Targeting DNA Repair, Cell Cycle, and Tumor Microenvironment in B Cell Lymphoma


1
Max Planck Research Group Mechanisms of DNA Repair, Max Planck Institute for Biology of Ageing, 50931 Cologne, Germany
2
Department I of Internal Medicine, Center for Integrated Oncology Aachen Bonn Cologne Duesseldorf (CIO ABCD), University of Cologne, 50937 Cologne, Germany
3
Department of Hematology, University Medical Center Groningen, University of Groningen, 9713 GZ Groningen, The Netherlands
4
Department of Pathology and Medical Biology, University Medical Center Groningen, University of Groningen, 9713 GZ Groningen, The Netherlands
5
Cologne Excellence Cluster on Cellular Stress Response in Aging-Associated Diseases (CECAD), University of Cologne, 50931 Cologne, Germany
6
Center for Molecular Medicine Cologne, University of Cologne, 50931 Cologne, Germany
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cells 2020, 9(10), 2287; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9102287
Submission received: 21 September 2020
/
Revised: 11 October 2020
/
Accepted: 12 October 2020
/
Published: 14 October 2020
(This article belongs to the Special Issue Cell Cycle Control and Cancer)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The DNA double-strand break (DSB) is the most cytotoxic lesion and compromises genome stability. In an attempt to efficiently repair DSBs, cells activate ATM kinase, which orchestrates the DNA damage response (DDR) by activating cell cycle checkpoints and initiating DSB repair pathways. In physiological B cell development, however, programmed DSBs are generated as intermediates for effective immune responses and the maintenance of genomic integrity. Disturbances of these pathways are at the heart of B cell lymphomagenesis. Here, we review the role of DNA repair and cell cycle control on B cell development and lymphomagenesis. In addition, we highlight the intricate relationship between the DDR and the tumor microenvironment (TME). Lastly, we provide a clinical perspective by highlighting treatment possibilities of defective DDR signaling and the TME in mantle cell lymphoma, which serves as a blueprint for B cell lymphomas.
1. Introduction
DNA damage activates a complex signaling network, commonly referred to as the DNA damage response (DDR) [1]. The DDR induces cell cycle checkpoints and therefore allows time for DNA repair, or—if the lesions are beyond repair capacity—leads to the activation of cell death pathways [1]. These checkpoints are the G1/S checkpoint, the S phase checkpoint, and the G2/M checkpoint. Once DNA damage occurs, DDR proteins will accumulate at damaged sites to stabilize broken ends, attract repair proteins, or even facilitate in the repair. Mammalian cells evolved a series of distinct DNA repair mechanisms to remove structurally distinct DNA aberrations throughout the different phases of the cell cycle [2]. These pathways include mismatch repair (MMR), base excision repair (BER), trans-lesion synthesis (TLS), the Fanconi anemia (FA) pathway, and two distinct dominant DNA double-strand break (DSB) repair pathways, namely, the error-prone classical non-homologous end joining (cNHEJ) pathway, and the error-free homologous recombination repair (HR) pathway [2,3,4,5]. Error-prone cNHEJ operates throughout the cell cycle and directly ligates the processed DSB ends, whereas error-free HR is restricted to late S- and G2, when a repair template is available [2]. HR involves the binding of the MRE11-RAD50-NBS1 (MRN) complex to tether DSB ends and to recruit and activate the proximal DDR kinase ataxia telangiectasia-mutated (ATM) [6]. ATM phosphorylates a plethora of substrates, such as CHK2, p53, UBQLN4, MDC1, and MRN complex components [6,7]. In particular, ATM-dependent phosphorylation of NBS1, RAD50 and MRE11, and downstream components of DNA end resection, such as CtIP contributes to the activation of HR and may also create a positive feedback loop that maintains ATM activity [6,8]. The overall performance of DNA repair pathways, such as the right choice between these pathways is essential for correct DNA repair [2]. At the same time, activation of the cell cycle checkpoints will set pathways in motion to halt cell cycle progression. An important example of a protein that bridges the gap between the DDR and the cell cycle is the WEE1 kinase. WEE1 phosphorylation of CDK1 and CDK2 keeps it in an inactive state, thereby preventing cell cycle progression [9]. Upon DNA damage, activation of the checkpoint kinase 1 (CHK1) by the DDR results in the activation of WEE1, halting the cell cycle and either allowing repair or inducing apoptosis [10].
Components of DNA repair signaling and the cell cycle machinery are among the most frequently mutated genes in human malignancies, including B cell lymphomas [2,11,12,13]. The fundamental process of B cell development is crucially dependent on functional DNA repair and cell cycle control mechanisms and thus deficiencies within these pathways do not only promote lymphomagenesis but are also frequently found in B cell lymphomas [14]. For example, ATM is recurrently mutated in up to 50% of mantle cell lymphoma (MCL) [15]. As a functional consequence, defects in ATM have been associated with resistance to conventional chemotherapy treatment in cancer [16]. In addition, Cyclin D1, a D-type cyclin which associates with CDK4 or CDK6 to control the progression to the G1 phase and the G1/S phase transition, is overexpressed typically due to a t(11;14) (q13;q32) translocation in the majority of MCL patients, which leads to uncontrolled cell proliferation [17]. While 1st-line therapy with intensive chemo-immunotherapy induces disease remissions, relapse is inevitable.
In this review, we will focus on the role of rewired DDR signaling and cell cycle control in B cell lymphomagenesis, and elucidate the therapeutic potential that these alterations reveal. We will place a specific emphasis on the intricate relationship between the cell cycle, the DDR, and the tumor microenvironment (TME). The TME is heterogeneous across cancer entities and composed largely of immune and stromal cells. It provides a protective niche for tumor cell proliferation and facilitates immune evasion, ultimately resulting in cancer growth and therapeutic resistance. Leveraging the TME regained attention with the advent of immune checkpoint blockade (ICB), which reverses inhibitory signaling and the anergic state of immune effector cells in the TME. An accumulating body of evidence implicates a direct link between DDR defects and responsiveness to ICB [18,19]. For instance, microsatellite instability/defective mismatch repair (MSI/dMMR) has been approved by the FDA as a pan-cancer DDR biomarker for predicting response to ICB [20]. An impaired DDR is also associated with decreased antitumor immunity [21]. Increasing evidence suggests that targeting cell cycle dysregulation has the potential to increase the efficacy of ICB in otherwise unresponsive tumors [22,23]. We review the existing evidence to develop novel therapeutic approaches by harnessing DDR deficiency and leveraging the increasingly understood TME features using MCL as a blueprint for B cell lymphomas.
2. Role of DNA Repair in B Cell Development and Lymphomagenesis
Initial B cell development takes place in the bone marrow in the absence of antigen. It is a highly regulated process in which DDR signaling plays an important role (Figure 1). More specifically, developing B cells undergo programmed DNA DSBs at precise loci within their genome as they assemble antigen receptor genes required for developmental processes [24]. B cell antigen receptors (BCRs) are composed of immunoglobulin heavy (IgH) and light (IgL) chains encoded by different genes, which are assembled from the recombination of variable (V), diversity (D), and joining (J) gene segments [24]. V(D)J recombination initiates when the recombination-activating gene 1 (RAG1) and RAG2 endonuclease cleaves DNA at the border of two Ig gene segments and their flanking recombination signal sequences in G1 phase B cells [25]. RAG DSBs are then repaired exclusively by error-prone cNHEJ to assemble a complete Ig gene, enabling Ig chain expression on the surface of maturing B cells which typically harbor insertion-deletion mutations (indels) [26]. This process leads to the activation of ATM, which in a positive feedback loop maintains the stability of RAG induced DSBs [27]. In response to BCR activation, mature B cells then again undergo developmentally programmed DSBs within the constant region of the IgH locus in a process termed class switch recombination (CSR) [28]. DNA repair deficiency during CSR leads to impaired immunoglobulin switching and to chromosomal translocations involving the IgH locus. To illustrate, B cells deficient for ATM display impaired CSR and frequently generate chromosomal translocations involving the IgH chain locus [29]. Similarly, in Atm−/− mice, CSR has been shown to be reduced by 75% [30]. Clinical and in vivo studies reveal a similar pattern: patients with inherited dysfunctional ATM alterations (termed Ataxia-telangiectasia, A-T) and mice lacking Atm function are immunocompromised and have an increased risk of developing lymphomas [31].
