LDAPred: A Method Based on Information Flow Propagation and a Convolutional Neural Network for the Prediction of Disease-Associated lncRNAs

Abstract
:1. Introduction
2. Result and Discussion
2.1. Parameter Settings
2.2. Evaluation Metrics
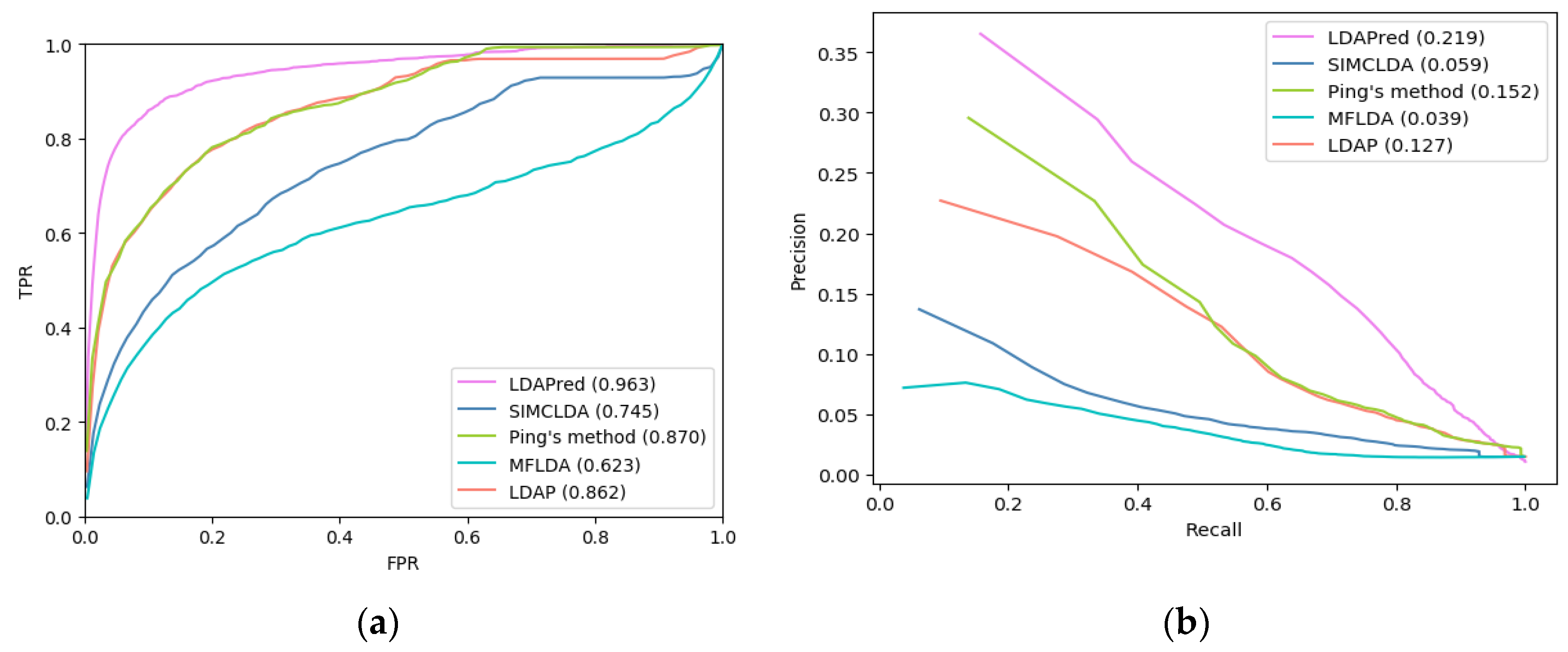
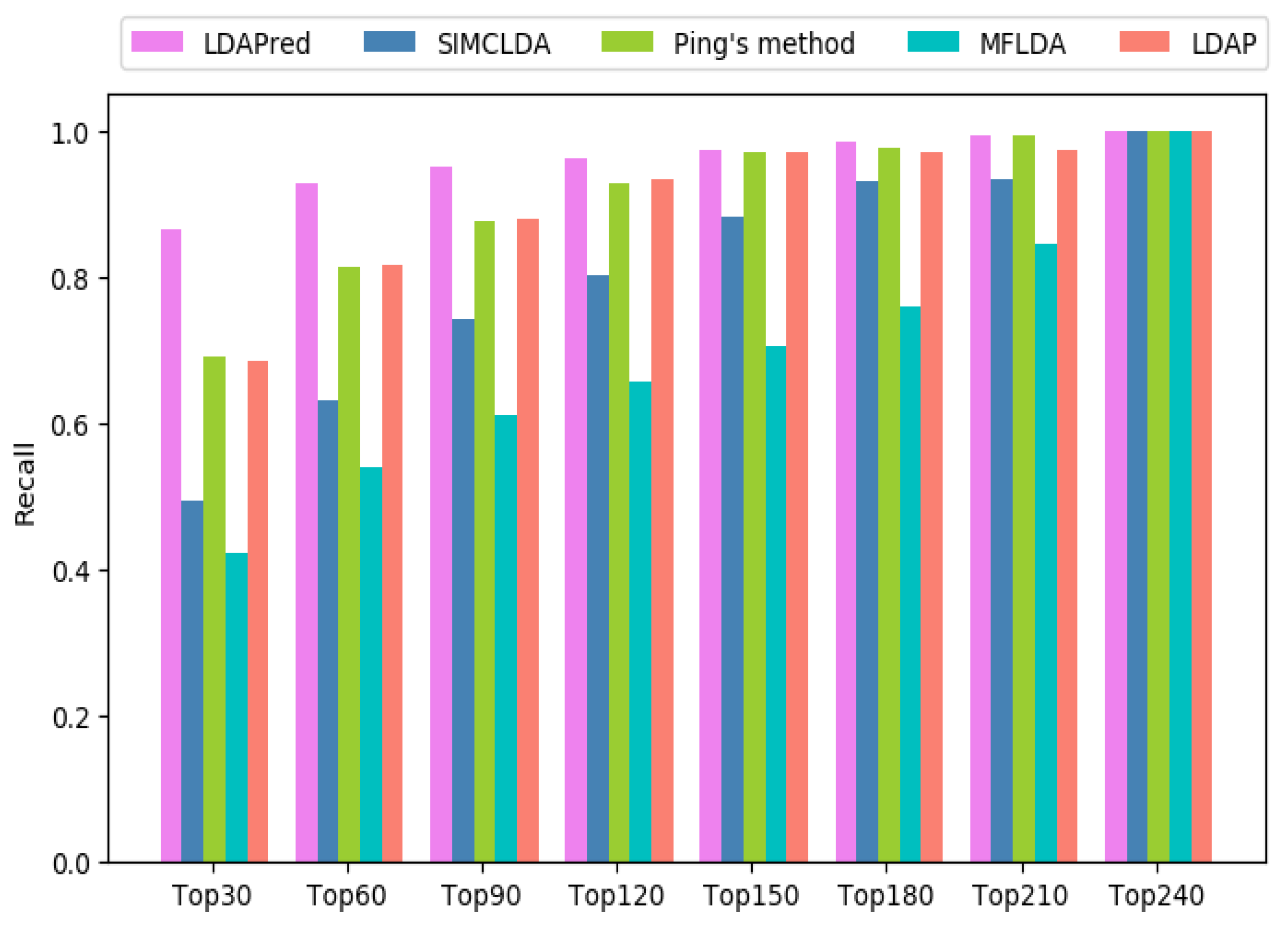
2.3. Comparison with Other Methods
2.4. Case Studies on Breast Cancer, Colon Cancer, and Osteosarcoma
3. Materials and Methods
3.1. Dataset
3.2. Similarity Calculation and Data Representation
3.2.1. Semantic Similarity of Diseases
3.2.2. Similarity of lncRNAs
3.2.3. Similarity of miRNAs
3.2.4. Interaction Matrix
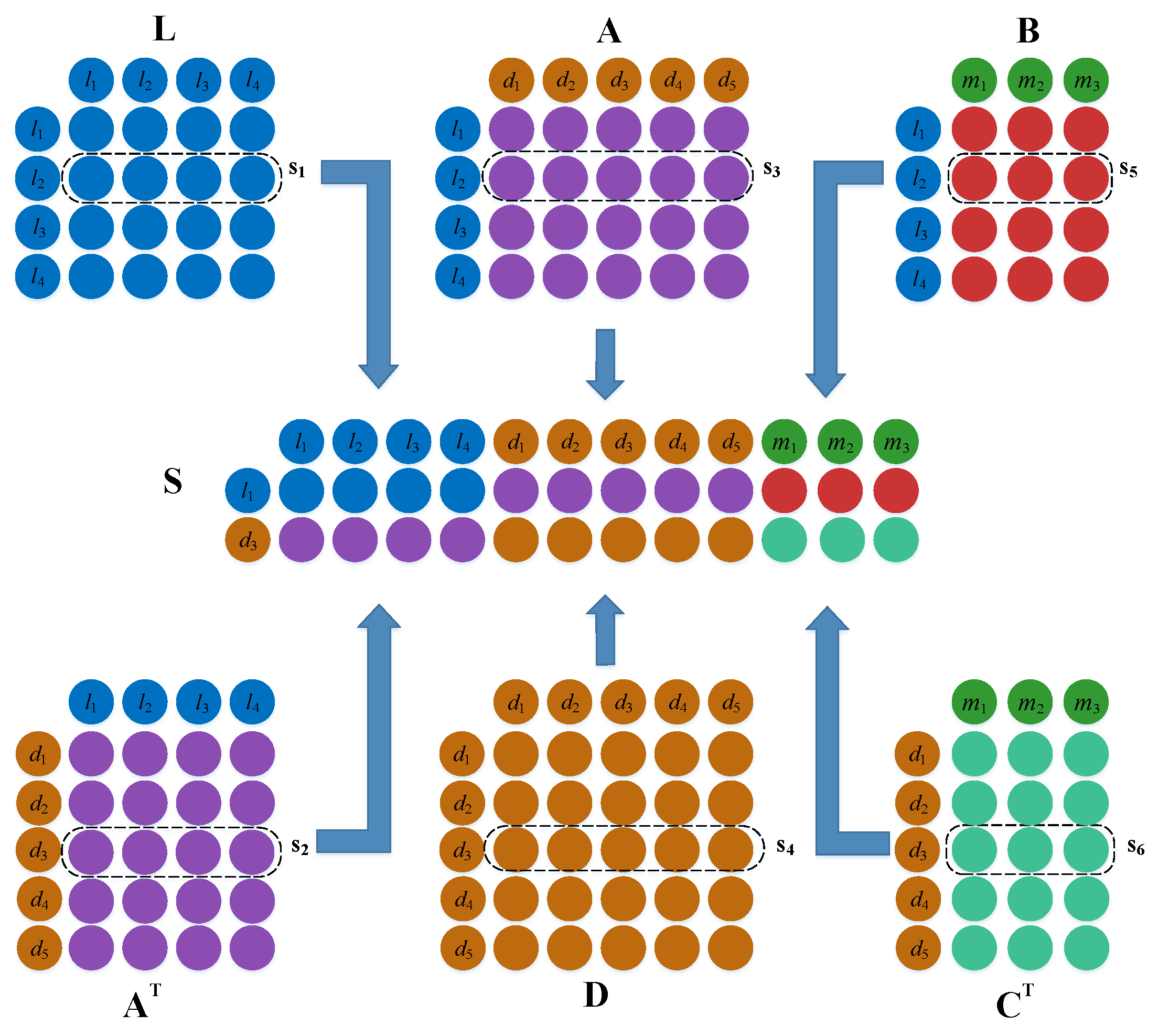
3.3. LncRNA–Disease Association Prediction Model Based on a Dual Convolutional Neural Network
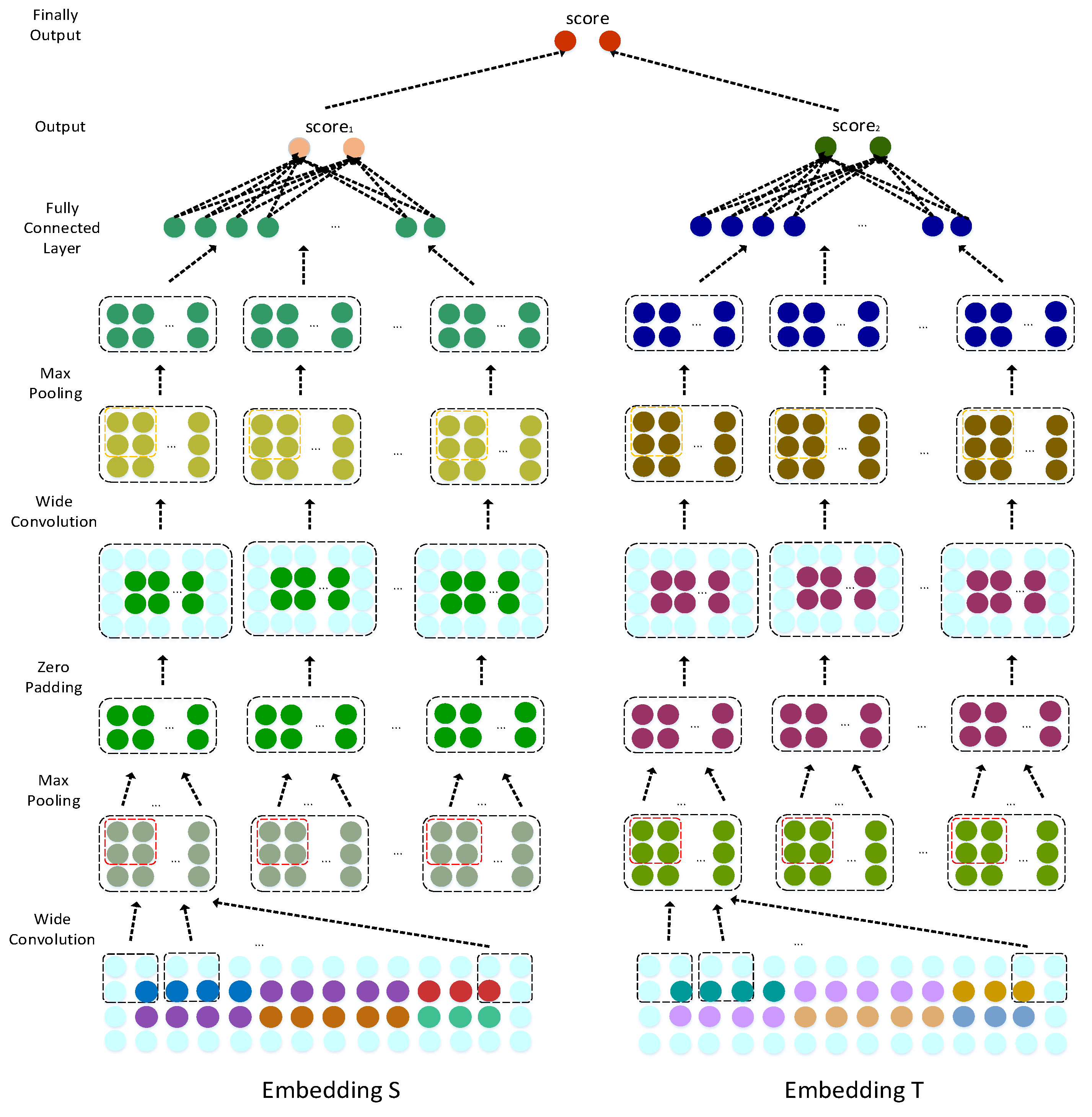
3.3.1. Embedded Layer
Establishment of the Left Feature Matrix
Establishment of the Right Side Topological Information Matrix
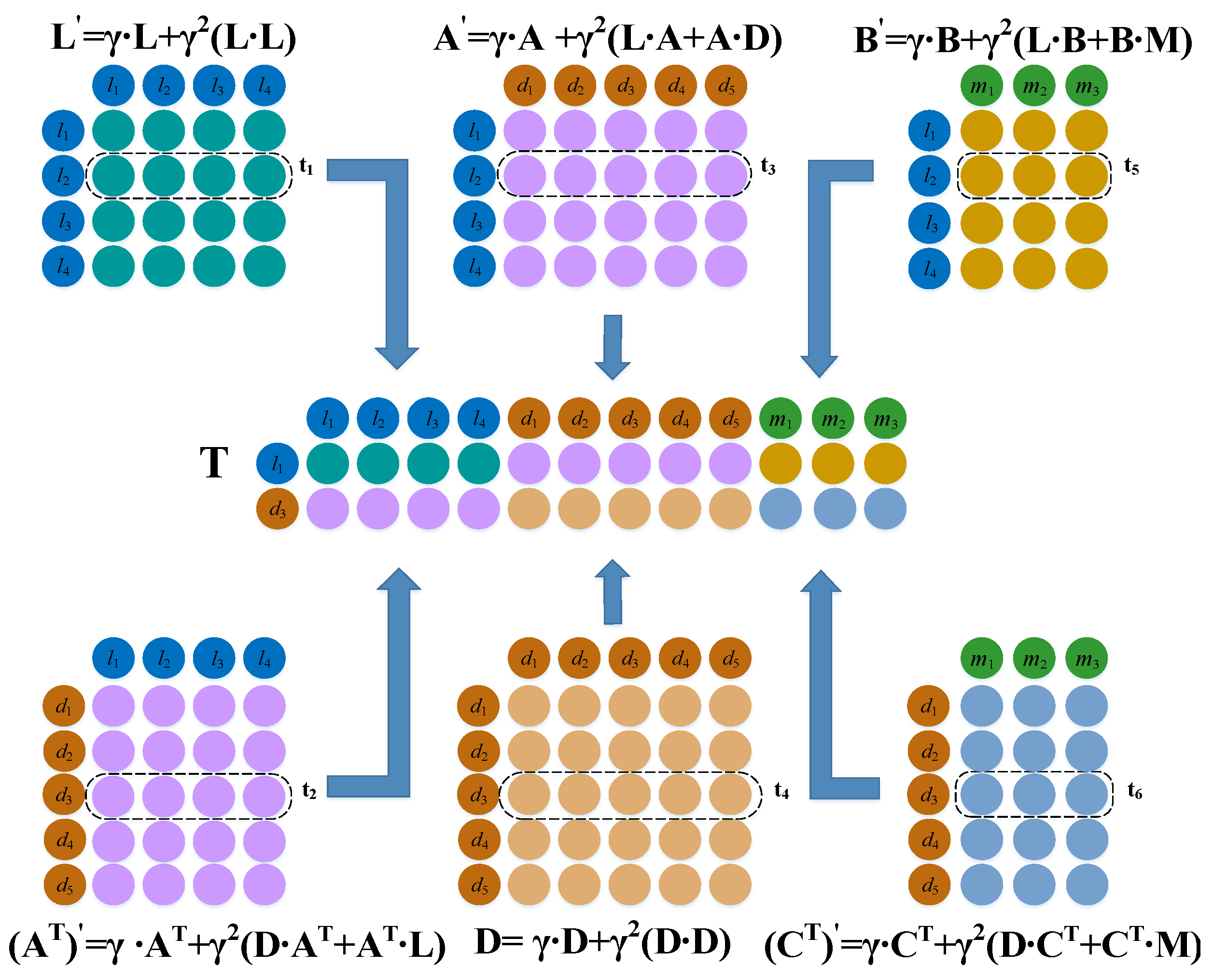
3.3.2. Convolution Module
3.3.3. Dual Combination Strategy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.P.; Weissman, S.M. Global Identification of Human Transcribed Sequences with Genome Tiling Arrays. Science 2004, 306, 2242–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L. RNA