Autophagy in Zika Virus Infection: A Possible Therapeutic Target to Counteract Viral Replication

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Autophagy
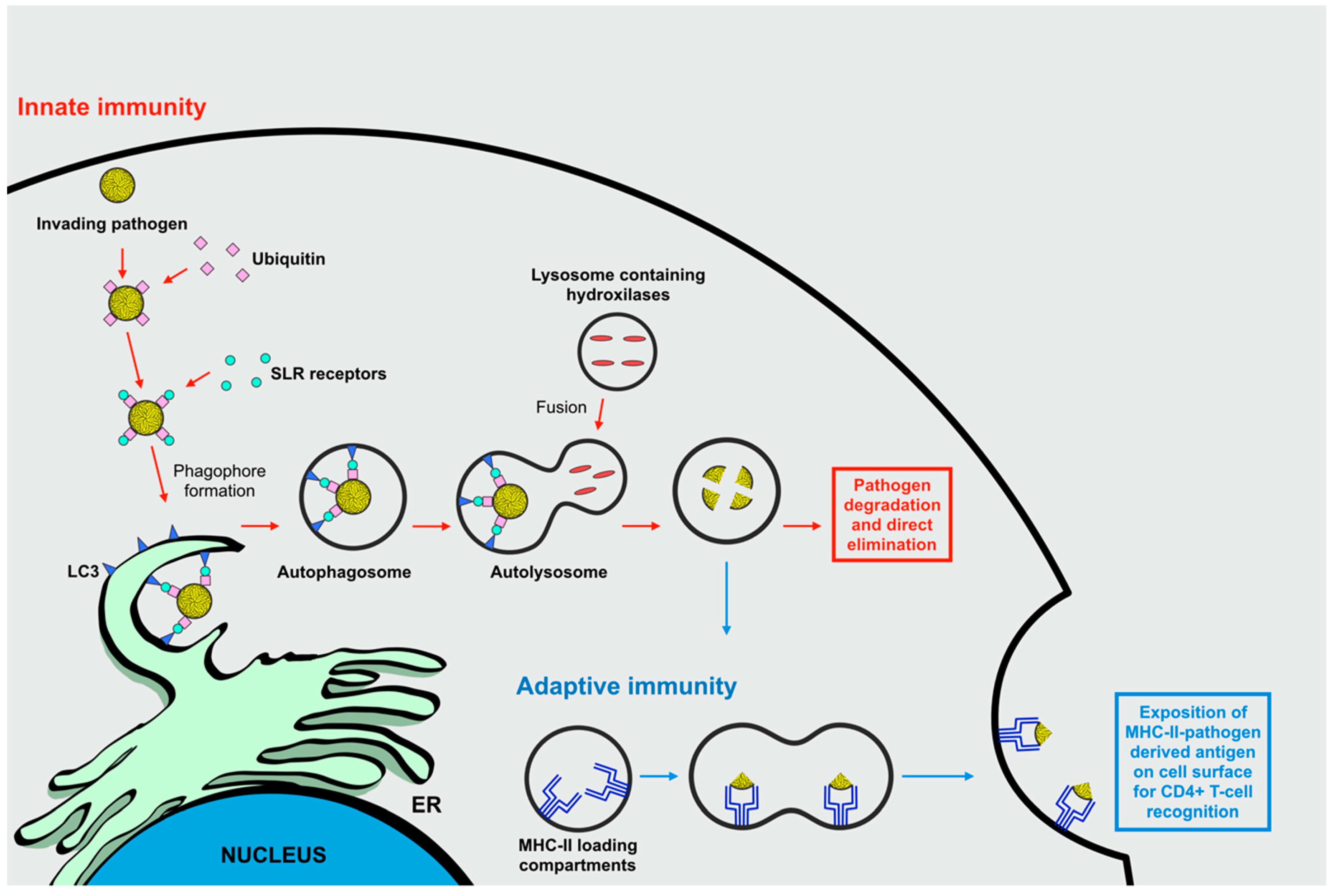
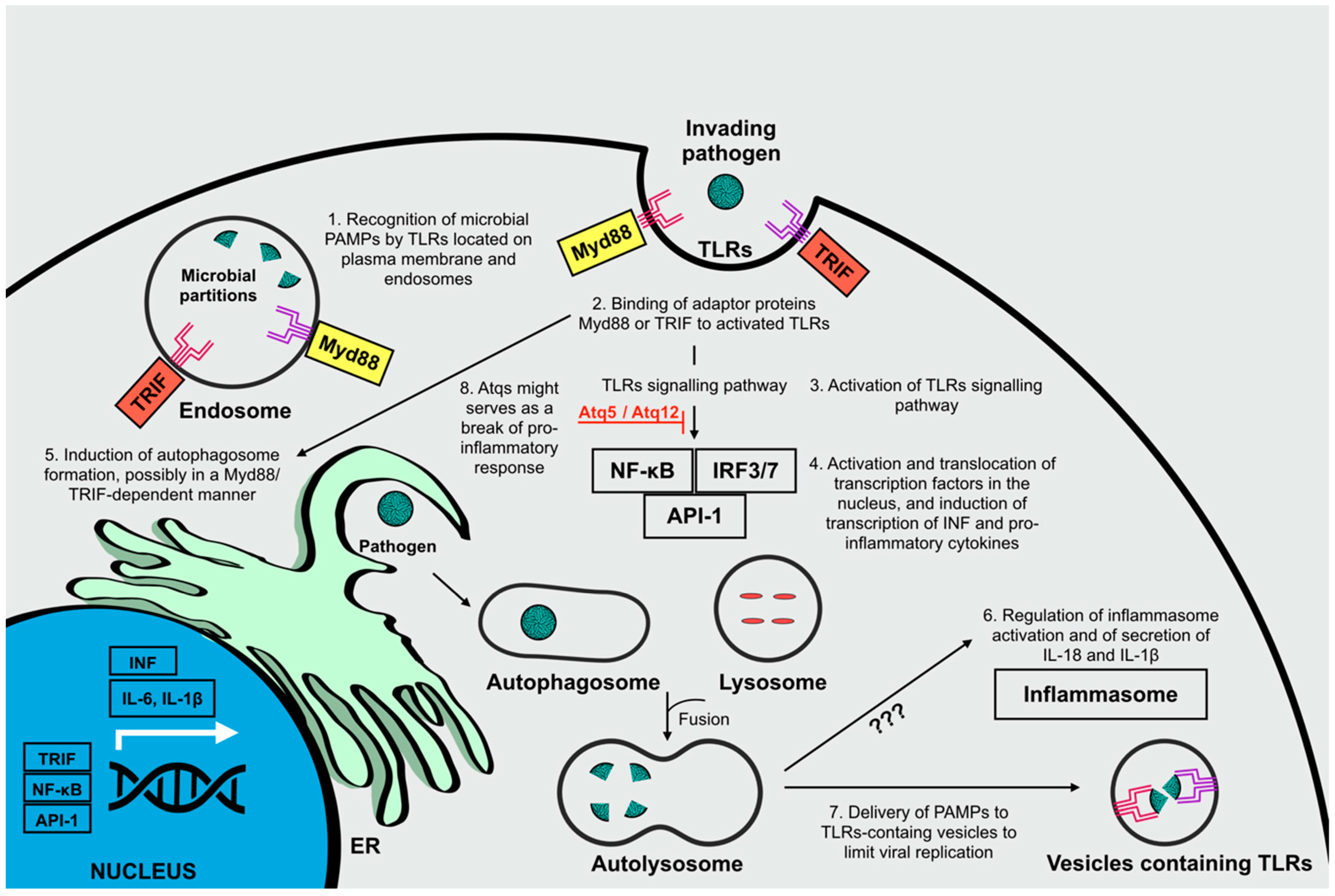
2.1. Autophagy and Its Role in the Immune Responses Against Invading Microorganisms
2.2. Regulation of Autophagic Flux
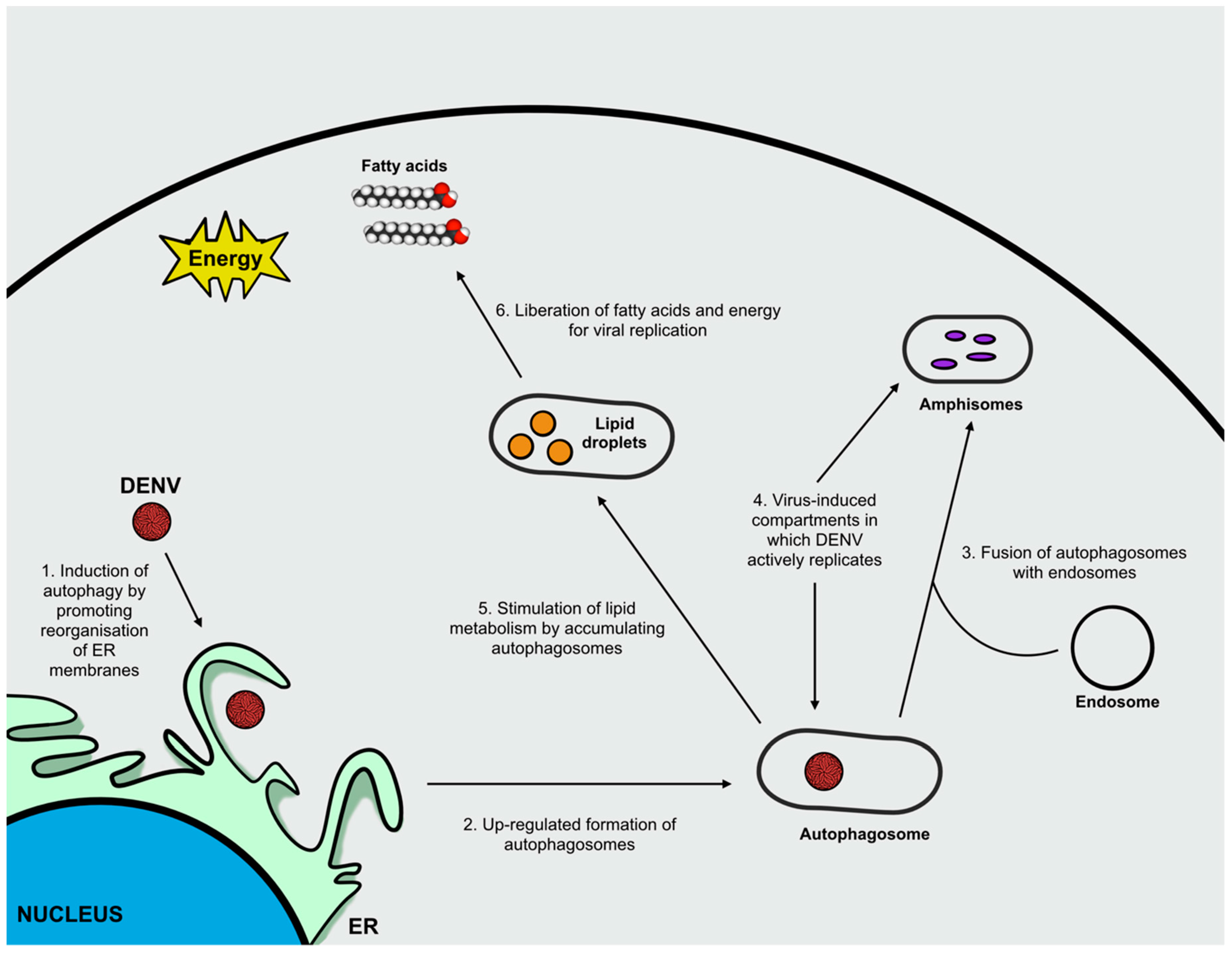
3. Flavivirus Infection and Autophagy
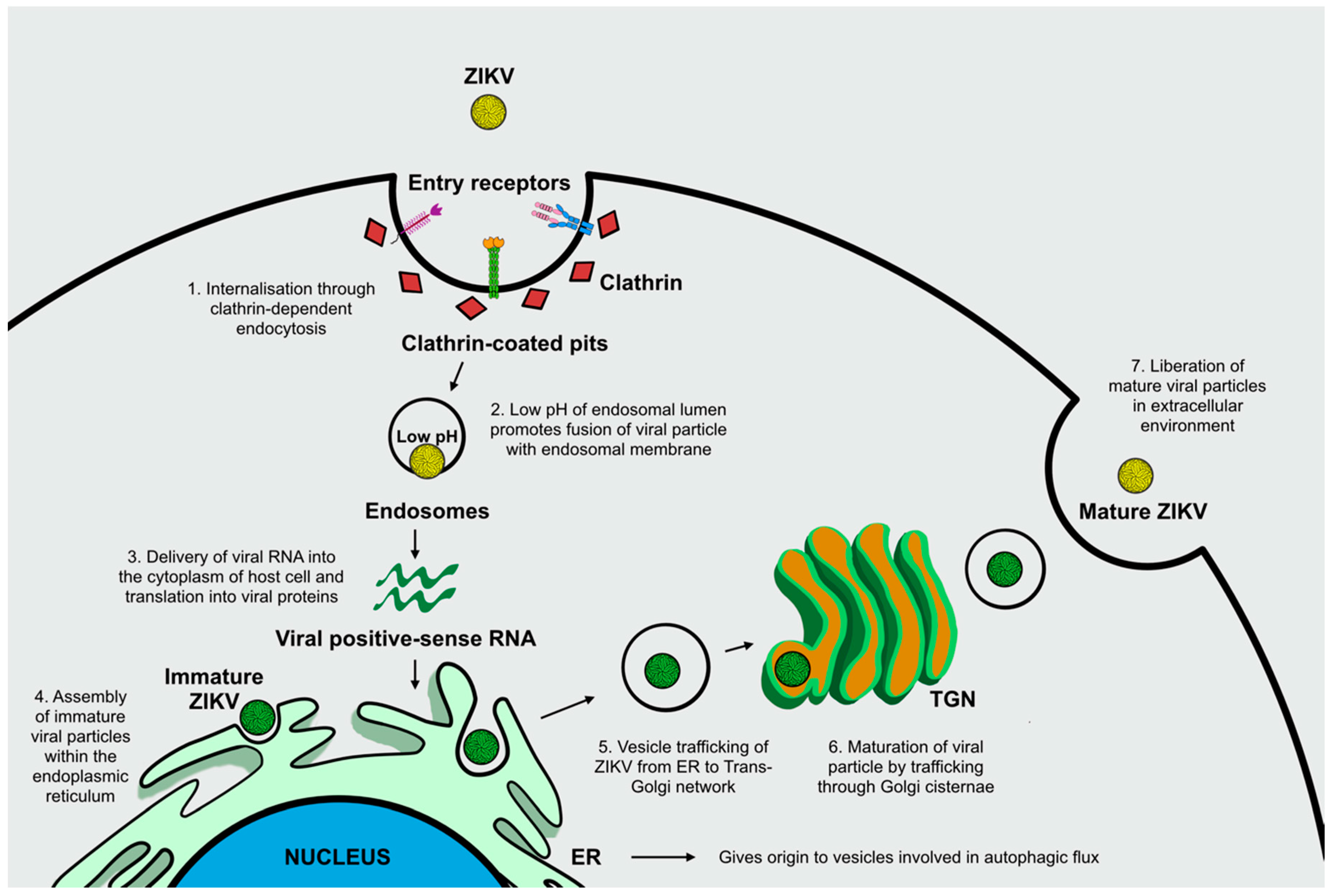
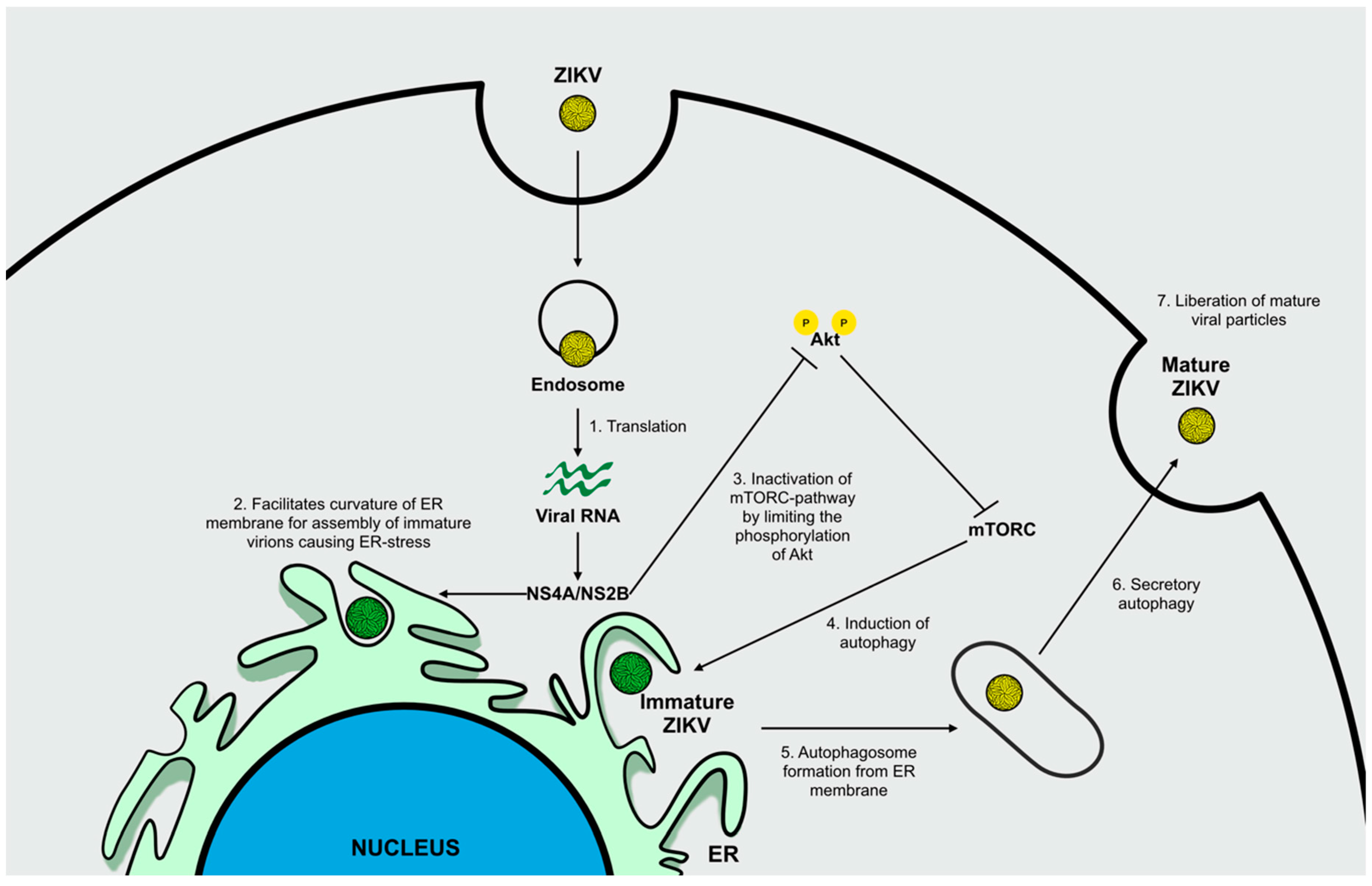
ZIKV Infection and Autophagy
4. Therapy Development and Approaches to Counteract ZIKV Infection
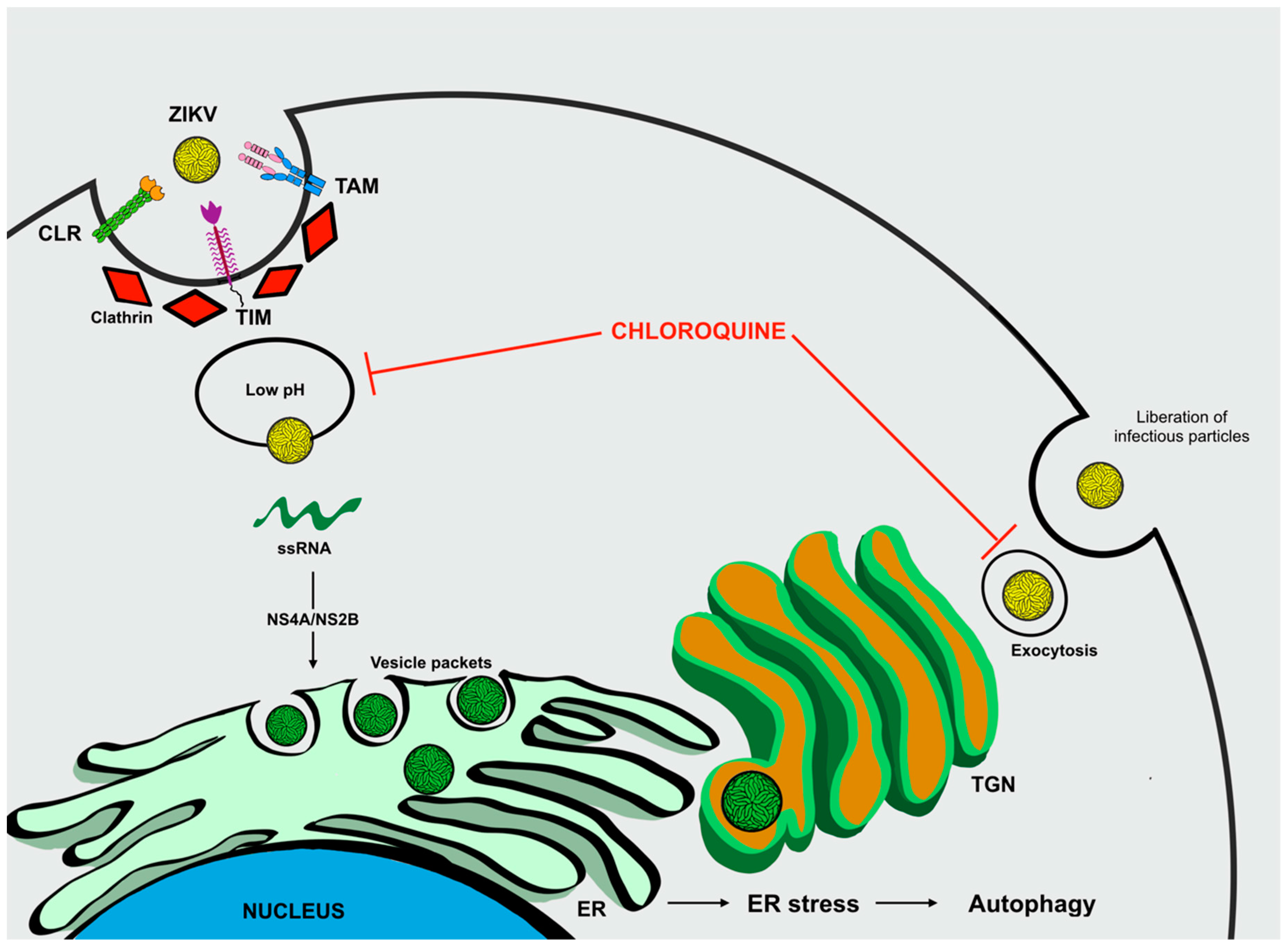
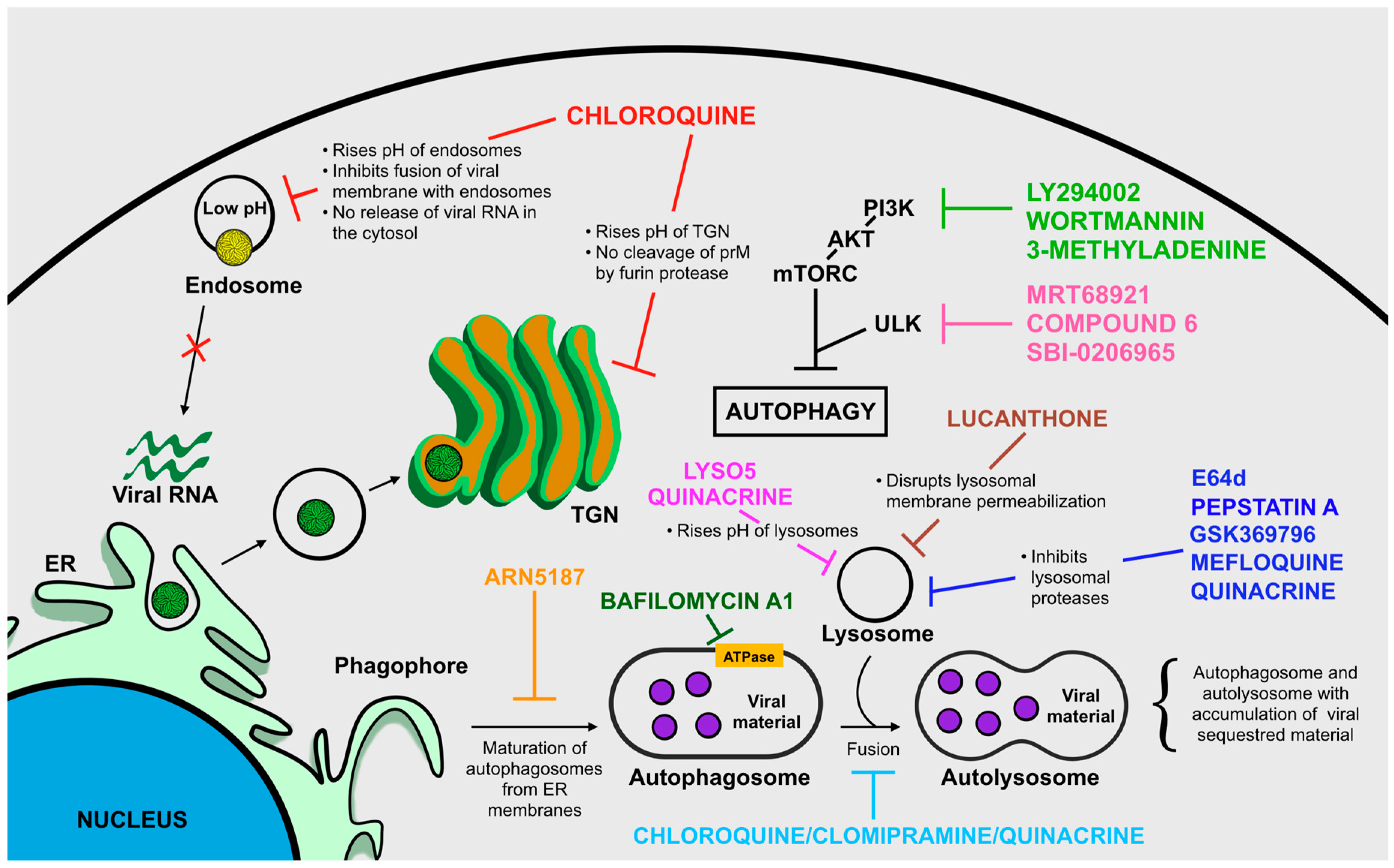
Targeting Autophagy to Limit ZIKV Infection: A Perspective Approach
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Maharajan, M.K.; Ranjan, A.; Chu, J.F.; Foo, W.L.; Chai, Z.X.; Lau, E.Y.; Ye, H.M.; Theam, X.J.; Lok, Y.L. Zika Virus Infection: Current Concerns ad Perspectives. Clin. Rev. Allergy Immunol. 2016, 51, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.D.; Garza, J.A.C.; Cuellar-Barboza, A. Going Viral 2019: Zika, Chikungunya, and Dengue. Dermatol. Clin. 2019, 37, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Cao-Lormeau, V.M. Is the Zika threat over? Clin. Microbiol. Infect. 2018, 24, 566–567. [Google Scholar] [CrossRef] [PubMed]
- Secretaria de Vigilância em Saúde, Ministério da Saúde. Monitoramento integrado de alterações no crescimento e desenvolvimento relacionadas à infecção pelo vírus Zika e outras etiologias infecciosas, até a Semana Epidemiológica 40 de 2018. Boletim Epidemiológico 2018, 49, 1–8. [Google Scholar]
- Lin, H.H.; Yip, B.S.; Huang, L.M.; Wu, S.C. Zika virus structural biology and progress in vaccine development. Biotechnol. Adv. 2018, 36, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Routhu, N.K.; Byrareddy, S.N. Virus Interaction of Zika Virus in Modulating Disease Pathogenesis. J. Neuroimmune Pharmacol. 2017, 12, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Esteves, E.; Rosa, N.; Correia, M.J.; Arrais, J.P.; Barros, M. New Targets for Zika Virus Determined by Human-Viral Interactomic: A Bioinformatics Approach. Biomed. Res. Int. 2017, 2017, 1734151. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, L.A.; Travassos, L.H. Autophagy and viral diseases transmitted by Aedes aegypti and Aedes albopictus. Microbes Infect. 2016, 18, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Bos, S.; Li, G.; Wang, S.; Gadea, G.; Desprès, P.; Zhao, R.Y. Probing Molecular Insightinto Zika Virus Host Interactions. Viruses 2018, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y. The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions. Int. J. Mol. Sci. 2018, 19, 3940. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Xavier, P.; Fransisco, R.; Palatini, F.; Pérez, R.; Ambrosio, S. LC3-I conversion to LC3-II does not necessarily result in complete autophagy. Int. J. Mol. Med. 2008, 22, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiramel, A.I.; Brady, N.R.; Bartenschlager, R. Divergent Roles of Autophagy in Virus Infection. Cells 2013, 2, 83–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation, and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Richetta, C.; Faure, M. Autophagy in antiviral innate immunity. Cell Microbiol. 2013, 15, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Gannage, M.; Münz, C. MHC presentation via autophagy and how viruses escape from it. Semin. Immunopathol. 2010, 32, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, D.S.; Gaviglio, E.A.; Peralta Ramos, J.M.; Bussi, C.; Rodriguez-Galan, M.C.; Iribarren, P. Autophagy in inflammation, infection, neurodegeneration and cancer. Int. Immunopharmacol. 2014, 18, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoji-Kawata, S.; Levine, B. Autophagy, antiviral immunity, and viral countermeasures. Biochim. Biophys. Acta 2009, 1793, 1478–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.M.; Iwasaki, A. Toll-Like Receptors Regulation of Viral Infection and Disease. Adv. Drug Deliv. Rev. 2008, 60, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Nabar, N.R.; Chi, C.S.; Kehrl, J.H. Chapter 6: Signaling by Toll-Like Receptors Induces Autophagy Through Modification in Beclin 1: Molecular Mechanism. In Immunotoxicology, Immunopathology and Immunotherapy, 1st ed.; Hyat, M.A., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 1, pp. 75–84. [Google Scholar]
- Ahmad, L.; Mostowy, S.; Sancho-Shimizu, V. Autophagy-Virus Interplay:From Cell Biology to Human Disease. Front. Cell Dev. Biol. 2018, 6, 155. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5–Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, V.; Dostert, C.; Muruve, D.A.; Tschopp, J. The inflammasome: A danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 2007, 19, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Lamkanfi, M.; Nunez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007, 27, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 2004, 36, 2445–2462. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb Binds and Regulates the mTOR Kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaya, C.; Marcelo Fader, C.; Colombo, M.I. Autophagy and proteins involved in vesicular trafficking. FEBS Lett. 2015, 589, 3343–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bento, C.F.; Puri, C.; Moreau, K.; Rubinsztein, D.C. The role of membrane-trafficking small GTPases in the regulation of autophagy. J. Cell Sci. 2013, 126, 1059–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updat. 2008, 11, 32–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blázquez, A.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.; Martín-Acebes, M.A. Stress responses in flavivirus-infected cells: Activation of unfolded protein response and autophagy. Front. Microbiol. 2014, 5, 266. [Google Scholar] [CrossRef] [PubMed]