Mature non-neoplastic B cells with successful V(D)J recombination and that express functional BCRs leave the bone marrow and migrate to peripheral lymphoid tissues. Upon exposure to an antigen in the secondary lymphoid organs, these cells enter the primary follicle to form the germinal centers (GCs) that comprise two different regions which are termed the dark zone (DZ) and light zone (LZ). GCs are specialized microenvironments and are critical for the formation of long-lived plasma cells and memory B cells. Within these structures, B cells undergo somatic hypermutation (SHM) and clonal selection based on the affinity of the BCR for the immunizing antigen [32]. Recent evidence suggests that CSR ceases upon the onset of SHM and—unlike SHM—mainly takes place before mature B cells differentiate in GCs [33]. SHM induced lesions are repaired through the DNA repair pathways MMR and BER, both of which are unaffected by ATM deletion [34]. The process of SHM allows the determination of whether a B cell has experienced the GC and provides some evidence for the cell-of-origin in situations in which faulty B cell development has led to B cell lymphoma. The intrinsic force of B cell development—the sequence of V(D)J recombination, CSR, and SHM—can lead to somatic mutations within numerous oncogenes that are associated with lymphomagenesis and their cell-of-origin stemming from the pre-GC, GC, or the post-GC [35,36]. The GC B cell, for instance, is at high risk of undergoing malignant transformation due to genetic lesions that occur in GC-specific DNA remodeling events for Ig affinity maturation [37]. Beyond mere B cell development, several alterations in DNA repair pathways have been linked to lymphoma susceptibility [38]. This is highlighted by human genome instability syndromes like A-T or Nijmegen-breakage syndrome (alterations in NBS1) which typically present with lymphomas at an early age [39]. Descriptive and functional genomic studies provide further evidence for the impact of DNA repair genes on lymphoma risk [40,41,42]. Despite its importance, the mechanism behind DDR alterations and lymphomagenesis remains unclear. Recently, Atm reactivation in initially Atm-deficient B cell lymphomas has been shown to induce tumor regression strongly corroborating the suppressive function of intact DDR on lymphomagenesis [43]. Thus, addressing the rewired DDR response for B cell lymphoma offers intriguing possibilities for genotype-stratified treatments.
3. Role of the Cell Cycle in B Cell Development and Lymphomagenesis
Cell division during B cell development is a strictly controlled process, which only occurs at specific moments during development or during an immune response after encountering antigens. DNA replication during proliferation is associated with HR, whereas VDJ rearrangement is associated with cNHEJ, making these two events mutually exclusive. Early B cell development is thus characterized by waves of V(D)J recombination followed by waves of proliferation. This separation ensures that no events of recombination will occur during DNA replication, thereby preventing an increase in the mutation rate. Expression of RAG2 is therefore mostly restricted to the G0 and G1 phase of the cell cycle and quickly degraded upon entering the S phase [44]. This degradation of RAG2 is initiated by the cyclin A/CDK2 complex, which promotes ubiquitination and degradation of RAG2 by Skp2-SCF [45]. Uncoupling this cell cycle regulation from RAG2 expression has been shown to promote aberrant recombination, and combined with p53-deficiency leads to genomic instability and lymphomagenesis [46]. A similar regulatory mechanism is in place for activation-induced cytidine deaminase (AID): the expression of nuclear AID and AID-induced DSBs are observed almost exclusively in the G1 phase of the cell cycle [47] and prolonged-expression of AID enhances DNA damage at off-target sites [48].
As one of the hallmarks of cancer, genetic alterations to cell cycle genes are found in most B cell lymphomas. The most commonly found genetic alteration is of the tumor suppressor gene TP53, with mutations found in MCL (30%), Burkitt lymphoma (30%), diffuse large B cell lymphoma (DLBCL) (8%), and follicular lymphoma (FL) (6%) [49], resulting in a loss of cell cycle arrest. A similar effect is induced by mutations in CDKN2A, which codes for tumor suppressor proteins p16 and p14, resulting in the simultaneous inactivation of retinoblastoma (Rb) through the loss of CDK4/6 inhibition by p16, and loss of p53 activation by p14. Deletions of CDKN2A are observed in 30% of MCL cases and combined with TP53 aberrations are associated with chemoresistance in MCL [50]. Adjacent to CDKN2A on chromosome 9 are CDKN2B and CDKN2C, which code for p15 and p18, respectively, and similarly inhibit CDK4/6 to prevent G1 phase cell cycle progression. Similarly, a loss of Rb protein expression (12% in DLBCL and 12% in FL) eliminates its suppressor effect in the G1/S-phase transition. Taken together, these aberrations are focused on enhancing cell cycle progression through the G1 phase, driving proliferation. Moreover, mutation of oncogenes with cell cycle regulator function such as BCL6 (30% in DLBCL) and MYC (70% in Burkitt lymphoma) also drive high proliferation levels of lymphoma cells [51]. Finally, many lymphomas harbor genetic aberrations in the AURKA and/or AURKB genes (40% in DLBCL and 80% in Burkitt lymphoma), which code for the mitotic aurora A and aurora B kinases. Aurora A is essential for proper formation and stabilization of the microtubule during mitosis, and overexpression has been shown to induce aneuploidy and spontaneous growth of lymphomas, Aurora B is essential for proper chromosome alignment and accurate chromosome segregation [52]. Taken together, these data present MCL, and B cell lymphomas in general, as a classical example for the most important tool of malignant transformation: bending and harnessing the power of the cell cycle to enable unlimited replication. Especially in B cell development, where cell division is more tightly controlled in order to prevent aberrant genetic alterations, mutations in proliferation genes have a much greater impact, making lymphomas the ideal candidate for cell cycle inhibitor therapies.
4. Harnessing DDR- and Cell Cycle Defects for the Treatment of B Cell Lymphoma
Based on the extensive knowledge on the cell cycle and genetic aberrations of cell cycle-related genes in B cell lymphomas, we are now in a position to explore treatment options that directly or indirectly target the cell cycle, of which the most recent and/or common treatments are summarized in Figure 2. As of today, many clinical trials are underway for CDK4/6 inhibitors, such as palbociclib/PD-0332991 (phase 2 in MCL), abemaciclib/LY2835219 (phase 2 in refractory/relapsed MCL), ribociclib/LEE011 (phase 1 in lymphomas), and voruciclib (phase 1 in relapsed/refractory MCL, B cell malignancies and AML). Promising results were also seen for palbociclib combined with BTK inhibitor ibrutinib [53] and proteasome inhibitor bortezomib in MCL [54], and combined with DNA damage-inducing treatments such as irradiation [55] or WEE1 inhibitor AZD1775 [56] in other cancers. Further investigation showed that CDK4/6 inhibition has the ability to shift the DDR response from HR to cNHEJ [57], in addition to reducing PARP1 expression levels [58], making it a potential candidate to use alongside DNA repair inhibitors or cancers with DNA damage deficiencies. Inhibitors that target CDK1/2 currently under investigation include dinaciclib/SCH727965 (phase 1 in MCL and non-Hodgkin lymphomas, NHL) and AG-024322 (phase 1 in NHL), in addition to specific CDK2 inhibitors SNS-032/BMS-387032 (phase 1 MCL and advanced B-lymphoid malignancies) and CYC065 (phase 1 in refractory/relapsed CLL). Besides specific CDK inhibitors, pan-CDK inhibitors such as alvocidib/flavopiridol (phase 1/2 in MCL and DLBCL), P276-00 (phase 2 in refractory/relapsed MCL), and AT7519M (phase 2 in refractory/relapsed MCL) are under investigation that target multiple CDKs at the same time, including CDK7 (part of the Cdk-activating kinase (CAK) complex) and CDK9 (part of the multiprotein elongation factor complex for RNA polymerase II). Pre-clinical experiments with CDK1/2 inhibitor seliciclib/roscovitine revealed that treatment of MCL cells resulted in cell cycle arrest at the G2/M phase followed by apoptosis, in addition to causing downregulation of cyclin D1 and MCL-1 protein levels [59]. Similar effects were induced in MCL by alvocidib [60], which also showed efficacy combined with bortezomib in MCL [61]. Moreover, alvocidib has been shown to enhance the radiosensitivity of ovarian carcinoma cells [62], likely caused by its ability to bind to DNA [63], making it another interesting compound for combination with DNA repair inhibitors. Other treatment options that target the cell cycle include inhibitors for aurora kinase A/B, with clinical trials on-going for alisertib/MLN8237 (phase 1 in MCL and NHL), AT9283 (phase 1 in NHL) and chiauranib (phase 1 in NHL), inhibitors for WEE1 with adavosertib/AZD1775 (phase 2 in NHL), and inhibitors for kinesin spindle protein (KSP) with ispinesib/SB-715992 (phase 1 in NHL). Research in other cancers has shown that treatment of ovarian carcinoma cells with alisertib decreased the expression of PARP and BRCA1/2 and enhanced DNA-PKcs activity, stimulating cNHEJ over HR [64] and showed synergism with CHK1 inhibitor rabusertib/LY2603618 [65]. In addition, alisertib showed promising results combined with docetaxel in MCL [66], further expanding its therapeutic potential. The observation that many cell cycle inhibitors alter the DDR, either by tilting the balance towards cNHEJ over HR or by directly influencing DDR protein expression levels, gives us novel therapeutic options. These may thus focus on the simultaneous targeting of both the DRR and cell cycle, especially in B cell lymphomas with additional DDR deficiencies.