Maps Reveal New RNA Classes and a Possible Function for Pervasive Transcription. Science 2007, 316, 1484–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Sandelin, A.; Lenhard, B.; Katayama, S.; Shimokawa, K.; Ponjavic, J.; Semple, C.A.; Taylor, M.S.; Engstrom, P.G.; Frith, M.C. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006, 38, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non–coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wapinski, O.L.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Spizzo, R.; Almeida, M.I.; Colombatti, A.; Calin, G.A. Long non-coding RNAs and cancer: A new frontier of translational research? Oncogene 2012, 31, 4577–4587. [Google Scholar] [CrossRef]
- van Poppel, H.; Haese, A.; Graefen, M.; de la Taille, A.; Irani, J.; de Reijke, T.; Remzi, M.; Marberger, M. The relationship between Prostate CAncer gene 3 (PCA3) and prostate cancer significance. Bju Int. 2012, 109, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, C.; Wang, Y.; Ma, W.; Tu, J.; Wang, J.; Chen, Z.; Kong, W.; Cui, Q. A bioinformatics method for predicting long noncoding RNAs associated with vascular disease. Science China. Life Sci. 2014, 57, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.X.; Chen, X.; Chen, G.; Cui, Q.H.; Yan, G.Y. A computational framework to infer human disease-associated long noncoding RNAs. PLoS One 2014, 9, e84408. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. KATZLDA: KATZ measure for the lncRNA-disease association prediction. Sci. Rep. 2015, 5, 16840. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, G.Y. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 2013, 29, 2617–2624. [Google Scholar] [CrossRef]
- Lan, W.; Li, M.; Zhao, K.; Liu, J.; Wu, F.X.; Pan, Y.; Wang, J. LDAP: A web server for lncRNA-disease association prediction. Bioinformatics 2017, 33, 458–460. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Z.; Chen, Z.; Deng, L. Integrating Multiple Heterogeneous Networks for Novel LncRNA-Disease Association Inference. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 396–406. [Google Scholar] [CrossRef]
- Yao, Q.; Wu, L.; Li, J.; guang Yang, L.; Sun, Y.; Li, Z.; He, S.; Feng, F.; Li, H.; Li, Y.J.S.r. Global prioritizing disease candidate lncRNAs via a multi-level composite network. Sci. Rep. 2017, 7, 39516. [Google Scholar]
- Sun, J.; Shi, H.; Wang, Z.; Zhang, C.; Liu, L.; Wang, L.; He, W.; Hao, D.; Liu, S.; Zhou, M. Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Mol. Biosyst. 2014, 10, 2074–2081. [Google Scholar] [CrossRef]
- Chen, X.; You, Z.H.; Yan, G.Y.; Gong, D.W. IRWRLDA: Improved random walk with restart for lncRNA-disease association prediction. Oncotarget 2016, 7, 57919–57931. [Google Scholar] [CrossRef]
- Gu, C.; Liao, B.; Li, X.; Cai, L.; Li, Z.; Li, K.; Yang, J.J., Sr. Global network random walk for predicting potential human lncRNA-disease associations. Sci. Rep. 2017, 7, 12442. [Google Scholar] [CrossRef] [PubMed]
- Xuan, P.; Cao, Y.; Zhang, T.; Kong, R.; Zhang, Z. Dual Convolutional Neural Networks With Attention Mechanisms Based Method for Predicting Disease-Related lncRNA Genes. Front. Genet. 2019, 10, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, X.; Shuxiang, P.; Tiangang, Z.; Yong, L.; Hao, S. Graph Convolutional Network and Convolutional Neural Network Based Method for Predicting lncRNA-Disease Associations. Cells 2019, 8, 12. [Google Scholar] [CrossRef]
- Ping, X.; Nan, S.; Tiangang, Z.; Yong, L.; Yahong, G. CNNDLP: A method based on convolutional autoencoder and convolutional neural network with adjacent edge attention for predicting lncRNA-disease associations. Int. J. Mol. Sci 2019, 20, 4260. [Google Scholar] [CrossRef]
