Electronegative LDL Promotes Inflammation and Triglyceride Accumulation in Macrophages

 , , , and
, , , and
Abstract
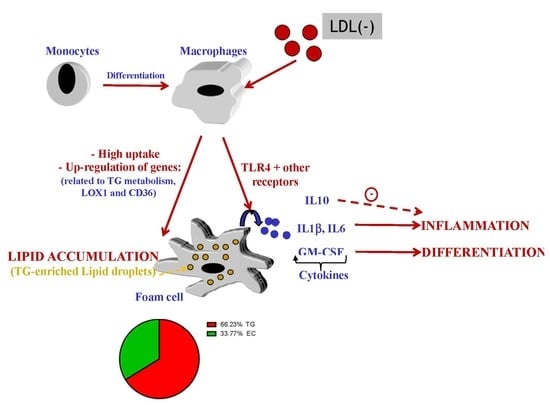
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Lipoprotein Isolation
2.2. Incubation of LDL with Monocytes/Macrophages
2.3. Flow Cytometry
2.4. Lactate Dehydrogenase (LDH) Test and ELISA
2.5. Uptake of DiI-Labeled LDL(−)
2.6. Intracellular Lipid Accumulation
2.7. Real-Time Polymerase Chain Reaction (RT-PCR)
2.8. Statistical Analysis
3. Results
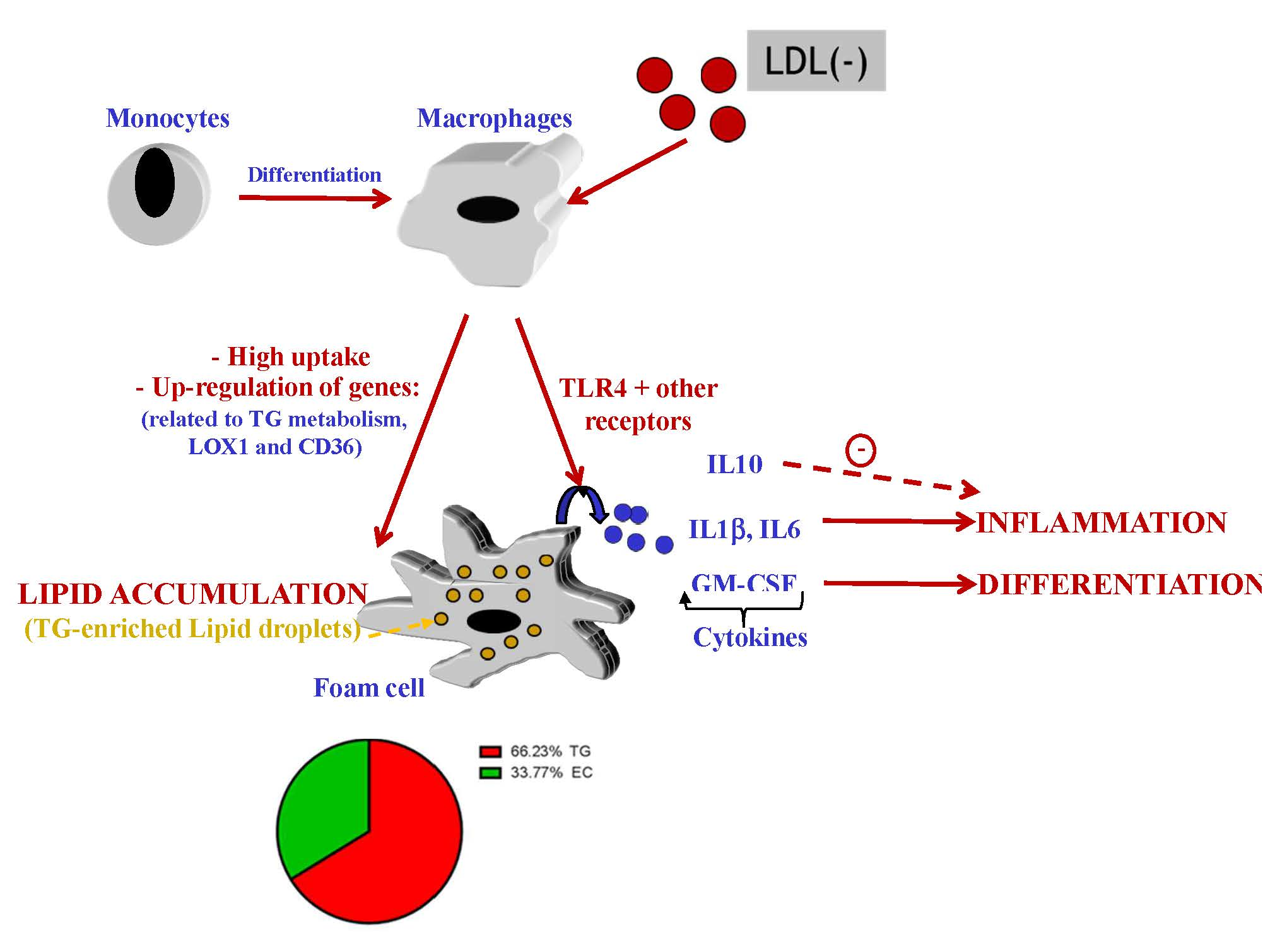
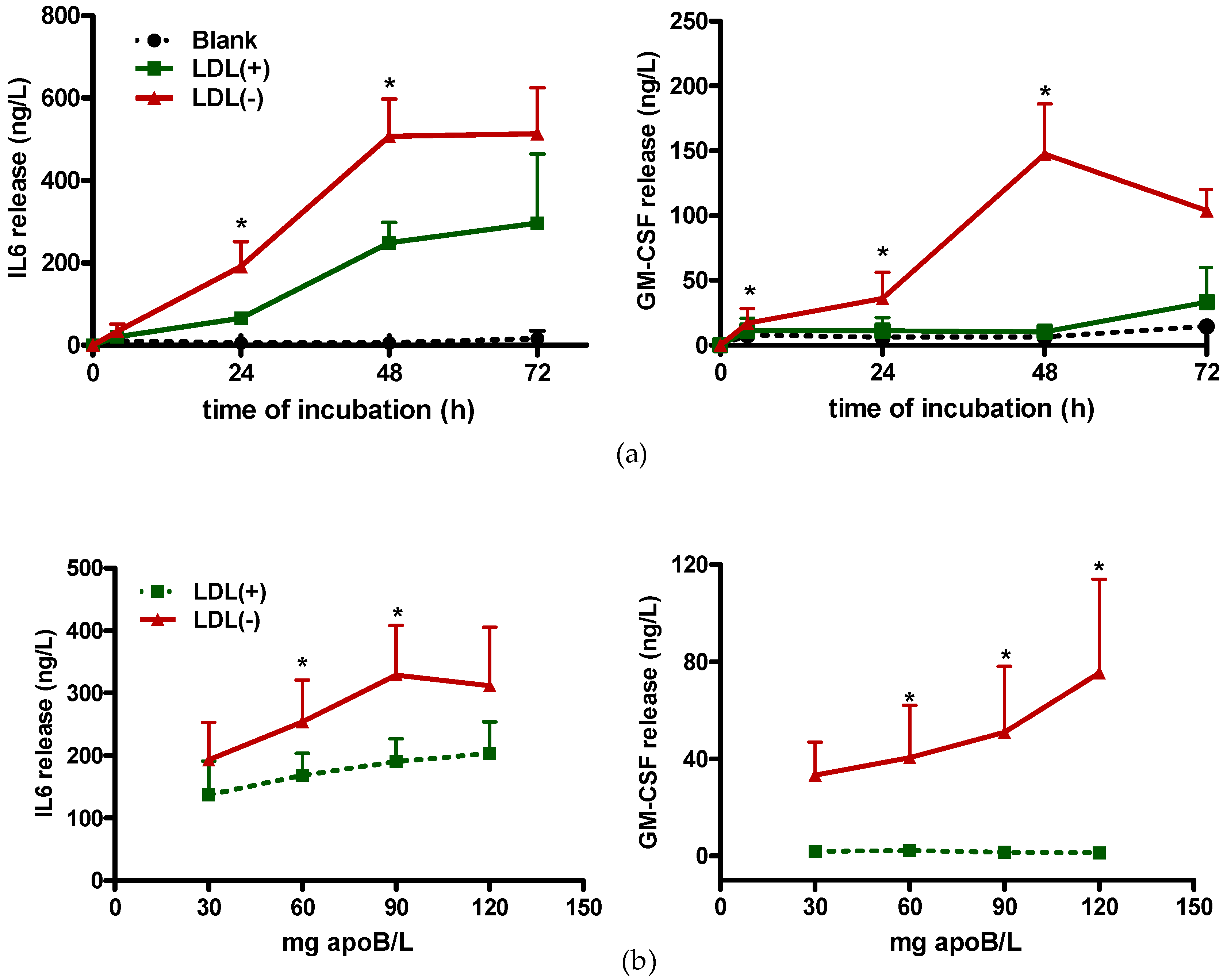
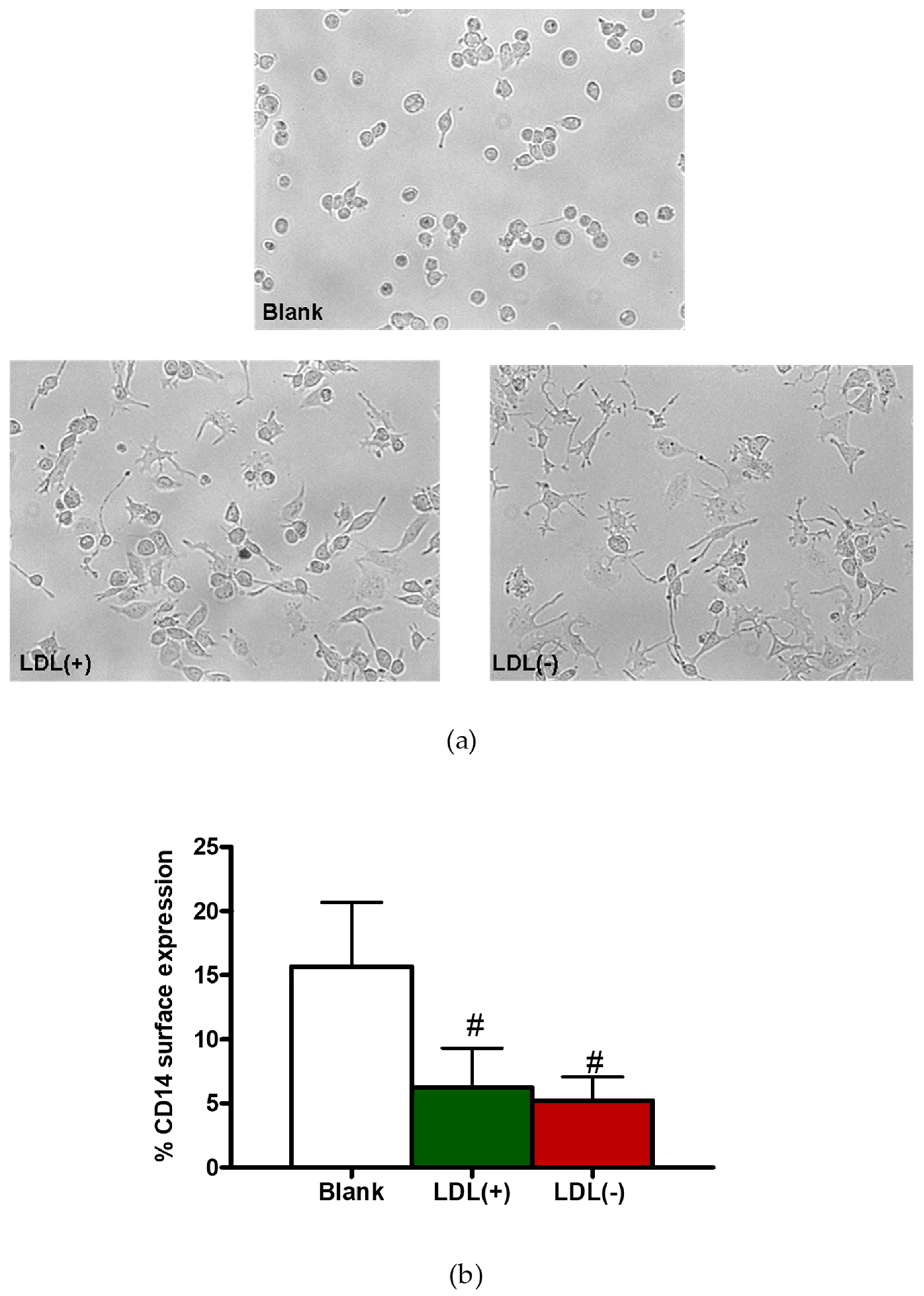
3.1. LDL(−) Induces Cytokine Release in Macrophages
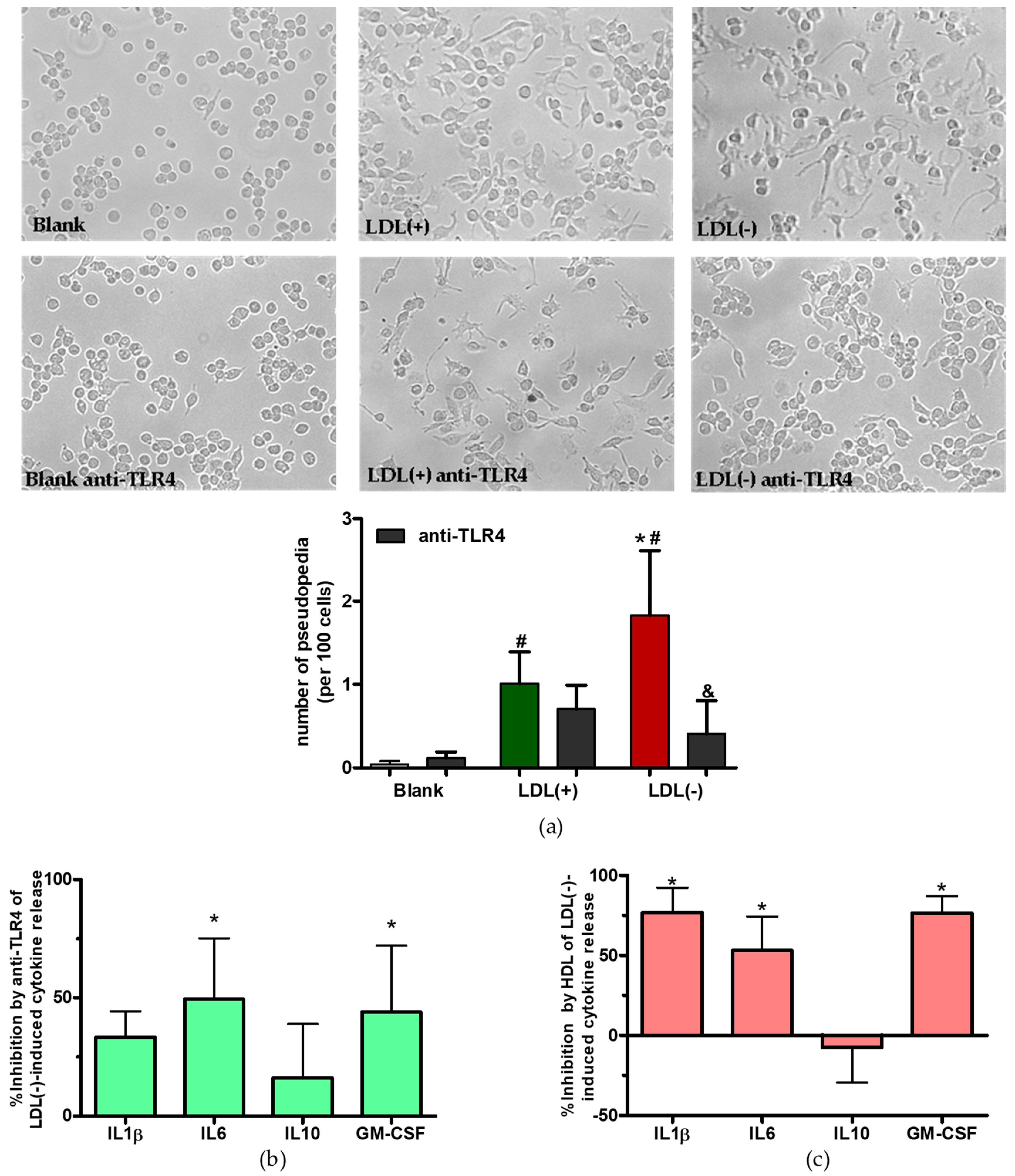
3.2. LDL(−) Induces Differentiation in Macrophages
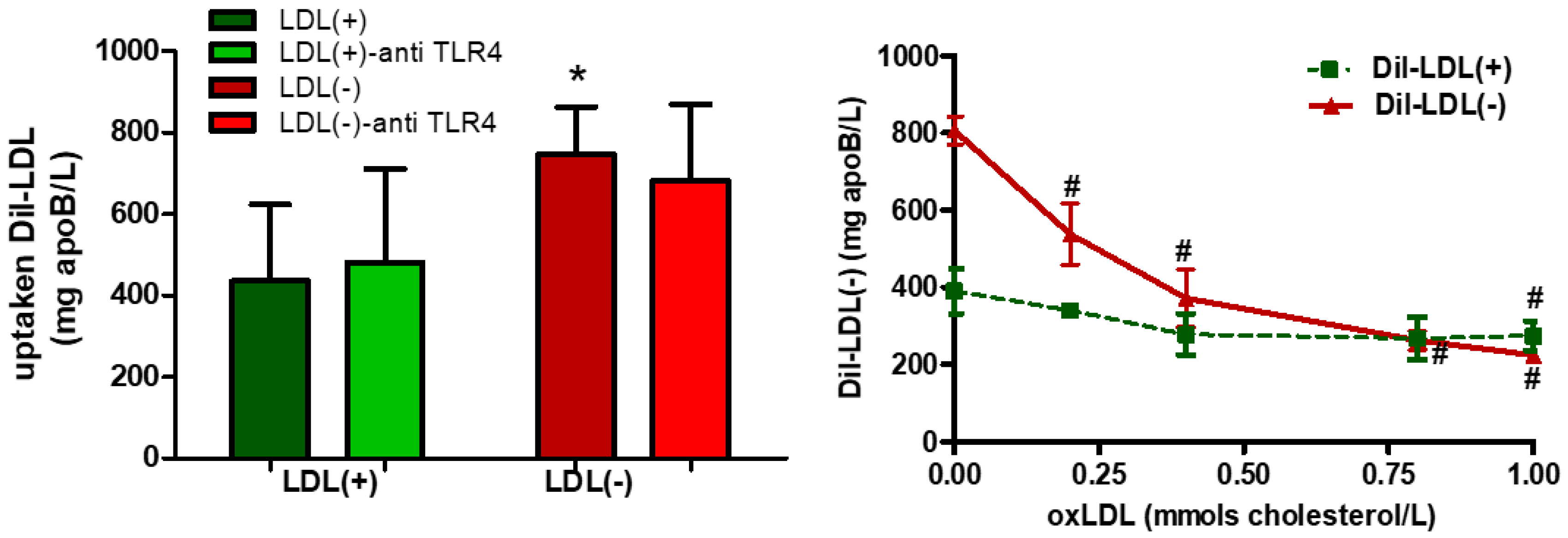
3.3. Role of TLR4 and HDL on LDL(−)-Induced Inflammation and Differentiation
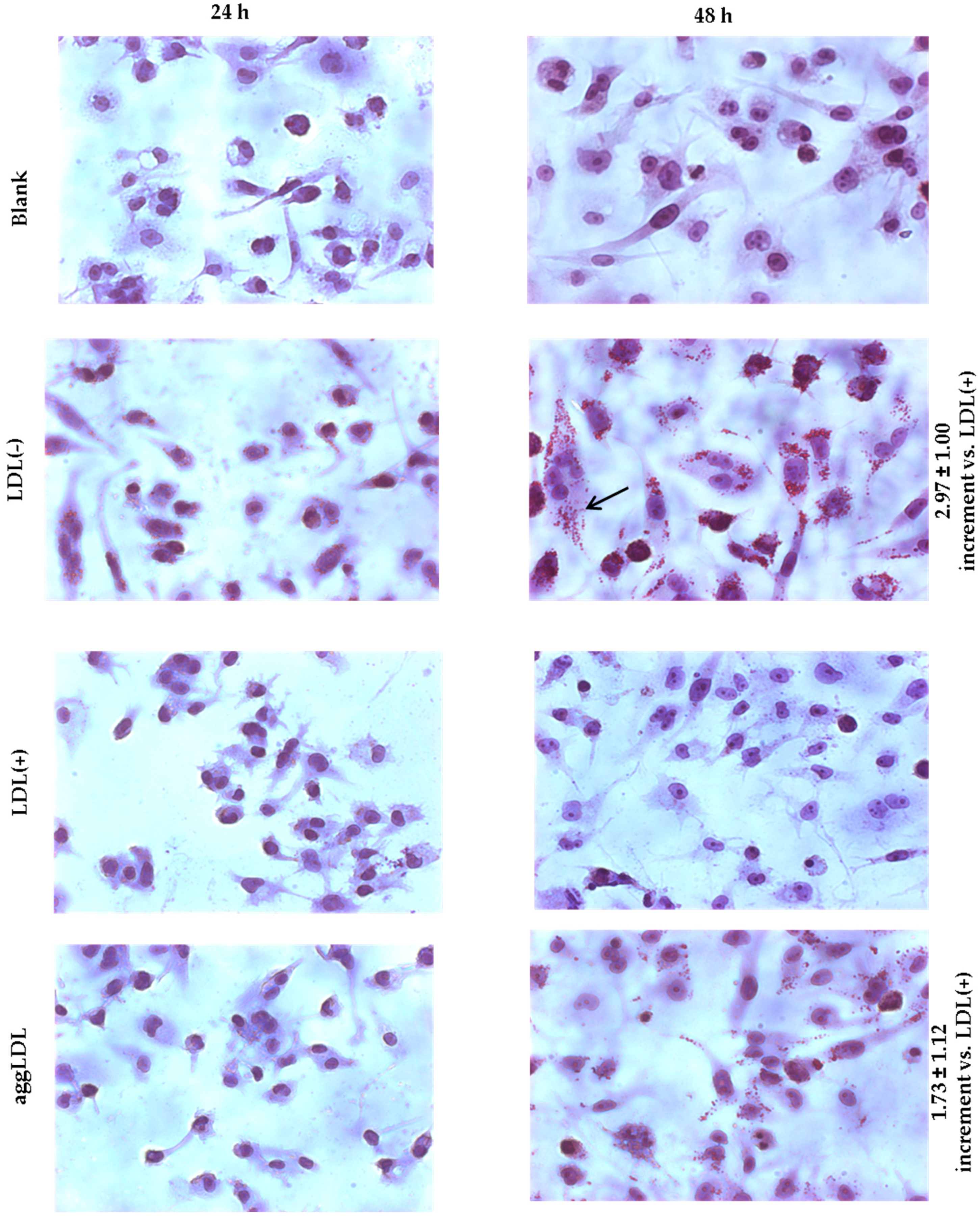
3.4. LDL(−) is More Avidly Internalized than LDL(+) and it Induces LD Formation
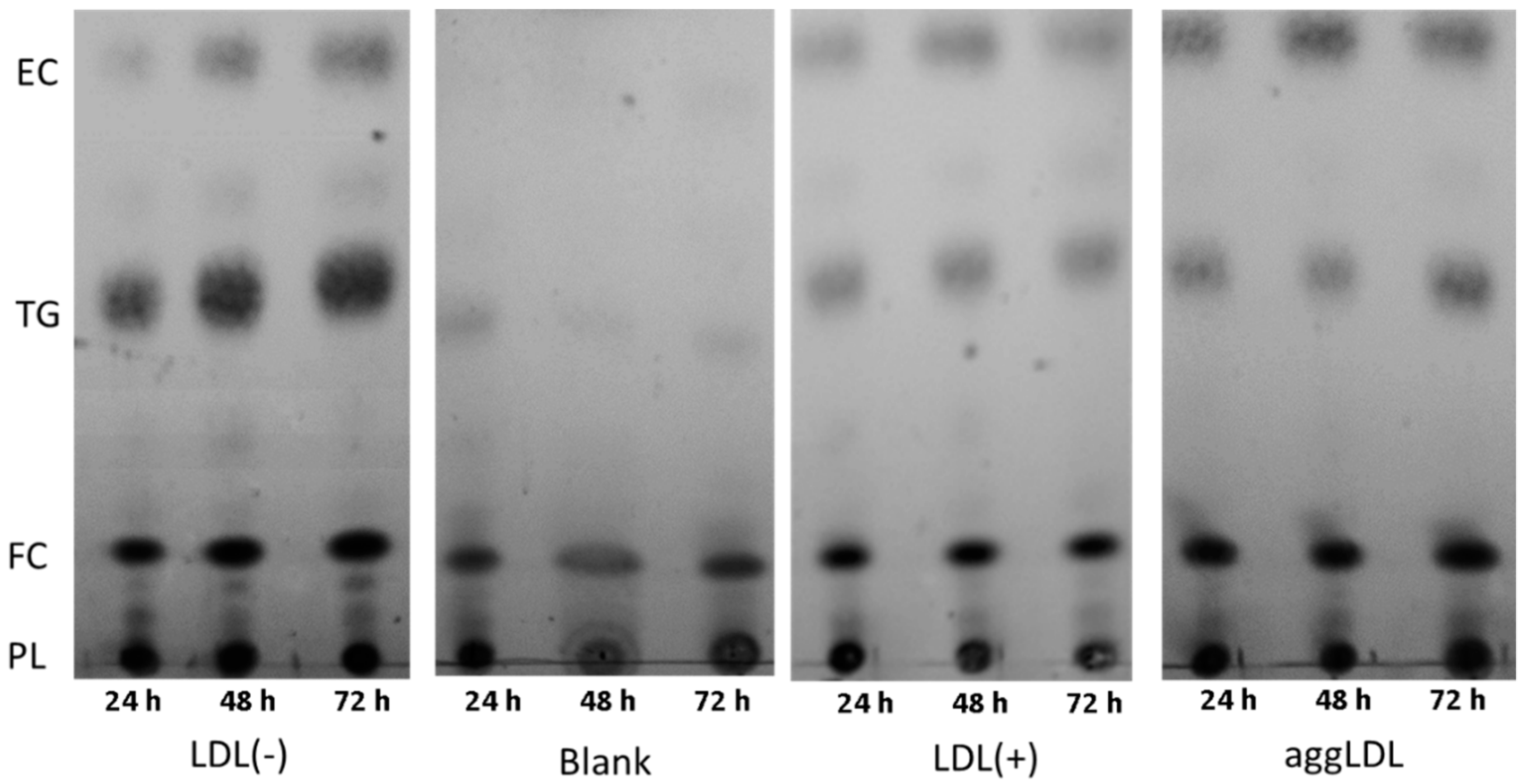
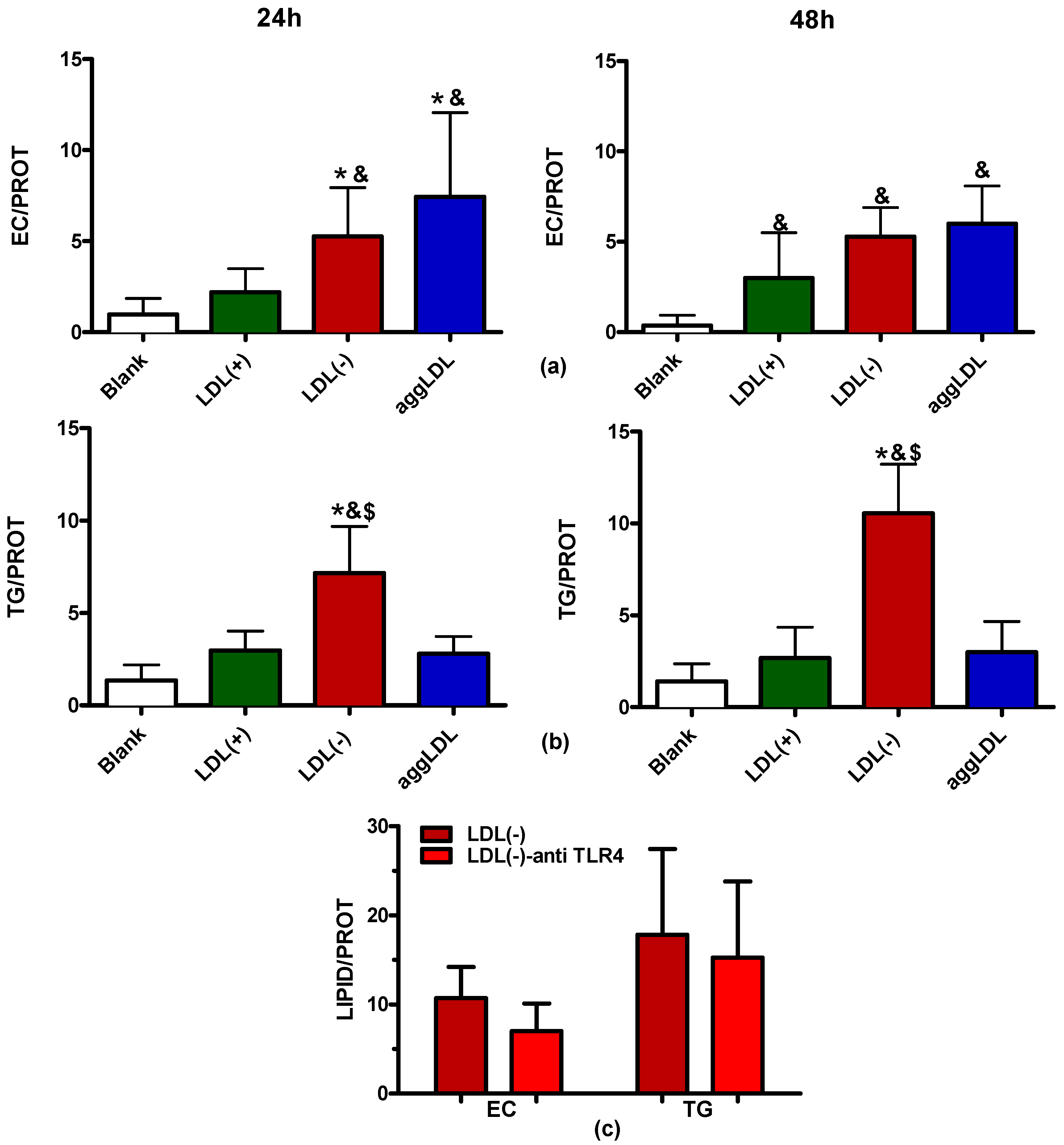
3.5. LDL(−) Induces Particularly Triglyceride (TG) Accumulation
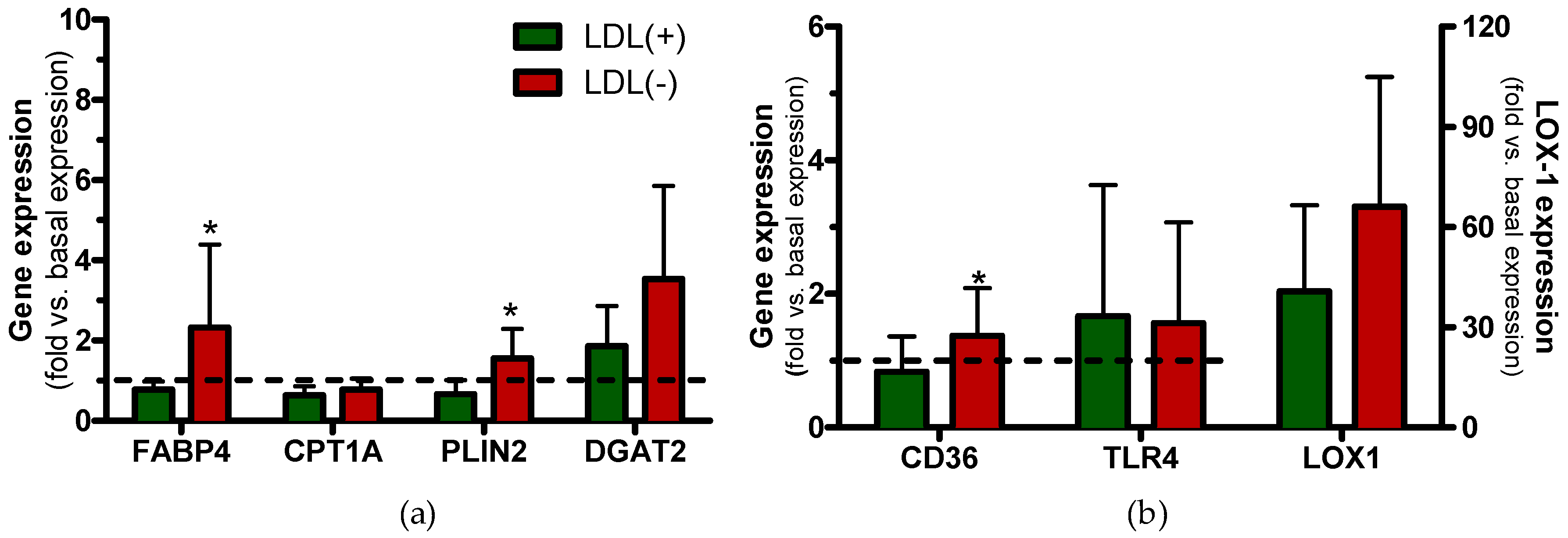
3.6. LDL(−) Promotes Changes in Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and their role in atherosclerosis: Pathophysiology and transcriptome analysis. BioMed Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.S.; Basu, S.K.; Falck, J.R.; Ho, Y.K.; Goldstein, J.L. The scavenger cell pathway for lipoprotein degradation: Specificity of the binding site that mediates the uptake of negatively-charged ldl by macrophages. J. Supramol. Struct. 1980, 13, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Zani, I.A.; Stephen, S.L.; Mughal, N.A.; Russell, D.; Homer-Vanniasinkam, S.; Wheatcroft, S.B.; Ponnambalam, S. Scavenger receptor structure and function in health and disease. Cells 2015, 4, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Orekhov, A.N.; Sobenin, I.A. Modified lipoproteins as biomarkers of atherosclerosis. Front. Biosci. 2018, 23, 1422–1444. [Google Scholar] [CrossRef] [Green Version]