5. Exploiting the Tumor Microenvironment for the Treatment of B Cell Lymphoma
B cell lymphomas usually arise in a TME reminiscent of lymphoid tissue which is heterogeneous and shaped by the genetic alterations and phenotype of the malignant cells. The TME of B cell malignancies is composed of variable types and numbers of immune cells, stromal cells, extracellular components, and blood vessels. Spatial arrangement and cross-talk between malignant and bystander cells and the host inflammatory response affect these protective niches [67]. Tumor cells are able to orchestrate the TME in their favor to evade an antitumor immune response or ICB treatment e.g., by downregulation of antigen-presenting features, upregulation of inhibitory proteins, or cytokine release [68]. The interaction between the immune infiltrate and malignant cells thereby shapes the TME, and as a result, the potential therapeutic implications of the interplay between DDR deficiencies and TME composition are increasingly revealed [19].
For example, recent preclinical studies suggest that targeting defective DNA repair may act synergistically with ICB, since the cell cycle dysregulation observed limits antitumor immunity: Mutations in TP53—found in up to 26% of MCL patients [69]—were associated with less immune cytolytic activity in one pan-cancer study [70]. An altered p53 pathway additionally results in recruitment of macrophages and CD4+ T cells while decreasing CD8+ T cell levels via dysregulated NF-κB or JAK-STAT signaling [71,72]. In a Kras and p53 driven pancreatic mouse model, signatures of cell cycle dysregulation (E2F, G2/M, DNA repair) correlated with low tumor T cell content and immunologically “cold” tumors, resulting in inferior efficacy of anti-PD1 blockade compared to T cell rich “hot” tumors [73]. Similarly, deletions or mutations of CDKN2A are more frequently observed in “cold” pancreatic cancers with a non-T cell-inflamed TME associated with reduced cytolytic activity [74]. Interestingly, CDKN2A mutations are also observed in 25% of MCL patients and result in cell cycle dysregulation due to increased CDK4/6 activity by lack of endogenous inhibition by the CDKN2A/B gene product p16 [75]. Cyclin D1 overexpression resulting in CDK4/6 activation is a hallmark of MCL, and recently, whole genome sequencing of different human tumors revealed a strong link between cyclin D1 amplification and ICB resistance [21]. Clinically, this aligns well with the disappointing response rates observed with single-agent anti-PD1 antibodies in relapsed or refractory MCL patients [76].
This feature of cyclin D1 overexpression is reversed by CDK4/6 inhibition, which results in increased antitumor immunity due to enhanced CD8+ T cell infiltration and activity with increased IL-2 secretion in lung cancer mouse models [77]. An improved antitumor immune response was also observed with the CDK4/6 inhibitor abemaciclib in a transgenic breast cancer mouse model [78]. By suppressing DNMT1 expression, abemaciclib leads to the expression of endogenous retroviral elements by the malignant cells and ultimately resulted in increased ICB responsiveness due to higher interferon and MHC-I signaling [22]. Similar observations were made in syngeneic mouse models and samples derived from melanoma patients [78,79]. Interestingly, CDK4/6 inhibition induces senescence of malignant cells activating senescence-associated secretory phenotypes (SASP) induced by NF-κB and STAT3 signaling, consecutively resulting in increased antitumor immunity [23,78]. In contrast, SASP induces recruitment and activation of immune effector cells such as NK cells, macrophages, and CD8+ T cells limiting tumor growth in tumors with p53 WT, activated MYC, active Wnt, and low Notch signaling. Such a SASP pattern is observed in the context of CDK4/6 inhibition in KRAS-mutant lung and pancreatic cancer models, leading to an effective antitumor immune response [23]. Importantly, alternative SASP may facilitate tumor growth by inducing a state of chronic inflammation establishing an immunosuppressive TME in tumors with p53 mutations or activated Notch signaling. Targeting the cell cycle machinery beyond CDK4/6, a recent study found enhanced ICB sensitivity due to increased CD4+ and CD8+ T cell immunity with CDK7 inhibition in small cell lung cancer models [80]. While these studies investigated solid tumor models, they provide a mechanistic rationale to investigate ICB combinations with CDK inhibitors in DDR deficient B cell lymphomas such as MCL.
Stimulator of interferon genes (STING) signaling is triggered by cytosolic DNA and is crucial for antitumor immunity e.g., by inducing type 1 interferon production, inducing cell death, and releasing tumor antigens or increased priming and activation of effector T cells [81]. B cell malignancy mouse models show that STING activation with the agonist 3′3′-cGAMP directly induces apoptosis and tumor regression of malignant B cells but not in solid tumors in an interferon independent fashion [82]. Inducing DNA damage in in vivo models of small cell lung cancer via PARP inhibition (PARPi) and/or CHK1 inhibition not only potentiate the efficacy of anti-PDL1 treatment by augmenting cytotoxic T cell infiltration but additionally activate the STING pathway. Notably, knockout of cGAS and STING reversed the observed synergies of ICB and DDR inhibition [83]. Further supporting activation of STING innate immunity by DDR inhibition, treatment of Brca1-deficient ovarian cancers with PARPi in mice resulted in STING activation, and an improved anti-tumor effect when combined with anti-PD1 ICB [84]. Additionally, DNA damage induced by targeting telomerase sensed by dendritic cells results in STING activation and overcomes ICB resistance in advanced tumor models [85]. Recently, small-molecule STING agonists with potent antitumor activity in various animal models synergizing with ICB were developed, providing promising drugs to further develop STING-based treatments in tumors harboring DDR deficiency [86,87].
As summarized previously, combinations of PARPi and anti-PD1 ICB leads to superior tumor control, and a similar effect was observed when combining PARPi with anti-CTLA4 antibodies in a T cell and IFN-γ dependent way [88]. The improved efficacy of PARPi in combination with ICB compared to single agent PARPi may additionally be due to the upregulation of PD-L1 expression observed in breast cancer models [89]. Supporting this evidence, Sato et al. demonstrated that PD-L1 upregulation is dependent on ATM/ATR/CHK1 kinases in response to DSBs [90]. In line with the previously summarized observation with CDK4/6 inhibitors, co-administration of ICB agents hence potentiates the potential of cell cycle or DDR targeting drugs by leveraging and amplifying the increasing antitumor immunity observed during treatment with these agents.
Taken together, these data provide a strong rationale to develop synergistic therapies in B cell lymphomas targeting the dysregulation of cell proliferation and impaired antitumor immunity. Figure 3 conceptualizes the current understanding of potential mechanisms of synergistic effects of cell cycle inhibitors and ICB, utilizing DDR deficient MCL as a blueprint for other B cell lymphomas. Further mechanistic dissection of the interplay between those two intertwined key features of malignancy by elaborate in vitro and in vivo models is crucial to identify targets and drug candidates for early clinical development. Innovative trial designs such as conditional multi-cohort studies may additionally help to ultimately translate these concepts into routine care and improve outcomes for our patients.