- Lu, C.; Yang, M.; Luo, F.; Wu, F.X.; Li, M.; Pan, Y.; Li, Y.; Wang, J. Prediction of lncRNA-disease associations based on inductive matrix completion. Bioinformatics 2018, 34, 3357–3364. [Google Scholar] [CrossRef]
- Ping, P.; Wang, L.; Kuang, L.; Ye, S.; Iqbal, M.F.B.; Pei, T. A Novel Method for LncRNA-Disease Association Prediction Based on an lncRNA-Disease Association Network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 688–693. [Google Scholar] [CrossRef]
- Fu, G.; Wang, J.; Domeniconi, C.; Yu, G. Matrix factorization-based data fusion for the prediction of lncRNA-disease associations. Bioinformatics 2018, 34, 1529–1537. [Google Scholar] [CrossRef]
- Ning, S.; Zhang, J.; Wang, P.; Zhi, H.; Wang, J.; Liu, Y.; Gao, Y.; Guo, M.; Yue, M.; Wang, L.; et al. Lnc2Cancer: A manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2016, 44, D980–D985. [Google Scholar] [CrossRef]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef]
- Huang, M.; Hou, J.; Wang, Y.; Xie, M.; Wei, C.; Nie, F.; Wang, Z.; Sun, M. Long Noncoding RNA LINC00673 Is Activated by SP1 and Exerts Oncogenic Properties by Interacting with LSD1 and EZH2 in Gastric Cancer. Mol. Ther. 2017, 25, 1014–1026. [Google Scholar] [CrossRef] [Green Version]
