
Altered Expression of TSPAN32 during B Cell Activation and Systemic Lupus Erythematosus

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
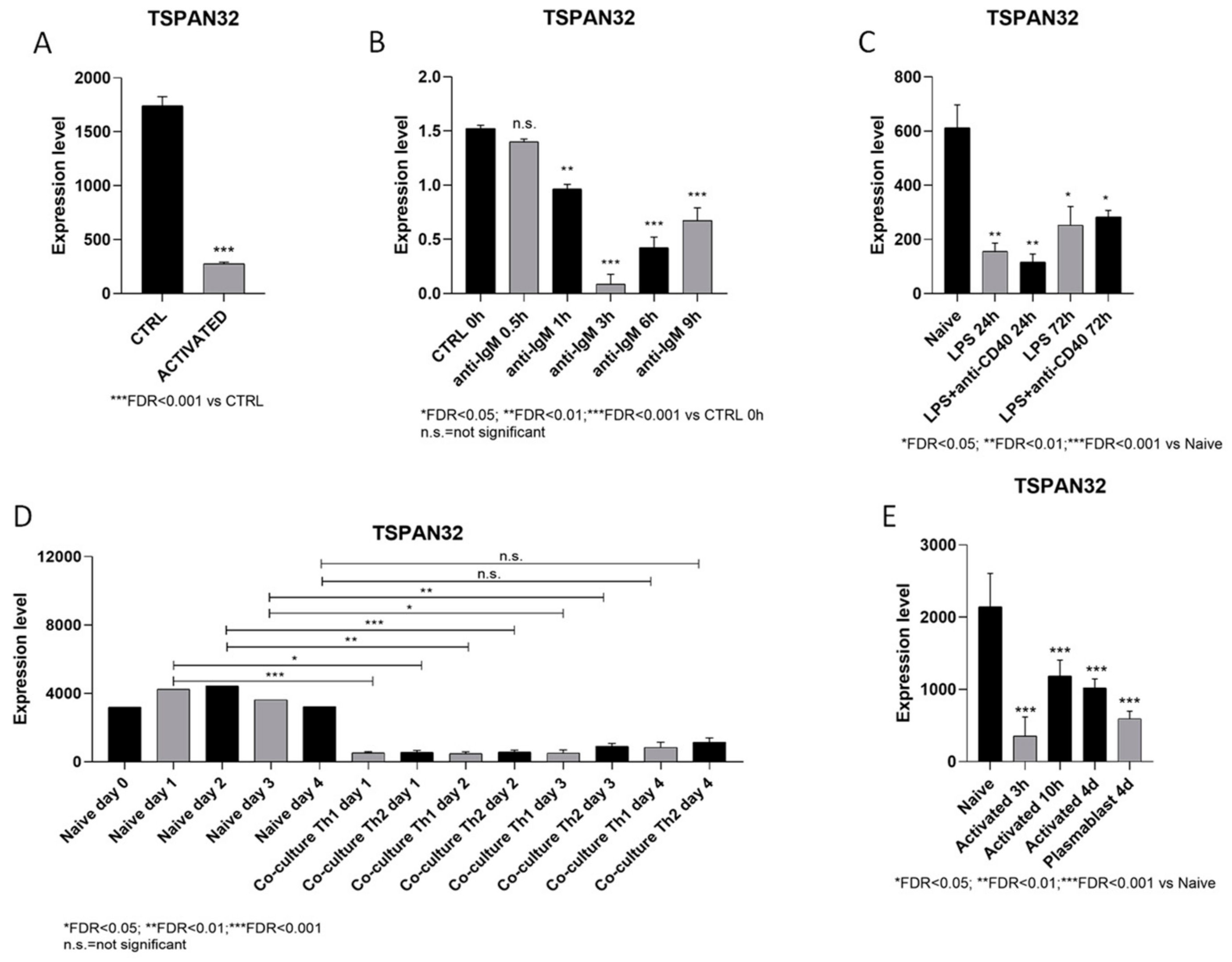
2.1. Profiling of TSPAN32 Transcription in B Cell Activation
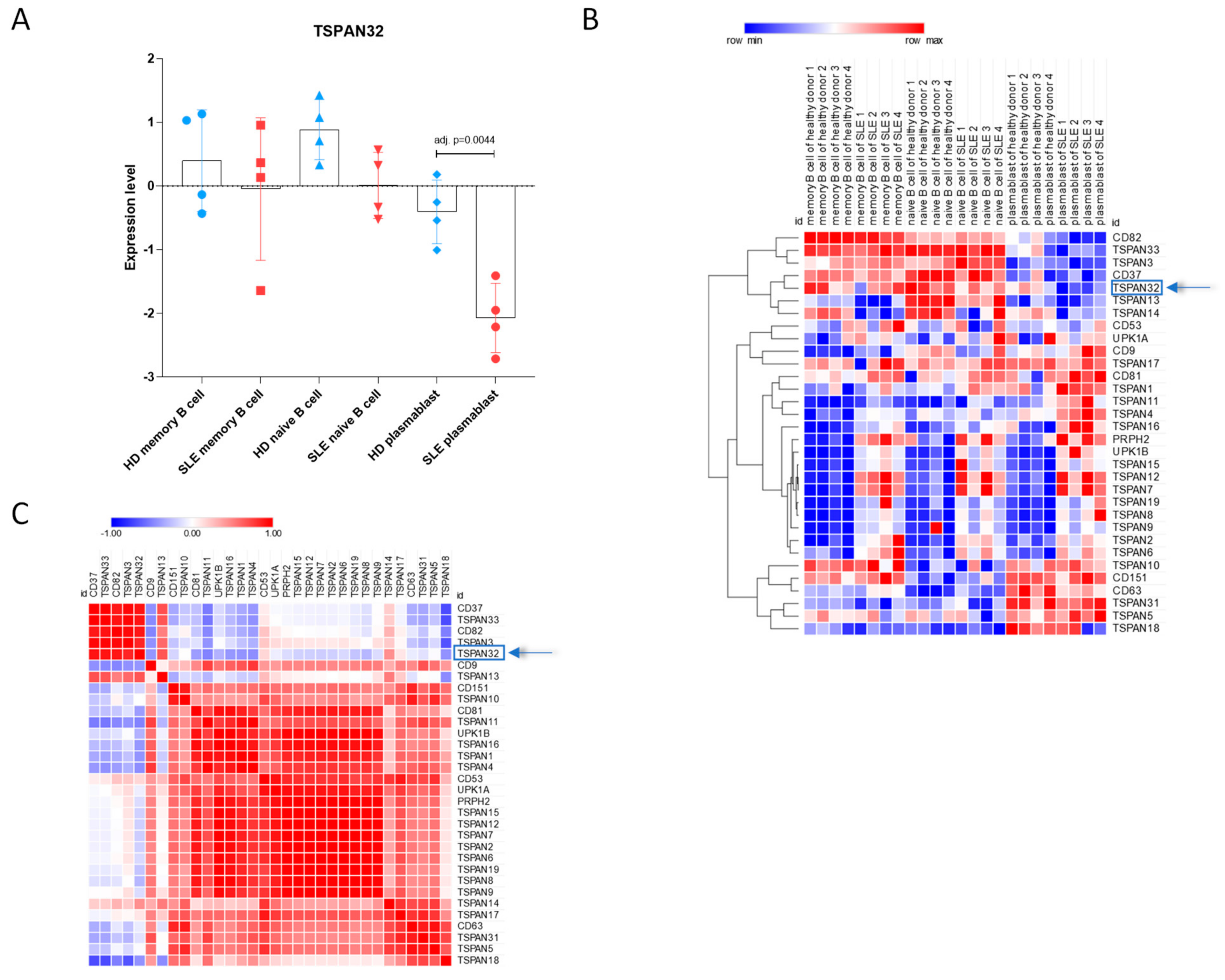
2.2. Evaluation of TSPAN32 Levels in SLE B Cells
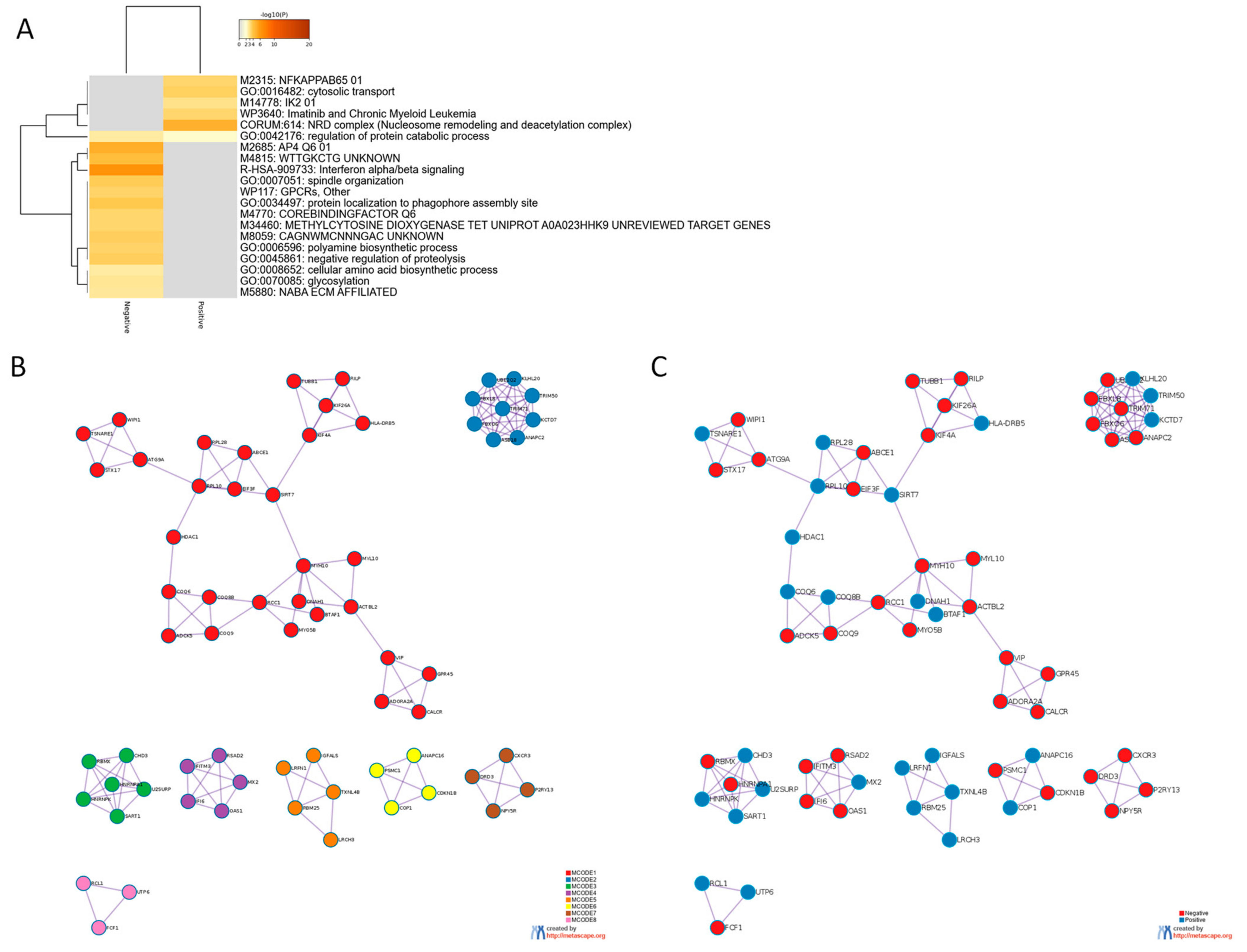
2.3. Gene Ontology Analysis
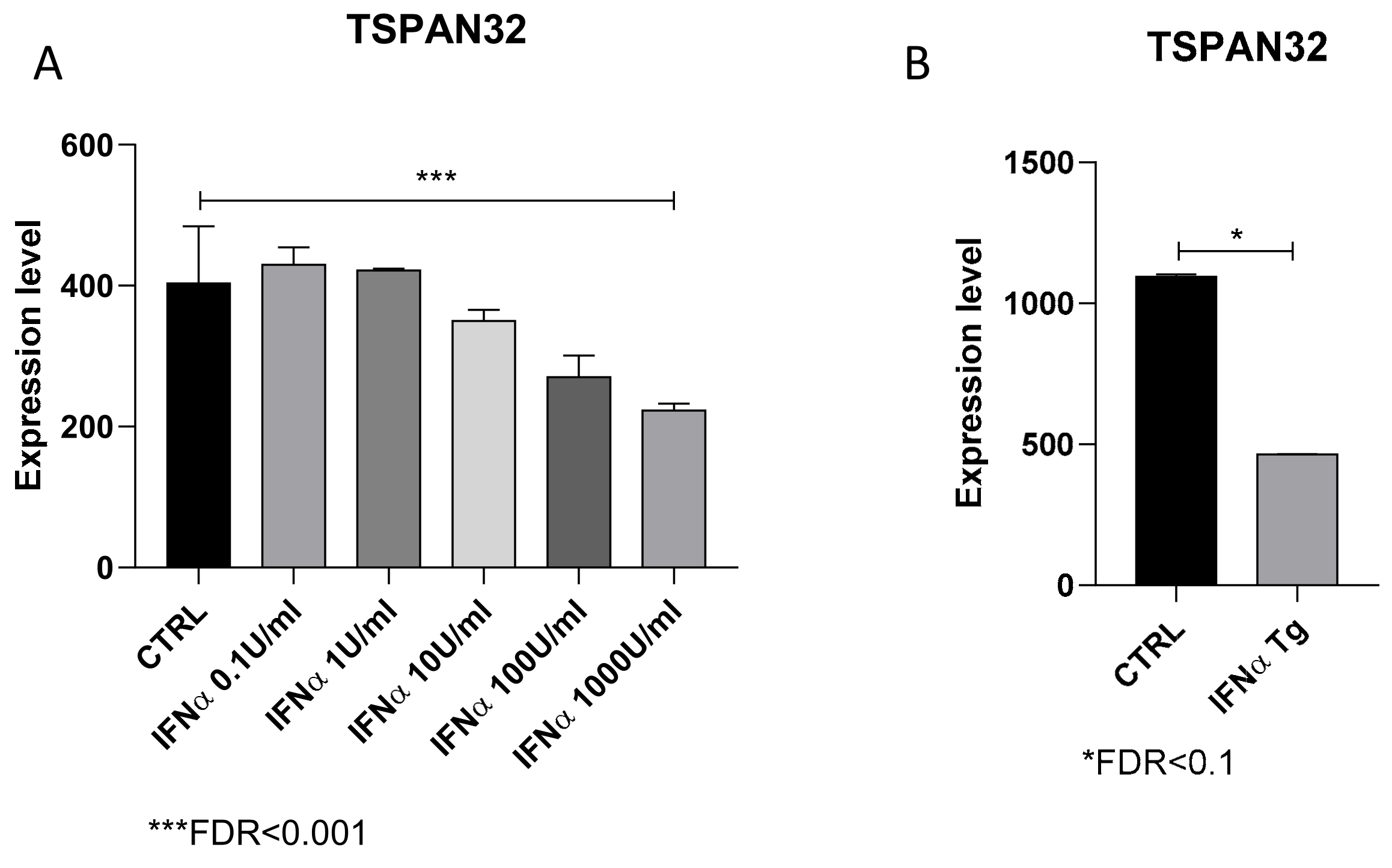
2.4. Evaluation of the Effect of IFN-α on TSPAN32 Levels
2.5. Statistical Analysis
3. Results
3.1. Modulation of TSPAN32 Expression in B Cell Activation
3.2. TSPAN32 Expression Levels in SLE B Cell
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tsokos, G.C. Systemic Lupus Erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chock, Y.P.; Moulinet, T.; Dufrost, V.; Erkan, D.; Wahl, D.; Zuily, S. Antiphospholipid antibodies and the risk of thrombocytopenia in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Autoimmun. Rev. 2019, 18, 102395. [Google Scholar] [CrossRef] [PubMed]
- Möckel, T.; Basta, F.; Weinmann-Menke, J.; Schwarting, A. B cell activating factor (BAFF): Structure, functions, autoimmunity and clinical implications in Systemic Lupus Erythematosus (SLE). Autoimmun. Rev. 2021, 20, 102736. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Zhao, L.; Zhang, X. B Cell Aberrance in Lupus: The Ringleader and the Solution. Clin. Rev. Allergy Immunol. 2021, 1–23. [Google Scholar] [CrossRef]