6. Remarks and Outlook
Defects in DDR pathways expose important vulnerabilities for the treatment of tumors with inhibitors targeting DNA-PKcs, ATR, and PARP1 [7,91,92]. Agents targeting the cell cycle promote antitumor immunity which is potentiated by simultaneous ICB in various in vivo models, providing a rationale to explore potentially synergistic therapies. Alternative error-prone DNA repair pathways such as cNHEJ, which generate potentially targetable dependencies, may bypass the defective DNA repair caused by ATM and/or TP53 mutations in MCL. For instance, the DNA-PKcs-dependent cNHEJ pathway becomes essential for the cellular survival of doxorubicin-treated B-NHL cells lacking ATM [93]. Identifying backup DNA repair pathways in ATM defective MCL is therefore critical for identifying targeted treatment options for these patients. Destabilization of the cell cycle regulator proteins during crucial events of genetic recombination has been shown to enhance genomic aberrations in B cells [45,47], making it the seemingly ideal strategy for the treatment of B cell lymphomas. Most of these strategies are focused either on (1) prolonging the G1 phase, thereby stimulating aberrant recombination; (2) stalling mitosis, thereby inducing mitotic catastrophe; or (3) abolishment of the cell cycle checkpoints and thereby allowing unscheduled cell cycle progression. However, many (pan-) CDK inhibitors have failed as monotherapy in the past because of a lack of a clear understanding of the biological pathways involved, many that go beyond simple cell division. A weakness of these inhibitors targeting proteins required for the survival of normal cells is a rather small therapeutic window leading to relevant toxicities in patients. The potential success of targeting the cell cycle as a therapeutic strategy will hence likely depend on identifying synergistic pathways and appropriate patient selection. Identification of synergistic drug partners in synthetic lethal screens could alleviate this problem by increasing the therapeutic window and efficacy. For instance, an siRNA screen in KRAS mutant tumors identified CDK1 as a synthetic lethal target [94]. In a similar approach utilizing a pharmacogenomic screen in T cell acute lymphoblastic leukemia, Pikman et al. identified a synergistic potential for CDK4/6 inhibitors with glucocorticoids and mTOR inhibition [95]. Leveraging the concept of synthetic lethality beyond siRNA- and pharmacogenomic screens, CRISPR-based screens are further able to identify novel drug combinations [96]. Combining a drug known to be effective in a certain disease with a CRISPR library, these so-called drug anchor screens, may be able to identify previously unmarked context-dependent oncogenes [97]. Applying an in vivo CRISPR screen geared to identify correlates of immune-mediated lethality, extends this approach beyond cell-autonomous targets [98]. A recent study evaluated 2700 candidate genes in anti-PD1 treated B16 syngeneic mouse models, identifying immune evasion genetic contexts such as JAK1 and B2M loss of function [99]. Applied ideally in paired syngeneic mouse models harboring loss of function in the identified gene and a wild-type control, this ultimately facilitates the discovery of drug targets able to counteract immune evasion in the previously identified context-specific immune evasion signature [96]. These concepts, such as immune-stimulating drug combination therapies, are of immediate relevance to future drug discovery and immunotherapy development in B cell lymphomas, such as for MCL, which is largely resistant to single-agent ICB [76]. In conclusion, the complex interplay of defective DNA repair, cell cycle control, and the TME raises intriguing possibilities for targeted treatment approaches in B cell lymphomas that to date represent tremendous challenges and cause high mortality.
Author Contributions
R.D.J. designed the study; P.J.B., M.R.W.d.J., and R.D.J. reviewed the literature data; P.J.B., M.R.W.d.J., and R.D.J. wrote and revised the manuscript. R.D.J., P.J.B., and M.R.W.d.J. made the figures. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Mildred Scheel School of Oncology Cologne-Bonn (MSSO 2020 Postgraduate Program to P.J.B.), the Bas Mulder grant of Alpe d’HuZes/Dutch Cancer Society (RUG 2014-6727) (M.R.W.d.J.), the Else Kröner-Fresenius Stiftung (2016_Kolleg.19 to R.D.J.), and the Exzellenz Initiieren—Stiftung Kölner Krebsforschung (R.D.J).
Acknowledgments
We apologize to those researchers whose work could not be described or cited due to space limitations.
Conflicts of Interest
P.J.B. declares honoraria by Bristol-Myers Squibb and research funding by BeiGene, Bristol-Myers Squibb, and MSD Sharp and Dohme. The other authors declare no potential conflict of interest.
References
- Reinhardt, H.C.; Yaffe, M.B. Phospho-Ser/Thr-binding domains: Navigating the cell cycle and DNA damage response. Nat. Rev. Mol. Cell Biol. 2013, 14, 563–580. [Google Scholar] [CrossRef]
- Dietlein, F.; Thelen, L.; Reinhardt, H.C. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014, 30, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nat. Cell Biol. 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nat. Cell Biol. 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Jachimowicz, R.D.; Beleggia, F.; Isensee, J.; Velpula, B.B.; Goergens, J.; Bustos, M.A.; Doll, M.A.; Shenoy, A.; Checa-Rodriguez, C.; Wiederstein, J.L.; et al. UBQLN4 Represses Homologous Recombination and Is Overexpressed in Aggressive Tumors. Cell 2019, 176, 505–519.e22. [Google Scholar] [CrossRef] [Green Version]