- Estruch, M.; Sanchez-Quesada, J.L.; Ordonez Llanos, J.; Benitez, S. Electronegative ldl: A circulating modified ldl with a role in inflammation. Mediat. Inflamm. 2013, 2013, 181324. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.C.; Ke, L.Y.; Chu, C.S.; Lee, A.S.; Shen, M.Y.; Cruz, M.A.; Hsu, J.F.; Cheng, K.H.; Chan, H.C.; Lu, J.; et al. Highly electronegative ldl from patients with st-elevation myocardial infarction triggers platelet activation and aggregation. Blood 2013, 122, 3632–3641. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.Y.; Chen, F.Y.; Hsu, J.F.; Fu, R.H.; Chang, C.M.; Chang, C.T.; Liu, C.H.; Wu, J.R.; Lee, A.S.; Chan, H.C.; et al. Plasma l5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood 2016, 127, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Niccoli, G.; Baca, M.; De Spirito, M.; Parasassi, T.; Cosentino, N.; Greco, G.; Conte, M.; Montone, R.A.; Arcovito, G.; Crea, F. Impact of electronegative low-density lipoprotein on angiographic coronary atherosclerotic burden. Atherosclerosis 2012, 223, 166–170. [Google Scholar] [CrossRef]
- Chen, W.Y.; Chen, F.Y.; Lee, A.S.; Ting, K.H.; Chang, C.M.; Hsu, J.F.; Lee, W.S.; Sheu, J.R.; Chen, C.H.; Shen, M.Y. Sesamol reduces the atherogenicity of electronegative l5 ldl in vivo and in vitro. J. Nat. Prod. 2015, 78, 225–233. [Google Scholar] [CrossRef]
- Lu, J.; Yang, J.H.; Burns, A.R.; Chen, H.H.; Tang, D.; Walterscheid, J.P.; Suzuki, S.; Yang, C.Y.; Sawamura, T.; Chen, C.H. Mediation of electronegative low-density lipoprotein signaling by lox-1: A possible mechanism of endothelial apoptosis. Circ. Res. 2009, 104, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Estruch, M.; Sanchez-Quesada, J.L.; Beloki, L.; Ordonez-Llanos, J.; Benitez, S. Cd14 and tlr4 mediate cytokine release promoted by electronegative ldl in monocytes. Atherosclerosis 2013, 229, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Ligi, D.; Benitez, S.; Croce, L.; Rivas-Urbina, A.; Puig, N.; Ordonez-Llanos, J.; Mannello, F.; Sanchez-Quesada, J.L. Electronegative ldl induces mmp-9 and timp-1 release in monocytes through cd14 activation: Inhibitory effect of glycosaminoglycan sulodexide. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3559–3567. [Google Scholar] [CrossRef] [PubMed]
- Bancells, C.; Villegas, S.; Blanco, F.J.; Benitez, S.; Gallego, I.; Beloki, L.; Perez-Cuellar, M.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Aggregated electronegative low density lipoprotein in human plasma shows a high tendency toward phospholipolysis and particle fusion. J. Biol. Chem. 2010, 285, 32425–32435. [Google Scholar] [CrossRef] [Green Version]
- Bancells, C.; Benitez, S.; Jauhiainen, M.; Ordonez-Llanos, J.; Kovanen, P.T.; Villegas, S.; Sanchez-Quesada, J.L.; Oorni, K. High binding affinity of electronegative ldl to human aortic proteoglycans depends on its aggregation level. J. Lipid Res. 2009, 50, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Pedrosa, A.M.; Faine, L.A.; Grosso, D.M.; de Las Heras, B.; Bosca, L.; Abdalla, D.S. Electronegative ldl induction of apoptosis in macrophages: Involvement of nrf2. Biochim. Biophys. Acta 2010, 1801, 430–437. [Google Scholar] [CrossRef]
- Yang, T.C.; Chang, P.Y.; Lu, S.C. L5-ldl from st-elevation myocardial infarction patients induces il-1beta production via lox-1 and nlrp3 inflammasome activation in macrophages. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H265–H274. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.C.; Chang, P.Y.; Kuo, T.L.; Lu, S.C. Electronegative l5-ldl induces the production of g-csf and gm-csf in human macrophages through lox-1 involving nf-kappab and erk2 activation. Atherosclerosis 2017, 267, 1–9. [Google Scholar] [CrossRef]
- Chang, P.Y.; Pai, J.H.; Lai, Y.S.; Lu, S.C. Electronegative ldl from rabbits fed with atherogenic diet is highly proinflammatory. Mediat. Inflamm. 2019, 2019, 6163130. [Google Scholar] [CrossRef] [Green Version]