- Gascard, P.; Bilenky, M.; Sigaroudinia, M.; Zhao, J.; Li, L.; Carles, A.; Delaney, A.; Tam, A.; Kamoh, B.; Cho, S.; et al. Epigenetic and transcriptional determinants of the human breast. Nat. Commun. 2015, 6, 6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Suzek, T.O.; Troup, D.B.; Wilhite, S.E.; Ngau, W.C.; Ledoux, P.; Rudnev, D.; Lash, A.E.; Fujibuchi, W.; Edgar, R. NCBI GEO: Mining millions of expression profiles--database and tools. Nucleic Acids Res. 2005, 33, D562–D566. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, Z.; Wang, D.; Qiu, C.; Liu, M.; Chen, X.; Zhang, Q.; Yan, G.; Cui, Q. LncRNADisease: A database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013, 41, D983–D986. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Cohen, K.B.; Hunter, L. GeneRIF QUALITY ASSURANCE AS SUMMARY REVISION. In Proceedings of the pacific symposium on Biocomputing; Grand Wailea, Maui, Hawaii, 3–7 January 2007; pp. 269–280. [Google Scholar]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014, 42, D1070–D1074. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Hu, Y.; Sun, J.; Zhou, M.; Jiang, Q. DincRNA: A comprehensive web-based bioinformatics toolkit for exploring disease associations and ncRNA function. Bioinformatics 2018, 34, 1953–1956. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Name | Percentage of Disease-Related lncRNAs | AUC | ||||