- Jachimowicz, R.D.; Reinhardt, H.C. UBQLN4 promotes non-homologous end joining by repressing DNA end-resection. Mol. Cell. Oncol. 2019, 6, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, T.N.; Qian, C.; Sugitani, N.; Osmanbeyoglu, H.U.; Bakkenist, C.J. WEE1 kinase inhibitor AZD1775 induces CDK1 kinase-dependent origin firing in unperturbed G1- and S-phase cells. Proc. Natl. Acad. Sci. USA 2019, 116, 23891–23893. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, M.J.; Raleigh, J.M.; Verkade, H.; Nurse, P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997, 16, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhardt, H.C.; Jiang, H.; Hemann, M.T.; Yaffe, M.B. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle 2009, 8, 3112–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knittel, G.; Rehkämper, T.; Nieper, P.; Schmitt, A.; Flümann, R.; Reinhardt, H.C. DNA damage pathways and B-cell lymphomagenesis. Curr. Opin. Hematol. 2018, 25, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Mareckova, A.; Malcikova, J.; Tom, N.; Pal, K.; Radova, L.; Salek, D.; Janikova, A.; Moulis, M.; Smardova, J.; Kren, L.; et al. ATM and TP53 mutations show mutual exclusivity but distinct clinical impact in mantle cell lymphoma patients. Leuk. Lymphoma 2019, 60, 1420–1428. [Google Scholar] [CrossRef]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Fernàndez, V.; Hartmann, E.; Ott, G.; Campo, E.; Rosenwald, A. Pathogenesis of Mantle-Cell Lymphoma: All Oncogenic Roads Lead to Dysregulation of Cell Cycle and DNA Damage Response Pathways. J. Clin. Oncol. 2005, 23, 6364–6369. [Google Scholar] [CrossRef]
- Li, J.; Stanger, B.Z. Cell Cycle Regulation Meets Tumor Immunosuppression. Trends Immunol. 2020, 41, 859–863. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; De Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [Green Version]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Vokes, N.I. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 2018, 50, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.-C.; Ho, Y.-J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell–mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassing, C.H.; Swat, W.; Alt, F.W. The Mechanism and Regulation of Chromosomal V(D)J Recombination. Cell 2002, 109, S45–S55. [Google Scholar] [CrossRef] [Green Version]
- Gellert, M. V(D)J Recombination: RAG Proteins, Repair Factors, and Regulation. Annu. Rev. Biochem. 2002, 71, 101–132. [Google Scholar] [CrossRef]
- Helmink, B.A.; Sleckman, B.P. The Response to and Repair of RAG-Mediated DNA Double-Strand Breaks. Annu. Rev. Immunol. 2012, 30, 175–202. [Google Scholar] [CrossRef] [Green Version]
- Bredemeyer, A.L.; Sharma, G.G.; Huang, C.-Y.; Helmink, B.A.; Walker, L.M.; Khor, K.C.; Nuskey, B.; Sullivan, K.E.; Pandita, T.K.; Bassing, C.H.; et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nat. Cell Biol. 2006, 442, 466–470. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Alt, F.W. Class-switch recombination: Interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 2004, 4, 541–552. [Google Scholar] [CrossRef]
- Callén, E.; Jankovic, M.; Difilippantonio, S.; Daniel, J.A.; Chen, H.-T.; Celeste, A.; Pellegrini, M.; McBride, K.; Wangsa, D.; Bredemeyer, A.L.; et al. ATM Prevents the Persistence and Propagation of Chromosome Breaks in Lymphocytes. Cell 2007, 130, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Lumsden, J.M.; Mccarty, T.; Petiniot, L.K.; Shen, R.; Barlow, C.; Wynn, T.A.; Morse, H.C.; Gearhart, P.J.; Wynshaw-Boris, A.; Max, E.E.; et al. Immunoglobulin Class Switch Recombination Is Impaired in Atm-deficient Mice. J. Exp. Med. 2004, 200, 1111–1121. [Google Scholar] [CrossRef] [Green Version]
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.; Eckhaus, M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.; et al. Atm-Deficient Mice: A Paradigm of Ataxia Telangiectasia. Cell 1996, 86, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Roco, J.A.; Mesin, L.; Binder, S.C.; Nefzger, C.; Gonzalez-Figueroa, P.; Canete, P.F.; Ellyard, J.; Shen, Q.; Robert, P.A.; Cappello, J.; et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 2019, 51, 337–350.e7. [Google Scholar] [CrossRef]
- Reina-San-Martin, B.; Chen, H.T.; Nussenzweig, A.; Nussenzweig, M.C. ATM Is Required for Efficient Recombination between Immunoglobulin Switch Regions. J. Exp. Med. 2004, 200, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Neumeister, P.; Goossens, T.; Nanjangud, G.; Chaganti, R.S.K.; Küppers, R.; Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nat. Cell Biol. 2001, 412, 341–346. [Google Scholar] [CrossRef]
- Jiang, Y.; Soong, T.D.; Wang, L.; Melnick, A.M.; Elemento, O. Genome-Wide Detection of Genes Targeted by Non-Ig Somatic Hypermutation in Lymphoma. PLoS ONE 2012, 7, e40332. [Google Scholar] [CrossRef] [Green Version]
- Mlynarczyk, C.; Fontán, L.; Melnick, A. Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol. Rev. 2019, 288, 214–239. [Google Scholar] [CrossRef] [Green Version]
- Economopoulou, P.; Pappa, V.; Papageorgiou, S.; Dervenoulas, J.; Economopoulos, T. Abnormalities of DNA repair mechanisms in common hematological malignancies. Leuk. Lymphoma 2011, 52, 567–582. [Google Scholar] [CrossRef]
- Kennedy, R.D.; D’Andrea, A.D. DNA Repair Pathways in Clinical Practice: Lessons from Pediatric Cancer Susceptibility Syndromes. J. Clin. Oncol. 2006, 24, 3799–3808. [Google Scholar] [CrossRef]
- Rendleman, J.; Antipin, Y.; Reva, B.; Adaniel, C.; Przybylo, J.A.; Dutra-Clarke, A.; Hansen, N.; Heguy, A.; Huberman, K.; Borsu, L.; et al. Genetic Variation in DNA Repair Pathways and Risk of Non-Hodgkin’s Lymphoma. PLoS ONE 2014, 9, e101685. [Google Scholar] [CrossRef]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Miranda, N.F.C.C.; Peng, R.; Georgiou, K.; Wu, C.; Sörqvist, E.F.; Berglund, M.; Chen, L.; Gao, Z.; Lagerstedt, K.; Lisboa, S.; et al. DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J. Exp. Med. 2013, 210, 1729–1742. [Google Scholar] [CrossRef] [PubMed]
- Riabinska, A.; Lehrmann, D.; Jachimowicz, R.D.; Knittel, G.; Fritz, C.; Schmitt, A.; Geyer, A.; Heneweer, C.; Wittersheim, M.; Frenzel, L.P.; et al. ATM activity in T cells is critical for immune surveillance of lymphoma in vivo. Leukemia 2019, 34, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Desiderio, S. Cell cycle regulation of V(D)J recombination-activating protein RAG-2. Proc. Natl. Acad. Sci. USA 1994, 91, 2733–2737. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Chang, F.-C.; Ross, A.E.; Lee, J.; Nakayama, K.; Nakayama, K.; Desiderio, S. Ubiquitylation of RAG-2 by Skp2-SCF Links Destruction of the V(D)J Recombinase to the Cell Cycle. Mol. Cell 2005, 18, 699–709. [Google Scholar] [CrossRef]