- Ponpuak, M.; Mandell, M.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretiv, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.W.; Li, Z.L.; Yuan, S. The Role of Secretory Autophagy in Zika Virus Transfer through the Placental Barrier. Front. Cell Infect. Microbiol. 2017, 6, 206. [Google Scholar] [CrossRef] [PubMed]
- Li, J.K.; Liang, J.J.; Liao, C.L.; Lin, Y.L. Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect. 2012, 14, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, U.; Ye, J.; Ruan, X.; Wan, S.; Zhu, B.; Cao, S. Usutu Virus: An Emerging Flavivirus in Europe. Viruses 2015, 7, 219–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.Y.; Hsu, Y.W.; Liao, C.L.; Lin, Y.L. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to facilitate replication and immune evasion. J. Virol. 2006, 80, 11868–11880. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, A.B.; Escribano-Romero, E.; Merino-Ramos, T.; Saiz, J.C.; Martín-Acebes, M.A. Infection with Usutu virus induces an autophagic response in mammalian cells. PLoS Negl. Trop. Dis. 2013, 7, e2509. [Google Scholar] [CrossRef] [PubMed]
- Martín-Acebes, M.A.; Blázquez, A.B.; Saiz, J.C. Reconciling West Nile virus with the autophagic pathway. Autophagy 2015, 11, 861–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blázquez, A.B.; Martín-Acebes, M.A.; Saiz, J.C. Amino acid substitutions in the non-structural proteins 4A or 4B modulate the induction of autophagy in West Nile virus infected cells independently of the activation of the unfolded protein response. Front. Microbiol. 2015, 5, 797. [Google Scholar] [CrossRef] [PubMed]
- Beatman, E.; Oyer, R.; Shives, K.D.; Headman, K.; Brault, A.C.; Tyler, K.L.; Beckham, J.D. West Nile virus growth is independent of autophagy activation. Virology 2012, 433, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandergaast, R.; Fredericksen, B.L. West Nile virus (WNV) replication is independent of autophagy in mammalian cells. PLoS ONE 2012, 7, e45800. [Google Scholar] [CrossRef] [PubMed]