- Chyuan, I.-T.; Tzeng, H.-T.; Chen, J.-Y. Signaling Pathways of Type I and Type III Interferons and Targeted Therapies in Systemic Lupus Erythematosus. Cells 2019, 8, 963. [Google Scholar] [CrossRef] [Green Version]
- Chasset, F.; Arnaud, L. Targeting interferons and their pathways in systemic lupus erythematosus. Autoimmun. Rev. 2018, 17, 44–52. [Google Scholar] [CrossRef]
- Nicholson, R.H.; Pantano, S.; Eliason, J.F.; Galy, A.; Weiler, S.; Kaplan, J.; Hughes, M.R.; Ko, M.S. Phemx, a Novel Mouse Gene Expressed in Hematopoietic Cells Maps to the Imprinted Cluster on Distal Chromosome 7. Genomics 2000, 68, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, S.D.; Mazzon, E.; Basile, M.S.; Campo, G.; Corsico, F.; Presti, M.; Bramanti, P.; Mangano, K.; Petralia, M.C.; Nicoletti, F.; et al. Modulation of Tetraspanin 32 (TSPAN32) Expression in T Cell-Mediated Immune Responses and in Multiple Sclerosis. Int. J. Mol. Sci. 2019, 20, 4323. [Google Scholar] [CrossRef] [Green Version]
- Basile, M.S.; Mazzon, E.; Mangano, K.; Pennisi, M.; Petralia, M.C.; Lombardo, S.D.; Nicoletti, F.; Fagone, P.; Cavalli, E. Impaired Expression of Tetraspanin 32 (TSPAN32) in Memory T Cells of Patients with Multiple Sclerosis. Brain Sci. 2020, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, J.; Verma-Gaur, J.; Wallenius, A.; Grundström, T. Initiation of Antigen Receptor-Dependent Differentiation into Plasma Cells by Calmodulin Inhibition of E2A. J. Immunol. 