- Faulin, T.; Kazuma, S.M.; Tripodi, G.L.; Cavalcante, M.F.; Wakasuqui, F.; Oliveira, C.L.P.; Degenhardt, M.F.S.; Michaloski, J.; Giordano, R.J.; Ketelhuth, D.F.J.; et al. Proinflammatory action of a new electronegative low-density lipoprotein epitope. Biomolecules 2019, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Avogaro, P.; Bon, G.B.; Cazzolato, G. Presence of a modified low density lipoprotein in humans. Arteriosclerosis 1988, 8, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avogaro, P.; Cazzolato, G.; Bittolo-Bon, G. Some questions concerning a small, more electronegative ldl circulating in human plasma. Atherosclerosis 1991, 91, 163–171. [Google Scholar] [CrossRef]
- Shimano, H.; Yamada, N.; Ishibashi, S.; Mokuno, H.; Mori, N.; Gotoda, T.; Harada, K.; Akanuma, Y.; Murase, T.; Yazaki, Y.; et al. Oxidation-labile subfraction of human plasma low density lipoprotein isolated by ion-exchange chromatography. J. Lipid Res. 1991, 32, 763–773. [Google Scholar] [PubMed]
- Holvoet, P.; Perez, G.; Zhao, Z.; Brouwers, E.; Bernar, H.; Collen, D. Malondialdehyde-modified low density lipoproteins in patients with atherosclerotic disease. J. Clin. Investig. 1995, 95, 2611–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estruch, M.; Sanchez-Quesada, J.L.; Ordonez-Llanos, J.; Benitez, S. Ceramide-enriched ldl induces cytokine release through tlr4 and cd14 in monocytes. Similarities with electronegative ldl. Clin. Investig. Arterioscler. 2014, 26, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Benitez, S.; Villegas, V.; Bancells, C.; Jorba, O.; Gonzalez-Sastre, F.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Impaired binding affinity of electronegative low-density lipoprotein (ldl) to the ldl receptor is related to nonesterified fatty acids and lysophosphatidylcholine content. Biochemistry 2004, 43, 15863–15872. [Google Scholar] [CrossRef] [PubMed]
- Daigneault, M.; Preston, J.A.; Marriott, H.M.; Whyte, M.K.; Dockrell, D.H. The identification of markers of macrophage differentiation in pma-stimulated thp-1 cells and monocyte-derived macrophages. PLoS ONE 2010, 5, e8668. [Google Scholar] [CrossRef]