- Randall, G. Lipid Droplet Metabolism during Dengue Virus Infection. Trends Microbiol. 2018, 26, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randall, G. Dengue Virus and Autophagy. Viruses 2011, 3, 1332–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiramel, A.I.; Best, S.M. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. 2018, 254, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Liu, B.; Yves, T.D.; He, Y.; Wang, S.; Tang, H.; Ren, H.; Zhao, P.; Qi, Z.; Qin, Z. Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells. Viruses 2018, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.; Lee, S.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abernathy, E.; Mateo, R.; Majzoub, K.; van Buuren, N.; Bird, S.W.; Carette, J.E.; Kirkegaard, K. Differential and convergent utilization of autophagy components by positive-strand RNA viruses. PLoS Biol. 2019, 17, e2006926. [Google Scholar] [CrossRef] [PubMed]
- Smit, J.M.; Moesker, B.; Rodenhuis-Zybert, I.; Wilschut, J. Flavivirus Cell Entry and Membrane Fusion. Viruses 2011, 3, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozach, P.Y.; Burleigh, L.; Staropoli, I.; Navarro-Sanchez, E.; Harriague, J.; Virelizier, J.L.; Rey, F.A.; Desprès, P.; Arenzana-Seisdedos, F.; Amara, A. Dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN)-mediated enhancement of dengue virus infection is independent of DC-SIGN internalization signals. J. Biol. Chem. 2005, 280, 23698–23708. [Google Scholar] [CrossRef] [PubMed]
- Tassaneetrithep, B.; Burgess, T.H.; Granelli-Piperno, A.; Trumpfheller, C.; Finke, J.; Sun, W.; Eller, M.A.; Pattanapanyasat, K.; Sarasombath, S.; Birx, D.L.; et al. DC-SIGN (CD209) Mediates Dengue Virus Infection of Human Dendritic Cells. J. Exp. Med. 2003, 197, 823–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacker, K.; White, L.; de Silva, A.M. N-linked glycans on dengue viruses grown in mammalian and insect cells. J. Gen. Virol. 2009, 90, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Sanchez, E.; Altmeyer, R.; Amara, A.; Schwartz, O.; Fieschi, F.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Desprès, P. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 2003, 4, 723–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Hu, K.; Luo, S.; Zhang, M.; Deng, X.; Li, C.; Jin, W.; Hu, B.; He, S.; Li, M.; et al. DC-SIGN as an attachment factor mediates Japanese encephalitis virus infection of human dendritic cells via interaction with a single high-mannose residue of viral E glycoprotein. Virology 2016, 488, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.L.; deWet, B.J.M.; Martinez-Pomares, L.; Radcliffe, C.M.; Dwek, R.A.; Rudd, P.M.; Gordon, S. The Mannose Receptor Mediates Dengue Virus Infection of Macrophages. PLoS Pathog. 2008, 4, e17. [Google Scholar] [CrossRef]