- Zhang, L.; Reynolds, T.L.; Shan, X.; Desiderio, S. Coupling of V(D)J Recombination to the Cell Cycle Suppresses Genomic Instability and Lymphoid Tumorigenesis. Immunity 2011, 34, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Schrader, C.E.; Guikema, J.E.J.; Linehan, E.K.; Selsing, E.; Stavnezer, J. Activation-Induced Cytidine Deaminase-Dependent DNA Breaks in Class Switch Recombination Occur during G1 Phase of the Cell Cycle and Depend upon Mismatch Repair. J. Immunol. 2007, 179, 6064–6071. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Kieffer-Kwon, K.-R.; Oliveira, T.Y.; Mayer, C.T.; Yao, K.; Pai, J.; Cao, Z.; Dose, M.; Casellas, R.; Jankovic, M.; et al. The cell cycle restricts activation-induced cytidine deaminase activity to early G1. J. Exp. Med. 2016, 214, 49–58. [Google Scholar] [CrossRef]
- O’Shea, D.; O’Riain, C.; Taylor, C.; Waters, R.; Carlotti, E.; MacDougall, F.; Gribben, J.; Rosenwald, A.; Ott, G.; Rimsza, L.M.; et al. The presence of TP53 mutation at diagnosis of follicular lymphoma identifies a high-risk group of patients with shortened time to disease progression and poorer overall survival. Blood 2008, 112, 3126–3129. [Google Scholar] [CrossRef]
- Malarikova, D.; Berkova, A.; Obr, A.; Blahovcova, P.; Svaton, M.; Forsterova, K.; Kriegova, E.; Prihodova, E.; Pavlistova, L.; Petrackova, A.; et al. Concurrent TP53 and CDKN2A Gene Aberrations in Newly Diagnosed Mantle Cell Lymphoma Correlate with Chemoresistance and Call for Innovative Upfront Therapy. Cancers 2020, 12, 2120. [Google Scholar] [CrossRef]
- Sánchez-Beato, M.; Sánchez-Aguilera, A.; Piris, M. Ángel Cell cycle deregulation in B-cell lymphomas. Blood 2003, 101, 1220–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Loyola, A.; Fernández-Miranda, G.; Trakala, M.; Partida, D.; Samejima, K.; Ogawa, H.; Cañamero, M.; De Martino, A.; Martínez-Ramírez, A.; De Cárcer, G.; et al. Aurora B Overexpression Causes Aneuploidy and p21Cip1Repression during Tumor Development. Mol. Cell. Biol. 2015, 35, 3566–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Bartlett, N.L.; Blum, K.A.; Park, S.; Maddocks, K.; Ruan, J.; Ridling, L.; Dittus, C.; Chen, Z.; Huang, X.; et al. A phase 1 trial of ibrutinib plus palbociclib in previously treated mantle cell lymphoma. Blood 2019, 133, 1201–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Ruan, J.; Furman, R.; Rutherford, S.; Allan, J.; Chen, Z.; Huang, X.; DiLiberto, M.; Chen-Kiang, S.; Leonard, J.P. A phase I trial of palbociclib plus bortezomib in previously treated mantle cell lymphoma. Leuk. Lymphoma 2019, 60, 2917–2921. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zheng, W.; Chen, T.; Lin, W.; Liao, Z.; Liu, J.; Ding, Y. CDK4/6 Inhibitor Palbociclib Amplifies the Radiosensitivity to Nasopharyngeal Carcinoma Cells via Mediating Apoptosis and Suppressing DNA Damage Repair. Onco Targets Ther. 2019, 12, 11107–11117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, A.M.; Alexander, A.; Liu, Y.; Vijayaraghavan, S.; Low, K.H.; Yang, D.; Bui, T.; Somaiah, N.; Ravi, V.; Keyomars, K.; et al. CDK4/6 Inhibitors Sensitize Rb-positive Sarcoma Cells to Wee1 Kinase Inhibition through Reversible Cell-Cycle Arrest. Mol. Cancer Ther. 2017, 16, 1751–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.L.; McClendon, A.K.; Knudsen, E.S. Modification of the DNA damage response by therapeutic CDK4/6 inhibition. J. Biol. Chem. 2012, 287, 29075–29087. [Google Scholar] [CrossRef] [Green Version]
- Tempka, D.; Tokarz, P.; Chmielewska, K.; Kluska, M.; Pietrzak, J.; Rygielska, Z.; Virág, L.; Robaszkiewicz, A. Downregulation of PARP1 transcription by CDK4/6 inhibitors sensitizes human lung cancer cells to anticancer drug-induced death by impairing OGG1-dependent base excision repair. Redox Biol. 2018, 15, 316–326. [Google Scholar] [CrossRef]
- Lacrima, K.; Valentini, A.; Lambertini, C.; Taborelli, M.; Rinaldi, A.; Zucca, E.; Catapano, C.; Cavalli, F.; Gianella-Borradori, A.; Maccallum, D.E.; et al. In vitro activity of cyclin-dependent kinase inhibitor CYC202 (Seliciclib, R-roscovitine) in mantle cell lymphomas. Ann. Oncol. 2005, 16, 1169–1176. [Google Scholar] [CrossRef]
- Venkataraman, G.; Maududi, T.; Ozpuyan, F.; Bahar, H.I.; Izban, K.F.; Qin, J.-Z.; Alkan, S. Induction of apoptosis and down regulation of cell cycle proteins in mantle cell lymphoma by flavopiridol treatment. Leuk. Res. 2006, 30, 1377–1384. [Google Scholar] [CrossRef]
- Holkova, B.; Kmieciak, M.; Perkins, E.B.; Bose, P.; Baz, R.C.; Roodman, G.D.; Stuart, R.K.; Ramakrishnan, V.; Wan, W.; Peer, C.J.; et al. Phase I Trial of Bortezomib (PS-341; NSC 681239) and “Non-Hybrid” (Bolus) Infusion Schedule of Alvocidib (Flavopiridol; NSC 649890) in Patients with Recurrent or Refractory Indolent B-cell Neoplasms. Clin. Cancer Res. 2014, 20, 5652–5662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raju, U.; Nakata, E.A.; Mason, K.; Ang, K.K.; Milas, L. Flavopiridol, a cyclin-dependent kinase inhibitor, enhances radiosensitivity of ovarian carcinoma cells. Cancer Res. 2003, 63, 3263–3267. [Google Scholar] [PubMed]
- Bible, K.C.; Bible, R.H.; Kottke, T.J.; Svingen, A.; Xu, K.P.; Pang, Y.P.; Hajdu, E.; Kaufmann, S.H. Flavopiridol binds to duplex DNA. Cancer Res. 2000, 60, 2419–2428. [Google Scholar] [PubMed]
- Do, T.-V.; Hirst, J.; Hyter, S.; Roby, K.F.; Godwin, A.K. Aurora A kinase regulates non-homologous end-joining and poly(ADP-ribose) polymerase function in ovarian carcinoma cells. Oncotarget 2017, 8, 50376–50392. [Google Scholar] [CrossRef]
- Alcaraz-Sanabria, A.; Nieto-Jimenez, C.; Corrales-Sanchez, V.; Serrano-Oviedo, L.; Andrés-Pretel, F.; Carlos Montero, J.; Burgos, M.; Llopis, J.; Galán-Moya, E.M.; Pandiella, A.; et al. Synthetic Lethality Interaction Between Aurora Kinases and CHEK1 Inhibitors in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 2552–2562. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Cooke, L.S.; Liu, X.; Rimsza, L.; Roe, D.J.; Persky, A.M.D.O.; Miller, T.P.; Mahadevan, D. Aurora inhibitor MLN8237 in combination with docetaxel enhances apoptosis and anti-tumor activity in mantle cell lymphoma. Biochem. Pharmacol. 2011, 81, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.W.; Gascoyne, R.D. The tumour microenvironment in B cell lymphomas. Nat. Rev. Cancer 2014, 14, 517–534. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2019, 20, 25–39. [Google Scholar] [CrossRef]