- Estruch, M.; Sanchez-Quesada, J.L.; Ordonez-Llanos, J.; Benitez, S. Inflammatory intracellular pathways activated by electronegative ldl in monocytes. Biochim. Biophys. Acta 2016, 1861, 963–969. [Google Scholar] [CrossRef]
- Bancells, C.; Sanchez-Quesada, J.L.; Birkelund, R.; Ordonez-Llanos, J.; Benitez, S. Hdl and electronegative ldl exchange anti- and pro-inflammatory properties. J. Lipid Res. 2010, 51, 2947–2956. [Google Scholar] [CrossRef] [Green Version]
- De Castellarnau, C.; Sanchez-Quesada, J.L.; Benitez, S.; Rosa, R.; Caveda, L.; Vila, L.; Ordonez-Llanos, J. Electronegative ldl from normolipemic subjects induces il-8 and monocyte chemotactic protein secretion by human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2281–2287. [Google Scholar] [CrossRef] [Green Version]
- Benitez, S.; Bancells, C.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Pro-inflammatory action of ldl(−) on mononuclear cells is counteracted by increased il10 production. Biochim. Biophys. Acta 2007, 1771, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Wang, Y.C.; Chang, S.S.; Lo, P.H.; Chang, C.M.; Lu, J.; Burns, A.R.; Chen, C.H.; Kakino, A.; Sawamura, T.; et al. Detection of a high ratio of soluble to membrane-bound lox-1 in aspirated coronary thrombi from patients with st-segment-elevation myocardial infarction. J. Am. Heart Assoc. 2020, 9, e014008. [Google Scholar] [CrossRef] [PubMed]
- Seeger, F.H.; Haendeler, J.; Walter, D.H.; Rochwalsky, U.; Reinhold, J.; Urbich, C.; Rossig, L.; Corbaz, A.; Chvatchko, Y.; Zeiher, A.M.; et al. P38 mitogen-activated protein kinase downregulates endothelial progenitor cells. Circulation 2005, 111, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacey, D.C.; Achuthan, A.; Fleetwood, A.J.; Dinh, H.; Roiniotis, J.; Scholz, G.M.; Chang, M.W.; Beckman, S.K.; Cook, A.D.; Hamilton, J.A. Defining gm-csf- and macrophage-csf-dependent macrophage responses by in vitro models. J. Immunol. 2012, 188, 5752–5765. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, J.; Degerman, E.; Nilsson, J.; Lindholm, M.W. Perilipin and adipophilin expression in lipid loaded macrophages. Biochem. Biophys. Res. Commun. 2007, 363, 1020–1026. [Google Scholar] [CrossRef]