2009, 183, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Damdinsuren, B.; Zhang, Y.; Khalil, A.; Wood, W.H.; Becker, K.G.; Shlomchik, M.J.; Sen, R. Single Round of Antigen Receptor Signaling Programs Naive B Cells to Receive T Cell Help. Immunity 2010, 32, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nat. Cell Biol. 2014, 507, 366–370. [Google Scholar] [CrossRef] [Green Version]
- Stone, S.L.; Peel, J.N.; Scharer, C.; Risley, C.A.; Chisolm, D.A.; Schultz, M.D.; Yu, B.; Ballesteros-Tato, A.; Wojciechowski, W.; Mousseau, B.; et al. T-bet Transcription Factor Promotes Antibody-Secreting Cell Differentiation by Limiting the Inflammatory Effects of IFN-γ on B Cells. Immunity 2019, 50, 1172–1187.e7. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, S.; Yoshida, H.; Moodley, D.; LeBoité, H.; Rothamel, K.; Raj, T.; Ye, C.J.; Chevrier, N.; Zhang, S.Y.; Feng, T.; et al. Immunological Genome Project Consortium. Parsing the Interferon Transcriptional Network and Its Disease Associations. Cell 2016, 164, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, C.; Tsumiyama, K.; Uchimura, C.; Honda, E.; Miyazaki, Y.; Sakurai, K.; Miura, Y.; Hashiramoto, A.; Felsher, D.W.; Shiozawa, S. Conditional Upregulation of IFN-α Alone Is Sufficient to Induce Systemic Lupus Erythematosus. J. Immunol. 2019, 203, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Shoham, T. The tetraspanin web modulates immune-signalling complexes. Nat. Rev. Immunol. 2005, 5, 136–148. [Google Scholar] [CrossRef]
