is required for activation of JNK but not of p38 or NF-kappaB. EMBO J. 2004, 23, 322–332. [Google Scholar] [CrossRef]
- Amschler, K.; Schön, M.P.; Pletz, N.; Wallbrecht, K.; Erpenbeck, L.; Schön, M. NF-kappaB inhibition through proteasome inhibition or IKKbeta blockade increases the susceptibility of melanoma cells to cytostatic treatment through distinct pathways. J. Investig. Dermatol. 2010, 130, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.M.; Kim, M.H.; Kim, B.H.; Jung, S.H.; Kim, Y.S.; Park, H.J.; Hong, J.T.; Min, K.R.; Kim, Y. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett. 2004, 571, 50–54. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Jenei, V.; Sherwood, V.; Howlin, J.; Linnskog, R.; Säfholm, A.; Axelsson, L.; Andersson, T. A t-butyloxycarbonyl-modified Wnt5a-derived hexapeptide functions as a potent antagonist of Wnt5a-dependent melanoma cell invasion. Proc. Natl. Acad. Sci. USA 2009, 106, 19473–19478. [Google Scholar] [CrossRef] [Green Version]
- Baarsma, H.A.; Skronska-Wasek, W.; Mutze, K.; Ciolek, F.; Wagner, D.E.; John-Schuster, G.; Heinzelmann, K.; Günther, A.; Bracke, K.R.; Dagouassat, M.; et al. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J. Exp. Med. 2017, 214, 143–163. [Google Scholar] [CrossRef]
- Weeraratna, A.T.; Jiang, Y.; Hostetter, G.; Rosenblatt, K.; Duray, P.; Bittner, M.; Trent, J.M. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 2002, 1, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Dissanayake, S.K.; Wade, M.; Johnson, C.E.; O’Connell, M.P.; Leotlela, P.D.; French, A.D.; Shah, K.V.; Hewitt, K.J.; Rosenthal, D.T.; Indig, F.E.; et al. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 17259–17271. [Google Scholar] [CrossRef] [PubMed]
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 2006, 19, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.T.; Kwon, S.J.; Kim, J.; Kwon, Y.S.; Lee, N.; Hong, J.H.; Jamieson, C.; Kim, W.J.; Kim, I.Y. WNT5A induces castration-resistant prostate cancer via CCL2 and tumour-infiltrating macrophages. Br. J. Cancer 2018, 118, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, W.; Zhang, N.; Ma, T.; Zhao, C. IL-1β mediates MCP-1 induction by Wnt5a in gastric cancer cells. BMC Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, E.J.; Bergenfelz, C.; von Bulow, V.; Serifler, F.; Carlemalm, E.; Jonsson, G.; Andersson, T.; Leandersson, K. Wnt5a induces release of exosomes containing pro-angiogenic and immunosuppressive factor from malignant melanoma cells. Mol. Cancer 2014, 13, 88. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, K.; Gosens, R. WNT-5A: Signaling and functions in health and disease. Cell. Mol. Life Sci. 2016, 73, 567–587. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.T.; Kang, D.I.; Ha, Y.S.; Jung, Y.S.; Chung, J.; Min, K.; Kim, T.H.; Moon, K.H.; Chung, J.M.; Lee, D.H.; et al. Prostate cancer bone metastases acquire resistance to androgen deprivation via WNT5A-mediated BMP-6 induction. Br. J. Cancer 2014, 110, 1634–1644. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Zhou, B.; Qu, Y.; Gao, B.; Xu, Y.; Chung, S.; Tanaka, H.; Yang, W.; Giuliano, A.E.; Cui, X. FOXC1-induced non-canonical WNT5A-MMP7 signaling regulates invasiveness in triple-negative breast cancer. Oncogene 2018, 37, 1399–1408. [Google Scholar] [CrossRef]
- Zeng, R.; Huang, J.; Zhong, M.Z.; Li, L.; Yang, G.; Liu, L.; Wu, Y.; Yao, X.; Shi, J.; Wu, Z. Multiple Roles of WNT5A in Breast Cancer. Med. Sci. Monit. 2016, 22, 5058–5067. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Chen, L.; Endo, T.; Tang, L.; Lu, D.; Castro, J.E.; Widhopf, G.F., 2nd; Rassenti, L.Z.; Cantwell, M.J.; Prussak, C.E.; et al. Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc. Natl. Acad. Sci. USA 2008, 105, 3047–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Horta, O.; Abad, C.; Sennaroglu, L.; Foster, J.; DeSmidt, A.; Bademci, G.; Tokgoz-Yilmaz, S.; Duman, D.; Cengiz, F.B.; Grati, M.; et al. ROR1 is essential for proper innervation of auditory hair cells and hearing in humans and mice. Proc. Natl. Acad. Sci. USA 2016, 113, 5993–5998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Zhao, Y.; Jiang, G.; Zhang, X.; Zhang, Y.; Dong, Q.; Luan, L.; Papavassiliou, P.; Wang, E.; Wang, E. Dishevelled-3 activates p65 to upregulate p120-catenin transcription via a p38-dependent pathway in non-small cell lung cancer. Mol. Carcinog. 2015, 54, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Kulikauskas, R.M.; Tamir, T.; Rizos, H.; Long, G.V.; von Euw, E.M.; Yang, P.T.; Chen, H.W.; Haydu, L.; Toroni, R.A.; et al. WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J. Clin. Investig. 2014, 124, 2877–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhawan, P.; Singh, A.B.; Ellis, D.L.; Richmond, A. Constitutive activation of Akt/protein kinase B in melanoma leads to up-regulation of nuclear factor-kappaB and tumor progression. Cancer Res. 2002, 62, 7335–7342. [Google Scholar] [PubMed]