- Revuelta-Lopez, E.; Cal, R.; Julve, J.; Rull, A.; Martinez-Bujidos, M.; Perez-Cuellar, M.; Ordonez-Llanos, J.; Badimon, L.; Sanchez-Quesada, J.L.; Llorente-Cortes, V. Hypoxia worsens the impact of intracellular triglyceride accumulation promoted by electronegative low-density lipoprotein in cardiomyocytes by impairing perilipin 5 upregulation. Int. J. Biochem. Cell Biol. 2015, 65, 257–267. [Google Scholar] [CrossRef]
- Feingold, K.R.; Shigenaga, J.K.; Kazemi, M.R.; McDonald, C.M.; Patzek, S.M.; Cross, A.S.; Moser, A.; Grunfeld, C. Mechanisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol. 2012, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.L.; Morales-Rosado, J.; Ray, J.; Myers, T.G.; Kho, T.; Lu, M.; Munford, R.S. Toll-like receptor agonists promote prolonged triglyceride storage in macrophages. J. Biol. Chem. 2014, 289, 3001–3012. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. Cd36 ligands promote sterile inflammation through assembly of a toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Tani, M.; Kawakami, A.; Mizuno, Y.; Imase, R.; Ito, Y.; Kondo, K.; Ishii, H.; Yoshida, M. Small dense ldl enhances thp-1 macrophage foam cell formation. J. Atheroscler. Thromb. 2011, 18, 698–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Kondratenko, N.; Green, S.; Steinberg, D.; Quehenberger, O. Identification of the lectin-like receptor for oxidized low-density lipoprotein in human macrophages and its potential role as a scavenger receptor. Biochem. J. 1998, 334 Pt 1, 9–13. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Quehenberger, O.; Kondratenko, N.; Green, S.; Steinberg, D. Minimally oxidized low-density lipoprotein increases expression of scavenger receptor a, cd36, and macrosialin in resident mouse peritoneal macrophages. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 794–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillis, A.P.; Muratoglu, S.C.; Au, D.T.; Migliorini, M.; Lee, M.J.; Fried, S.K.; Mikhailenko, I.; Strickland, D.K. Ldl receptor-related protein-1 (lrp1) regulates cholesterol accumulation in macrophages. PLoS ONE 2015, 10, e0128903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chellan, B.; Reardon, C.A.; Getz, G.S.; Hofmann Bowman, M.A. Enzymatically modified low-density lipoprotein promotes foam cell formation in smooth muscle cells via macropinocytosis and enhances receptor-mediated uptake of oxidized low-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M. Fatty acid-binding protein 4 in cardiovascular and metabolic diseases. J. Atheroscler. Thromb. 2019, 26, 216–232. [Google Scholar] [CrossRef] [Green Version]
- Larigauderie, G.; Bouhlel, M.A.; Furman, C.; Jaye, M.; Fruchart, J.C.; Rouis, M. Perilipin, a potential substitute for adipophilin in triglyceride storage in human macrophages. Atherosclerosis 2006, 189, 142–148. [Google Scholar] [CrossRef]
- Estruch, M.; Sanchez-Quesada, J.L.; Beloki, L.; Ordonez-Llanos, J.; Benitez, S. The induction of cytokine release in monocytes by electronegative low-density lipoprotein (ldl) is related to its higher ceramide content than native ldl. Int. J. Mol. Sci. 2013, 14, 2601–2616. [Google Scholar] [CrossRef] [Green Version]
- Puig, N.; Estruch, M.; Jin, L.; Sanchez-Quesada, J.L.; Benitez, S. The role of distinctive sphingolipids in the inflammatory and apoptotic effects of electronegative ldl on monocytes. Biomolecules 2019, 9, 300. [Google Scholar] [CrossRef] [Green Version]
- Lehti, S.; Nguyen, S.D.; Belevich, I.; Vihinen, H.; Heikkila, H.M.; Soliymani, R.; Kakela, R.; Saksi, J.; Jauhiainen, M.; Grabowski, G.A.; et al. Extracellular lipids accumulate in human carotid arteries as distinct three-dimensional structures and have proinflammatory properties. Am. J. Pathol. 