- Tarrant, J.M.; Groom, J.; Metcalf, D.; Li, R.; Borobokas, B.; Wright, M.D.; Tarlinton, D.; Robb, L. The Absence of Tssc6, a Member of the Tetraspanin Superfamily, Does Not Affect Lymphoid Development but Enhances In Vitro T-Cell Proliferative Responses. Mol. Cell. Biol. 2002, 22, 5006–5018. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, M.; Edström, M.; Gawel, D.; Nestor, C.E.; Wang, H.; Zhang, H.; Barrenäs, F.; Tojo, J.; Kockum, I.; Olsson, T.; et al. Integrated genomic and prospective clinical studies show the importance of modular pleiotropy for disease susceptibility, diagnosis and treatment. Genome Med. 2014, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, S.; Mazzon, E.; Basile, M.; Cavalli, E.; Bramanti, P.; Nania, R.; Fagone, P.; Nicoletti, F.; Petralia, M. Upregulation of IL-1 Receptor Antagonist in a Mouse Model of Migraine. Brain Sci. 2019, 9, 172. [Google Scholar] [CrossRef] [Green Version]
- Fagone, P.; Mazzon, E.; Chikovani, T.; Saraceno, A.; Mammana, S.; Colletti, G.; Mangano, K.; Bramanti, P.; Nicoletti, F. Decitabine induces regulatory T cells, inhibits the production of IFN-gamma and IL-17 and exerts preventive and therapeutic efficacy in rodent experimental autoimmune neuritis. J. Neuroimmunol. 2018, 321, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangano, K.; Cavalli, E.; Mammana, S.; Basile, M.S.; Caltabiano, R.; Pesce, A.; Puleo, S.; Atanasov, A.G.; Magro, G.; Nicoletti, F.; et al. Involvement of the Nrf2/HO-1/CO axis and therapeutic intervention with the CO-releasing molecule CORM-A1, in a murine model of autoimmune hepatitis. J. Cell. Physiol. 2018, 233, 4156–4165. [Google Scholar] [CrossRef] [PubMed]