- Hussain, A.; Ahmed, S.; Ahmed, M.; Khan, O.; AbdulMohsen, S.; Platanias, L.; Al-Kuraya, K.S.; Uddin, S. Cross-Talk between NFkB and the PI3-Kinase/AKT Pathway Can Be Targeted in Primary Effusion Lymphoma (PEL) Cell Lines for Efficient Apoptosis. PLoS ONE 2012, 7, e39945. [Google Scholar] [CrossRef]
- Chen, S.; Chen, W.; Zhang, X.; Lin, S.; Chen, Z. Overexpression of KiSS-1 reduces colorectal cancer cell invasion by downregulating MMP-9 via blocking PI3K/Akt/NF-kB signal pathway. Int. J. Oncol. 2016, 48, 1391–1398. [Google Scholar] [CrossRef]
- Gu, Y.; Liu, H.; Kong, F.; Ye, J.; Jia, X.; Zhang, Z.; Li, N.; Yin, J.; Zheng, G.; He, Z. miR-22/KAT6B axis is a chemotherapeutic determiner via regulation of PI3k-Akt-NF-kB pathway in tongue squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 164. [Google Scholar] [CrossRef]
- Madrid, L.V.; Mayo, M.W.; Reuther, J.Y.; Baldwin, A.S. Akt stimulates the transactivation potential of RelA/p65 subunit of NF-κB through the utilization of IκB kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 2001, 276, 18934–18940. [Google Scholar] [CrossRef]
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.; Baldwin, A.S. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes. Dev. 2008, 22, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; London, N.; Schueler-Furman, O.; Ben-Neriah, Y. Ubiquitination and degradation of the inhibitors of NF-kappaB. Cold Spring Harb. Perspect. Biol. 2010, 2, a000166. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.; Kim, D.W.; Ha, Y.; Ihm, M.H.; Kim, H.; Song, K.; Lee, I. Wnt5a induces endothelial inflammation via beta-catenin-independent signaling. J. Immunol. 2010, 185, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Chang, W.; Jung, Y.; Song, K.; Lee, I. Wnt5a activates THP-1 monocytic cells via a β-catenin-independent pathway involving JNK and NF-κB activation. Cytokine 2012, 60, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Rauner, M.; Stein, N.; Winzer, M.; Goettsch, C.; Zwerina, J.; Schett, G.; Distler, J.H.; Albers, J.; Schulze, J.; Schinke, T. WNT5A is induced by inflammatory mediators in bone marrow stromal cells and regulates cytokine and chemokine production. J. Bone Miner. Res. 2012, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, C.L.; Li, R.M.; Hou, T.Q.; Su, Y.Y.; Yuan, Q. Wnt5a Promotes Inflammatory Responses via NF-κB and MAPK Pathways in Human Dental Pulp Cells. J. Biol. Chem. 2014, 289, 21028–21039. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Masjedi, A.; Hashemi, V.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. Biomed. Pharmacother. 2018, 108, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-Associated Neutrophils in Cancer: Going Pro. Cancers (Basel) 2019, 11, 564. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M.; Katoh, M. Transcriptional mechanisms of WNT5A based on NF-kappaB, Hedgehog, TGFbeta, and Notch signaling cascades. Int. J. Mol. Med. 2009, 23, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, S.K.; Olkhanud, P.B.; O’Connell, M.P.; Carter, A.; French, A.D.; Camilli, T.C.; Emeche, C.D.; Hewitt, K.J.; Rosenthal, D.T.; Leotlela, P.D. Wnt5A regulates expression of tumor-associated antigens in melanoma via changes in signal transducers and activators of transcription 3 phosphorylation. Cancer Res. 2008, 68, 10205–10214. [Google Scholar] [CrossRef]
- Linnskog, R.; Mohapatra, P.; Moradi, F.; Prasad, C.P.; Andersson, T. Demonstration of a WNT5A-IL-6 positive feedback loop in melanoma cells: Dual interference of this loop more effectively impairs melanoma cell invasion. Oncotarget 2016, 7, 37790–37802. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbero, G.; Castro, M.V.; Villanueva, M.B.; Quezada, M.J.; Fernández, N.B.; DeMorrow, S.; Lopez-Bergami, P. An Autocrine Wnt5a Loop Promotes NF-κB Pathway Activation and Cytokine/Chemokine Secretion in Melanoma. Cells 2019, 8, 1060. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8091060
Barbero G, Castro MV, Villanueva MB, Quezada MJ, Fernández NB, DeMorrow S, Lopez-Bergami P. An Autocrine Wnt5a Loop Promotes NF-κB Pathway Activation and Cytokine/Chemokine Secretion in Melanoma. Cells. 2019; 8(9):1060. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8091060
Chicago/Turabian StyleBarbero, Gastón, María Victoria Castro, María Belén Villanueva, María Josefina Quezada, Natalia Brenda Fernández, Sharon DeMorrow, and Pablo Lopez-Bergami. 2019. "An Autocrine Wnt5a Loop Promotes NF-κB Pathway Activation and Cytokine/Chemokine Secretion in Melanoma" Cells 8, no. 9: 1060. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8091060