2018, 188, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orekhov, A.N.; Oishi, Y.; Nikiforov, N.G.; Zhelankin, A.V.; Dubrovsky, L.; Sobenin, I.A.; Kel, A.; Stelmashenko, D.; Makeev, V.J.; Foxx, K.; et al. Modified ldl particles activate inflammatory pathways in monocyte-derived macrophages: Transcriptome analysis. Curr. Pharm. Des. 2018, 24, 3143–3151. [Google Scholar] [CrossRef] [PubMed]
- Waldo, S.W.; Li, Y.; Buono, C.; Zhao, B.; Billings, E.M.; Chang, J.; Kruth, H.S. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am. J. Pathol. 2008, 172, 1112–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spann, N.J.; Garmire, L.X.; McDonald, J.G.; Myers, D.S.; Milne, S.B.; Shibata, N.; Reichart, D.; Fox, J.N.; Shaked, I.; Heudobler, D.; et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 2012, 151, 138–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozza, P.T.; Magalhaes, K.G.; Weller, P.F. Leukocyte Lipid Bodies-Biogenesis and Functions in Inflammation. Biochim. Biophys. Acta 2009, 1791, 540–551. [Google Scholar] [CrossRef] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puig, N.; Montolio, L.; Camps-Renom, P.; Navarra, L.; Jiménez-Altayó, F.; Jiménez-Xarrié, E.; Sánchez-Quesada, J.L.; Benitez, S. Electronegative LDL Promotes Inflammation and Triglyceride Accumulation in Macrophages. Cells 2020, 9, 583. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030583
Puig N, Montolio L, Camps-Renom P, Navarra L, Jiménez-Altayó F, Jiménez-Xarrié E, Sánchez-Quesada JL, Benitez S. Electronegative LDL Promotes Inflammation and Triglyceride Accumulation in Macrophages. Cells. 2020; 9(3):583. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030583
Chicago/Turabian StylePuig, Núria, Lara Montolio, Pol Camps-Renom, Laia Navarra, Francesc Jiménez-Altayó, Elena Jiménez-Xarrié, Jose Luis Sánchez-Quesada, and Sonia Benitez. 2020. "Electronegative LDL Promotes Inflammation and Triglyceride Accumulation in Macrophages" Cells 9, no. 3: 583. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9030583