- Mangano, K.; Mazzon, E.; Basile, M.S.; Di Marco, R.; Bramanti, P.; Mammana, S.; Petralia, M.C.; Fagone, P.; Nicoletti, F. Pathogenic role for macrophage migration inhibitory factor in glioblastoma and its targeting with specific inhibitors as novel tailored therapeutic approach. Oncotarget 2018, 9, 17951–17970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcellini, W.; Rizzardi, G.; Borghi, M.; Nicoletti, F.; Fain, C.; Del Papa, N.; Meroni, P. In vitro type-1 and type-2 cytokine production in systemic lupus erythematosus: Lack of relationship with clinical disease activity. Lupus 1996, 5, 139–145. [Google Scholar] [CrossRef]
- Piantoni, S.; Regola, F.; Masneri, S.; Merletti, M.; Lowin, T.; Airò, P.; Tincani, A.; Franceschini, F.; Andreoli, L.; Pongratz, G. Characterization of B- and T-Cell Compartment and B-Cell Related Factors Belonging to the TNF/TNFR Superfamily in Patients with Clinically Active Systemic Lupus Erythematosus: Baseline BAFF Serum Levels Are the Strongest Predictor of Response to Belimumab after Twelve Months of Therapy. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Jacobi, A.M.; Mei, H.; Hoyer, B.F.; Mumtaz, I.M.; Thiele, K.; Radbruch, A.; Burmester, G.-R.; Hiepe, F.; Dörner, T. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2009, 69, 305–308. [Google Scholar] [CrossRef]