- Chen, S.T.; Lin, Y.L.; Huang, M.T.; Wu, M.F.; Cheng, S.C.; Lei, H.Y.; Lee, C.K.; Chiou, T.W.; Wong, C.H.; Hsieh, S.L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature 2008, 453, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelensky, A.N.; Gready, J.E. The C-type lectin-like domain superfamily. FEBS J. 2005, 272, 6179–6217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambi, A.; Koopman, M.; Figdor, C.G. How C-type lectins detect pathogens. Cell Microbiol. 2005, 7, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Li, B.; Linhardt, R.J. Pathogenesis and Inhibition of Flaviviruses from a Carbohydrate Perspective. Pharmaceuticals 2017, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Delalio, L.J.; Isakson, B.E.; Wang, T.T. AXL-Mediated Productive Infection of Human Endothelial Cells by Zika Virus. Circ. Res. 2016, 119, 1183–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retallack, H.; Di, E.; Arias, C.; Knopp, K.A.; Laurie, M.T. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Tabata, T.; Petitt, M.; Puerta-Guardo, H.; Fang-Hoover, J.; Harris, E.; Tabata, T.; Petitt, M.; Puerta-Guardo, H.; Michlmayr, D.; Wang, C.; et al. Zika Virus Targets Different Primary Human Placental Cells, Suggesting Two Routes for Vertical Transmission. Cell Host Microbe 2016, 20, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.F.; Salick, M.R.; Wiskow, O.; Ho, D.J.; Worringer, K.A.; Ihry, R.J.; Kommineni, S.; Bilican, B.; Klim, J.R.; Hill, E.J.; et al. Genetic Ablation of AXL Does Not Protect Human Neural Progenitor Cells and Cerebral Organoids from Zika Virus Infection. Cell Stem Cell 2016, 19, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Perera-Lecoin, M.; Meertens, L.; Carnec, X.; Amara, A. Flavivirus Entry Receptors: An Update. Viruses 2013, 6, 69–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM Families of Phosphatidylserine Receptors Mediate Dengue Virus Entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rey, A.; Stiasny, K.; Heinz, F.X. Flavivirus structural heterogeneity: Implications for cell entry. Curr. Opin. Virol. 2017, 24, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafizi, S.; Dahlbäck, B. Gas6 and protein S Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. FEBS J. 2006, 273, 5231–5244. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.M.; Camenisch, T.D.; Lemke, G.; Earp, S.; Matsushima, G.K.; Seitz, H.M.; Camenisch, T.D.; Lemke, G.; Earp, H.S.; Matsushima, G.K. Macrophages and Dendritic Cells Use Different Axl/Mertk/Tyro3 Receptors in Clearance of Apoptotic Cells. J. Immunol. 2007, 78, 5635–5642. [Google Scholar] [CrossRef]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis Targeting vector. Nature 1999, 398, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Prasad, D.; Rothlin, C.V.; Burrola, P.; Burstyn-Cohen, T.; Lu, Q.; Garcia de Frutos, P.; Lemke, G. TAM receptor function in the retinal pigment epithelium. Mol. Cell Neurosci. 2006, 33, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Prieto, A.L.; Weber, J.L.; Lai, C. Expression of the receptor protein-tyrosine kinases Tyro-3, Axl, and mer in the developing rat central nervous system. J. Comp. Neurol. 2000, 425, 295–314. [Google Scholar] [CrossRef]