|---|---|---|---|---|---|---|
| LDAPred | SIMCLDA | Ping’s Method | MFLDA | LDAP | ||
| Respiratory system cancer | 1.1% | 0.913 | 0.789 | 0.911 | 0.719 | 0.891 |
| Organ system cancer | 1.6% | 0.958 | 0.820 | 0.950 | 0.729 | 0.884 |
| Intestinal cancer | 2.3% | 0.963 | 0.811 | 0.909 | 0.559 | 0.905 |
| Prostate cancer | 1.0% | 0.951 | 0.873 | 0.826 | 0.553 | 0.711 |
| Lung cancer | 1.1% | 0.833 | 0.790 | 0.911 | 0.676 | 0.883 |
| Breast cancer | 0.1% | 0.970 | 0.742 | 0.871 | 0.517 | 0.830 |
| Reproductive organ cancer | 1.1% | 0.993 | 0.707 | 0.818 | 0.741 | 0.742 |
| Gastrointestinal system cancer | 0.1% | 0.985 | 0.784 | 0.896 | 0.582 | 0.867 |
| Liver cancer | 1.5% | 0.911 | 0.799 | 0.910 | 0.634 | 0.898 |
| Hepatocellular carcinoma | 1.5% | 0.867 | 0.765 | 0.903 | 0.688 | 0.902 |
| Disease Name | AUPR | ||||
|---|---|---|---|---|---|
| LDAPred | SIMCLDA | Ping’s Method | MFLDA | LDAP | |
| Respiratory system cancer | 0.178 | 0.149 | 0.414 | 0.072 | 0.303 |
| Organ system cancer | 0.029 | 0.411 | 0.765 | 0.338 | 0.628 |
| Intestinal cancer | 0.271 | 0.141 | 0.252 | 0.042 | 0.246 |