- Soni, C.; Perez, O.A.; Voss, W.N.; Pucella, J.; Serpas, L.; Mehl, J.; Ching, K.; Goike, J.; Georgiou, G.; Ippolito, G.C.; et al. Plasmacytoid Dendritic Cells and Type I Interferon Promote Extrafollicular B Cell Responses to Extracellular Self-DNA. Immunity 2020, 52, 1022–1038.e7. [Google Scholar] [CrossRef]
- Guthridge, J.M.; Lu, R.; Tran, L.T.-H.; Arriens, C.; Aberle, T.; Kamp, S.; Munroe, M.E.; Dominguez, N.; Gross, T.; DeJager, W.; et al. Adults with systemic lupus exhibit distinct molecular phenotypes in a cross-sectional study. EClinicalMedicine 2020, 20, 100291. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; De, S.; Nelson, V.; Chopra, S.; LaPan, M.; Kampta, K.; Sun, S.; He, M.; Thompson, C.D.; Li, D.; et al. Inhibition of IRF5 hyperactivation protects from lupus onset and severity. J. Clin. Investig. 2020, 130, 6700–6717. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network | Annotation |
|---|---|
| MCODE_1 | GO:0034497|protein localization to phagophore assembly site|-6.7;R-HSA-2132295|MHC class II antigen presentation|-6.7;GO:1901663|quinone biosynthetic process|-6.1 |
| MCODE_2 | R-HSA-983168|Antigen processing: Ubiquitination & Proteasome degradation|-17.7;R-HSA-983169|Class I MHC mediated antigen processing & presentation|-17.0;R-HSA-1280218|Adaptive Immune System|-14.2 |
| MCODE_3 | ko03040|Spliceosome|-10.9;hsa03040|Spliceosome|-10.9;R-HSA-72163|mRNA Splicing—Major Pathway|-10.2 |