- Freeman, G.J.; Casanovas, J.M.; Umetsu, D.T.; DeKryuff, R.H. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Su, E.W.; Lin, J.Y.; Kane, L.P. TIM-1 and TIM-3 Proteins in Immune Regulation. Cytokine 2008, 44, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Heinz, F.X.; Stiasny, K. Flaviviruses and their antigenic structure. J. Clin. Virol. 2012, 55, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kaufmann, B.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G. Structure of Immature West Nile Virus. J. Virol. 2007, 81, 6141–6145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Huang, X.Y.; Liu, Z.Y.; Zhang, F.; Zhu, X.L.; Yu, J.Y.; Ji, X.; Xu, Y.P.; Li, G.; Li, C.; et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017, 358, 933–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavi, S.A.; Malerød, L.; Berg, T.; Kjeken, R. Clathrin-dependent endocytosis. Biochem. J. 2004, 377, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus Entry by Endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.M.; Zhang, W.; Holdaway, H.A.; Li, L.; Kostyuchenko, V.A.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G.; Chen, J. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 2008, 319, 1834–1837. [Google Scholar] [CrossRef] [PubMed]
- Munjal, A.; Khandia, R.; Dhama, K.; Sachan, S.; Karthik, K.; Ruchi, T.; Malik, Y.S.; Kumar, D.; Singh, R.K.; Igbal, H.M.N.; et al. Advances in Developing Therapies to Combat Zika Virus: Current Knowledge and Future Perspectives. Front. Microbiol. 2017, 8, 1469. [Google Scholar] [CrossRef] [PubMed]
- Ojha, C.R.; Rodriguez, M.; Lapierre, J.; Karuppan, M.K.M.; Branscome, H.; Kashanchi, F.; El-Hage, N. Complementary Mechanisms Potentially Involved in the Pathology of Zika Virus. Front. Immunol. 2018, 9, 2340. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.G.; Koivusalo, M.; Ma, X.; Wohland, T.; Grinstein, S.; Gruenberg, J.E. Phosphatidylserine dynamics in cellular membranes. Mol. Biol. Cell 2012, 23, 2198–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Mlakar, J.; Korva, M.; Tul, N.; Popović, M.; Poljak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodušek, V.; et al. Zika virus associated with microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Carteaux, G.; Maquart, M.; Bedet, A.; Contou, D.; Brugières, P.; Fourati, S.; Cleret de Langavant, L.; de Broucker, T.; Brun-Buisson, C.; Leparc-Goffart, I.; et al. Zika Virus Associated with Meningoencephalitis. N. Engl. J. Med. 2016, 374, 1595–1596. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Era, N. Zika virus disease: Global concerns and making way through it. Communit. Acquir. Infect. 2016, 3, 31–35. [Google Scholar] [CrossRef]