- Greiner, T.C.; Dasgupta, C.; Ho, V.V.; Weisenburger, D.D.; Smith, L.M.; Lynch, J.C.; Vose, J.M.; Fu, K.; Armitage, J.O.; Braziel, R.M.; et al. Mutation and genomic deletion status ofataxia telangiectasia mutated(ATM) andp53confer specific gene expression profiles in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2006, 103, 2352–2357. [Google Scholar] [CrossRef] [Green Version]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wörmann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2–STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balli, D.; Rech, A.J.; Stanger, B.Z.; Vonderheide, R.H. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin. Cancer Res. 2016, 23, 3129–3138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delfau-Larue, M.-H.; Klapper, W.; Berger, F.; Jardin, F.; Briere, J.; Salles, G.; Casasnovas, O.; Feugier, P.; Haioun, C.; Ribrag, V.; et al. High-dose cytarabine does not overcome the adverse prognostic value of CDKN2A and TP53 deletions in mantle cell lymphoma. Blood 2015, 126, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov 2018, 8, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.-J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997. [Google Scholar] [CrossRef] [Green Version]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chinet, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Christensen, C.L.; Dries, R.; Oser, M.G.; Deng, J.; Diskin, B.; Li, F.; Pan, Y.; Zhang, X.; Yin, Y.; et al. CDK7 Inhibition Potentiates Genome Instability Triggering Anti-tumor Immunity in Small Cell Lung Cancer. Cancer Cell 2020, 37, 37–54. [Google Scholar] [CrossRef]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.-H.A.; Zundell, J.A.; Ranatunga, S.; Lin, C.; Nefedova, Y.; Del Valle, J.R.; Hu, C.-C.A. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res. 2016, 76, 2137–2152. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Della Corte, C.M.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mender, I.; Zhang, A.; Ren, Z.; Han, C.; Deng, Y.; Siteni, S.; Li, H.; Zhu, J.; Vemula, A.; Shay, J.W.; et al. Telomere Stress Potentiates STING-Dependent Anti-tumor Immunity. Cancer Cell 2020, 38, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.N.; Yu, C.; Vartabedian, V.F.; Jia, J.; Kumar, M.; Gamo, A.M.; Vernier, W.; Ali, S.H.; Kissai, M.; Lazar, D.C.; et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 2020, 369, 993–999. [Google Scholar]
- Pan, B.S.; Perera, S.A.; Piesvaux, J.A.; Presland, J.P.; Schroeder, G.K.; Cumming, J.N.; Trotter, B.W.; Altman, M.D.; Buevich, A.V.; Cash, B.; et al. An orally available non-nucleotide STING agonist with antitumor activity. Science 2020, 369, eaba6098. [Google Scholar]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2016, 7, 20–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jachimowicz, R.D.; Goergens, J.; Reinhardt, H.C. DNA double-strand break repair pathway choice—From basic biology to clinical exploitation. Cell Cycle 2019, 18, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Reinhardt, H.C.; Bartkova, J.; Tommiska, J.; Blomqvist, C.; Nevanlinna, H.; Bartek, J.; Yaffe, M.B.; Hemann, M.T. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009, 23, 1895–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Cabral, S.; Brough, R.; Konde, A.; Aarts, M.; Campbell, J.; Marinari, E.; Riffell, J.; Bardelli, A.; Torrance, C.; Lord, C.J.; et al. CDK1 Is a Synthetic Lethal Target for KRAS Mutant Tumours. PLoS ONE 2016, 11, e0149099. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Alexe, G.; Roti, G.; Conway, A.S.; Furman, A.; Lee, E.S.; Place, A.E.; Kim, S.; Saran, C.; Modiste, R.; et al. Synergistic Drug Combinations with a CDK4/6 Inhibitor in T-cell Acute Lymphoblastic Leukemia. Clin Cancer Res. 2017, 23, 1012–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2019, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nat. Cell Biol. 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Chow, R.D.; Zhu, L.; Bai, Z.; Ye, L.; Zhang, F.A.; Renauer, P.; Dong, M.B.; Dai, X.; Zhang, X.; et al. CRISPR-GEMM pooled mutagenic screening identifies KMT2D as a major modulator of immune checkpoint blockade. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nat. Cell Biol. 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
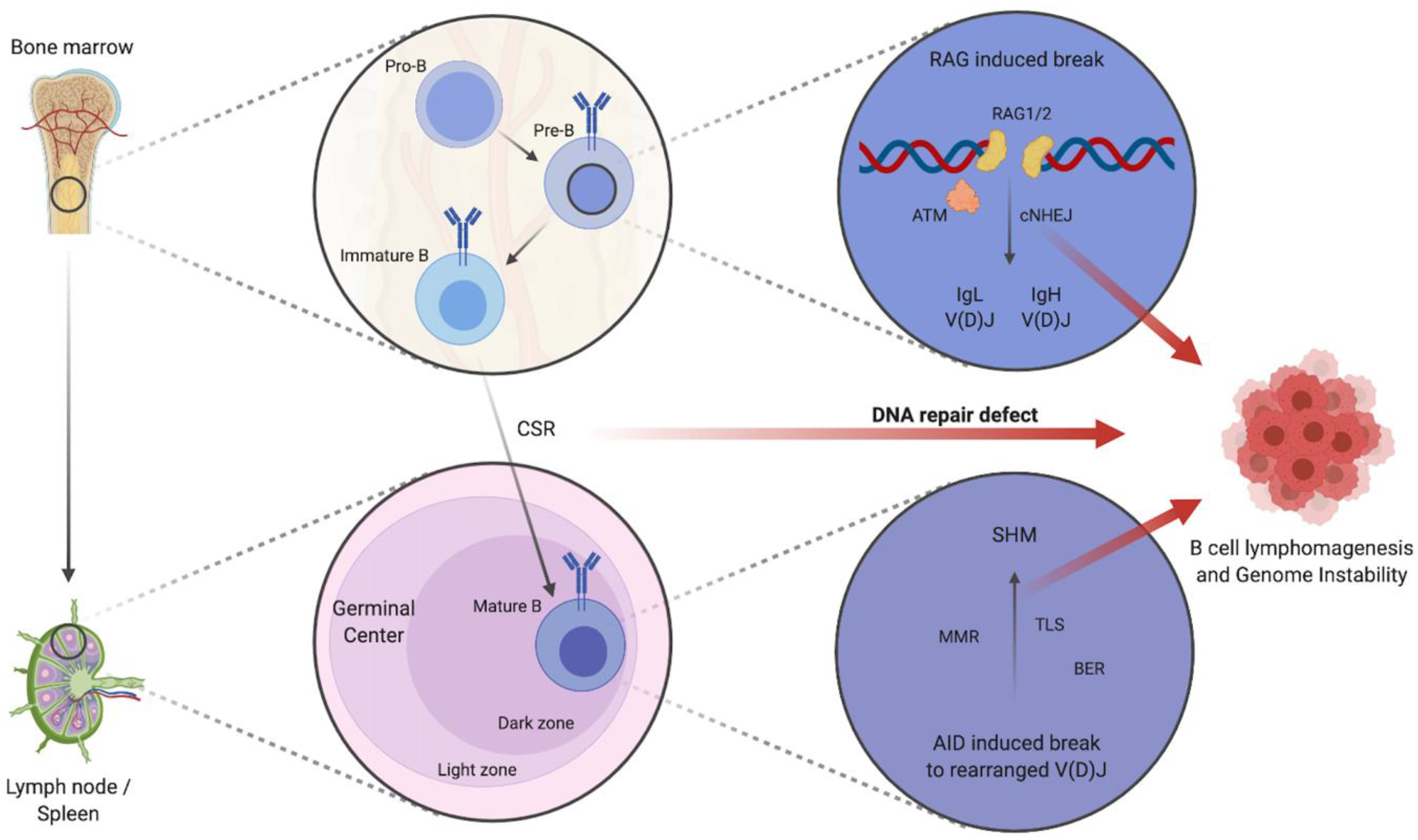
Figure 1.
Schematic depiction of B cell development and impaired DNA repair contributing to B cell lymphomagenesis and increased genome instability. Schematic figure depicting the maturation of pro-B cells to immature B cells in the bone marrow by cNHEJ mediated IgL and IgH V(D)J. CSR and consequent SHM of mature B cells in the dark zone of GCs is mediated by further DNA repair mechanisms: e.g., MMR, BER, and TLS. Defects in DNA repair pathways contribute to B cell lymphomagenesis and lead to genome instability with the increased occurrence of translocations, fusions, deletions, or breaks/gaps. Abbreviations: IgL: immunoglobulin light chain, IgH: immunoglobulin heavy chain, V(D)J: variable (V), diversity (D), and joining (J) gene segments, cNHEJ: classical non-homologous end joining, ATM: ataxia telangiectasia mutated, CSR: class switch recombination, SHM: somatic hypermutation, MMR: mismatch repair, BER: base excision repair, TLS: trans-lesion synthesis, AID: activation-induced cytidine deaminase, RAG1/2: recombination-activation gene 1/2. Created with BioRender.com.
Figure 1.
Schematic depiction of B cell development and impaired DNA repair contributing to B cell lymphomagenesis and increased genome instability. Schematic figure depicting the maturation of pro-B cells to immature B cells in the bone marrow by cNHEJ mediated IgL and IgH V(D)J. CSR and consequent SHM of mature B cells in the dark zone of GCs is mediated by further DNA repair mechanisms: e.g., MMR, BER, and TLS. Defects in DNA repair pathways contribute to B cell lymphomagenesis and lead to genome instability with the increased occurrence of translocations, fusions, deletions, or breaks/gaps. Abbreviations: IgL: immunoglobulin light chain, IgH: immunoglobulin heavy chain, V(D)J: variable (V), diversity (D), and joining (J) gene segments, cNHEJ: classical non-homologous end joining, ATM: ataxia telangiectasia mutated, CSR: class switch recombination, SHM: somatic hypermutation, MMR: mismatch repair, BER: base excision repair, TLS: trans-lesion synthesis, AID: activation-induced cytidine deaminase, RAG1/2: recombination-activation gene 1/2. Created with BioRender.com.

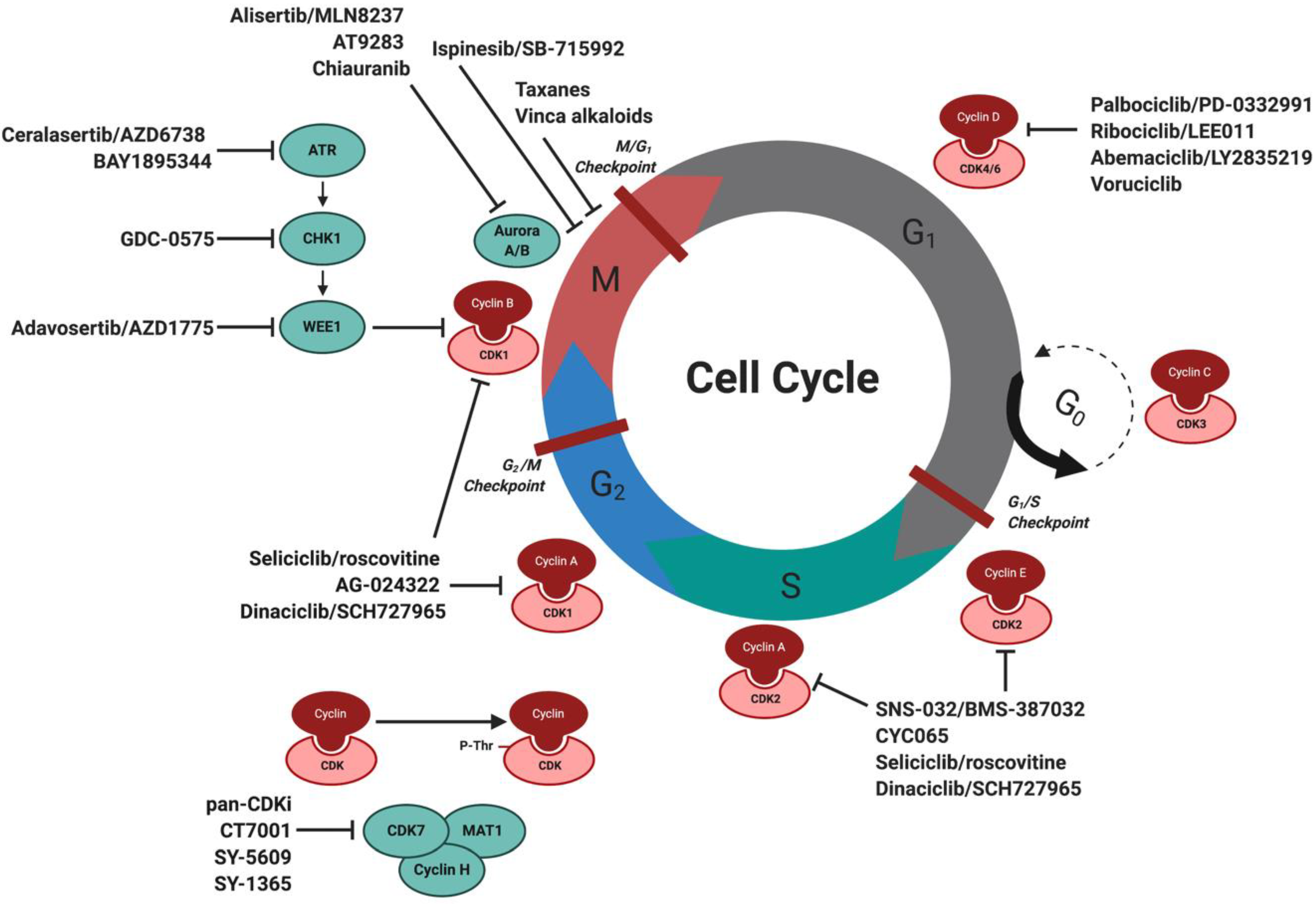
Figure 2.