| MCODE_4 | R-HSA-909733|Interferon α/β signaling|-13.1;GO:0060337|type I interferon signaling pathway|-12.4;GO:0071357|cellular response to type I interferon|-12.4 |
| MCODE_6 | R-HSA-69620|Cell Cycle Checkpoints|-7.9;R-HSA-69580|p53-Dependent G1/S DNA damage checkpoint|-7.3;R-HSA-69563|p53-Dependent G1 DNA Damage Response|-7.3 |
| MCODE_7 | WP455|GPCRs, Class A Rhodopsin-like|-8.2;R-HSA-373076|Class A/1 (Rhodopsin-like receptors)|-7.7;R-HSA-418594|G α (i) signalling events|-7.4 |
| MCODE_8 | R-HSA-6790901|rRNA modification in the nucleus and cytosol|-8.0;ko03008|Ribosome biogenesis in eukaryotes|-7.3;hsa03008|Ribosome biogenesis in eukaryotes|-7.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fagone, P.; Mangano, K.; Di Marco, R.; Reyes-Castillo, Z.; Muñoz-Valle, J.F.; Nicoletti, F. Altered Expression of TSPAN32 during B Cell Activation and Systemic Lupus Erythematosus. Genes 2021, 12, 931. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060931
Fagone P, Mangano K, Di Marco R, Reyes-Castillo Z, Muñoz-Valle JF, Nicoletti F. Altered Expression of TSPAN32 during B Cell Activation and Systemic Lupus Erythematosus. Genes. 2021; 12(6):931. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060931
Chicago/Turabian StyleFagone, Paolo, Katia Mangano, Roberto Di Marco, Zyanya Reyes-Castillo, José Francisco Muñoz-Valle, and Ferdinando Nicoletti. 2021. "Altered Expression of TSPAN32 during B Cell Activation and Systemic Lupus Erythematosus" Genes 12, no. 6: 931. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060931