- Wahid, B.; Ali, A.; Rafique, S.; Idrees, M. Current status of therapeutic and vaccine approaches against Zika virus. Eur. J. Intern. Med. 2017, 44, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Shiryaev, S.A.; Mesci, P.; Pinto, A.; Fernandes, I.; Sheets, N.; Shresta, S.; Farhy, C.; Huang, C.T.; Strongin, A.Y.; Muotri, A.R.; et al. Repurposing of the anti-malaria drug chloroquine for Zika Virus treatment and prophylaxis. Sci. Rep. 2017, 7, 15771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zhu, X.; Ji, X.; Quanquin, N.; Deng, Y.Q.; Tian, M.; Aliyari, R.; Zuo, X.; Yuan, L.; Afridi, S.K.; et al. Chloroquine, a FDA-approved Drug, Prevents Zika Virus Infection and its Associated Congenital Microcephaly in Mice. EBioMedicine 2017, 24, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Boleaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases? Lancet Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef]
- Legssyer, R.; Josse, C.; Piette, J.; Ward, R.J.; Crichton, R.R. Changes in function of iron-loaded alveolar macrophages after in vivo administration of desferrioxamine and/or chloroquine. J. Inorg. Biochem. 2003, 94, 36–42. [Google Scholar] [CrossRef]
- Byrd, T.F.; Horwitz, M.A. Chloroquine inhibits the intracellular multiplication of Legionella pneumophila by limiting the availability of iron. A potential new mechanism for the therapeutic effect of chloroquine against intracellular pathogens. J. Clin. Investig. 1991, 88, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.F.; Santo, M.P.; Rebello, M.A.; Rebello, M.C. Weak bases affect late stages of Mayaro virus replication cycle in vertebrate cells. J. Med. Microbiol. 2000, 49, 313–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.P.; Nara, P.L.; Kung, H.F.; Oroszlan, S. Inhibition of human immunodeficiency virus infectivity by chloroquine. AIDS Res. Hum. Retroviruses 1990, 6, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Solitro, A.R.; MacKeigan, J.P. Leaving the lysosome behind: Novel developments in autophagy inhibition. Future Med. Chem. 2016, 8, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Adcock, R.S.; Chu, Y.K.; Golden, J.E.; Chung, D.H. Evaluation of anti-Zika virus activities of broad-spectrum antivirals and NIH clinical collection compounds using a cell-based, high-throughput screen assay. Antivir. Res. 2017, 138, 47–56. [Google Scholar] [CrossRef] [PubMed]