Schematic overview of the cell cycle and cell cycle targeting inhibitors in MCL and other B cell lymphomas. Progression of the cell cycle is facilitated by specific interaction and activation of CDKs with cyclins to allow cell cycle progression. Listed are cell cycle inhibitors currently under investigation for the treatment of MCL and B cell lymphomas, in addition to upstream targeted inhibitors against DDR proteins. Abbreviations: CDK: cyclin-dependent kinases, MCL: mantle cell lymphoma, DDR: DNA damage response. Created with BioRender.com.
Figure 2.
Schematic overview of the cell cycle and cell cycle targeting inhibitors in MCL and other B cell lymphomas. Progression of the cell cycle is facilitated by specific interaction and activation of CDKs with cyclins to allow cell cycle progression. Listed are cell cycle inhibitors currently under investigation for the treatment of MCL and B cell lymphomas, in addition to upstream targeted inhibitors against DDR proteins. Abbreviations: CDK: cyclin-dependent kinases, MCL: mantle cell lymphoma, DDR: DNA damage response. Created with BioRender.com.

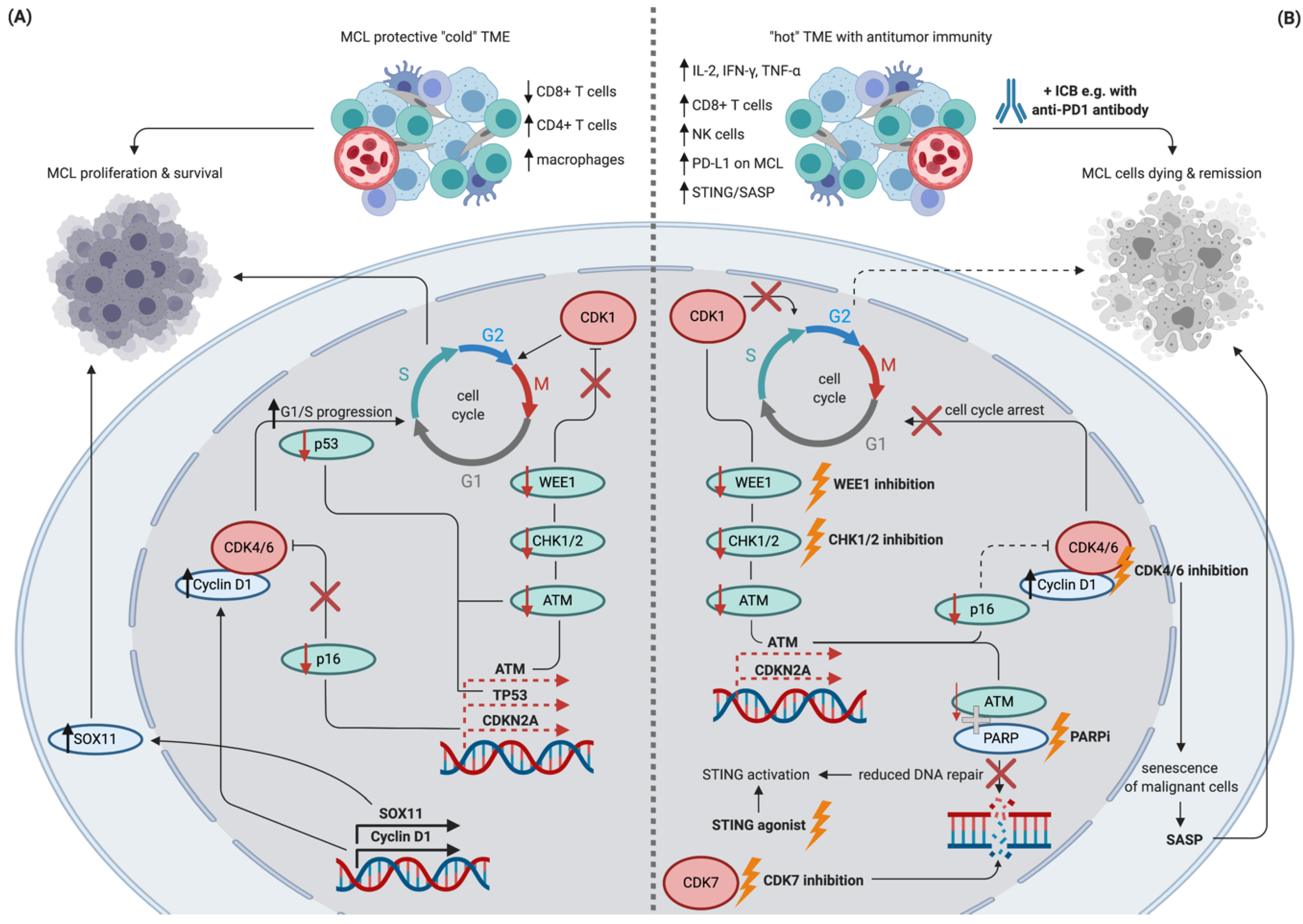
Figure 3.
Schematic depiction of potential synergistic effects of DNA repair inhibition, cell cycle inhibition, and immune checkpoint blockade in MCL. Schematic figure showing selected features of treatment naïve MCL resulting in the proliferation of malignant cells (panel A) in contrast to potentially synergistic effects of the currently available treatments targeting cell cycle inhibition in combination with ICB (panel B). Dashed lines indicate a lower than normal transcription/function. Targetable cell cycle machinery and respective interventions are highlighted in bold and by lightning symbols. Abbreviations: MCL: mantle cell lymphoma, TME: tumor microenvironment, ICB: immune checkpoint blockade, SASP: senescence-associated secretory phenotype, STING: stimulator of interferon genes, PARPi: PARP inhibition. Created with BioRender.com.
Figure 3.
Schematic depiction of potential synergistic effects of DNA repair inhibition, cell cycle inhibition, and immune checkpoint blockade in MCL. Schematic figure showing selected features of treatment naïve MCL resulting in the proliferation of malignant cells (panel A) in contrast to potentially synergistic effects of the currently available treatments targeting cell cycle inhibition in combination with ICB (panel B). Dashed lines indicate a lower than normal transcription/function. Targetable cell cycle machinery and respective interventions are highlighted in bold and by lightning symbols. Abbreviations: MCL: mantle cell lymphoma, TME: tumor microenvironment, ICB: immune checkpoint blockade, SASP: senescence-associated secretory phenotype, STING: stimulator of interferon genes, PARPi: PARP inhibition. Created with BioRender.com.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bröckelmann, P.J.; de Jong, M.R.W.; Jachimowicz, R.D. Targeting DNA Repair, Cell Cycle, and Tumor Microenvironment in B Cell Lymphoma. Cells 2020, 9, 2287. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9102287
AMA Style
Bröckelmann PJ, de Jong MRW, Jachimowicz RD. Targeting DNA Repair, Cell Cycle, and Tumor Microenvironment in B Cell Lymphoma. Cells. 2020; 9(10):2287. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9102287
Chicago/Turabian StyleBröckelmann, Paul J., Mathilde R. W. de Jong, and Ron D. Jachimowicz. 2020. "Targeting DNA Repair, Cell Cycle, and Tumor Microenvironment in B Cell Lymphoma" Cells 9, no. 10: 2287. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9102287
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.