- McAfee, Q.; Zhang, Z.; Samanta, A.; Levi, S.M.; Ma, X.H.; Piao, S.; Lynch, J.P.; Uehara, T.; Sepulveda, A.R.; Davis, L.E.; et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Mei, C.; Ercolani, L.; Parodi, C.; Veronesi, M.; Lo Vecchio, C.; Bottegoni, G.; Torrente, E.; Scarpelli, R.; Marotta, R.; Ruffili, R.; et al. Dual inhibition of REV-ERBb and autophagy as a novel pharmaco- logical approach to induce cytotoxicity in cancer cells. Oncogene 2015, 34, 2597–2608. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, A.; Teramoto, T.; Kulkarni, A.A.; Bhattacharjee, A.K.; Padmanabhan, R. Antiviral activities of selected antimalarials against dengue virus type 2 and Zika virus. Antiv. Res. 2017, 137, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small molecule inhibition of the autophagy kinase ULK1 and identi- fication of ULK1 substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Petherick, K.J.; Conway, O.J.L.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycine (mTOR)- dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Bockus, A.; Wozniak, A.L.; Jones, K.; Weinman, S.; Yin, X.M.; Ding, W.X. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 2011, 7, 188–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Munarriz, E.R.; Bartesaghi, S.; Milanese, M.; Dinsdale, D.; Guerra-Martin, M.A.; Bampton, E.T.; Glynn, P.; Bonanno, G.; Knight, R.A.; et al. Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. J. Cell Sci. 2009, 122, 3330–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carew, J.S.; Espitia, C.M.; Esquivel, J.A., 2nd; Mahalingam, D.; Kelly, K.R.; Reddy, G.; Giles, F.J.; Nawrocki, S.T. Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J. Biol. Chem. 2011, 286, 6602–6613. [Google Scholar] [CrossRef] [PubMed]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Gordon, P.B. 3-methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Vinod, V.; Padmakrishnan, C.J.; Vijayan, B.; Gopala, S. How can I halt thee? The puzzles involved in autophagic inhibition. Pharmacol. Res. 2014, 82, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Blommaart, E.F.; Krause, U.; Schellens, J.P.; Vreeling-Sindelárová, H.; Meijer, A.J. The phosphatidylinositol 3-kinase inhbitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gratton, R.; Agrelli, A.; Tricarico, P.M.; Brandão, L.; Crovella, S. Autophagy in Zika Virus Infection: A Possible Therapeutic Target to Counteract Viral Replication. Int. J. Mol. Sci. 2019, 20, 1048. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051048
Gratton R, Agrelli A, Tricarico PM, Brandão L, Crovella S. Autophagy in Zika Virus Infection: A Possible Therapeutic Target to Counteract Viral Replication. International Journal of Molecular Sciences. 2019; 20(5):1048. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051048
Chicago/Turabian StyleGratton, Rossella, Almerinda Agrelli, Paola Maura Tricarico, Lucas Brandão, and Sergio Crovella. 2019. "Autophagy in Zika Virus Infection: A Possible Therapeutic Target to Counteract Viral Replication" International Journal of Molecular Sciences 20, no. 5: 1048. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051048