| Prostate cancer | 0.338 | 0.176 | 0.333 | 0.095 | 0.297 |
| Lung cancer | 0.655 | 0.138 | 0.334 | 0.008 | 0.094 |
| Breast cancer | 0.125 | 0.445 | 0.803 | 0.476 | 0.629 |
| Reproductive organ cancer | 0.490 | 0.047 | 0.403 | 0.031 | 0.396 |
| Gastrointestinal system cancer | 0.500 | 0.130 | 0.271 | 0.104 | 0.238 |
| Liver cancer | 0.672 | 0.201 | 0.526 | 0.086 | 0.498 |
| Hepatocellular carcinoma | 0.198 | 0.096 | 0.239 | 0.082 | 0.303 |
| p-Value and Other Methods | SIMCLDA | Ping’s Method | MFLDA | LDAP |
|---|---|---|---|---|
| p-values of AUCs | 2.4816 × 10−17 | 0.0079 × 10−15 | 1.2144 × 10−15 | 0.0033 × 10−14 |
| p-values of AUPRs | 0.0118 × 10−14 | 0.3000 × 10−13 | 0.0030 × 10−14 | 0.9211 × 10−11 |
| Disease Name | Rank | LncRNA Name | Description | Rank | LncRNA Name | Description |
|---|---|---|---|---|---|---|
| Breast cancer | 1 | AFAP1-AS1 | Lnc2Cancer, lncRNADisease | 9 | CECR7 | Unconfirmed |
| 2 | LINC00675 | Literature | 10 | DBET | lncRNADisease_P | |
| 3 | H19 | Lnc2Cancer, lncRNADisease_P | 11 | CARMN | lncRNADisease_P | |
| 4 | HOTTIP | Lnc2Cancer, lncRNADisease_P | 12 | DISC1FP1 | lncRNADisease_P | |
| 5 | HCG9 | lncRNADisease_P | 13 | VLDLR-AS1 | lncRNADisease_P | |
| 6 | MEG8 | Literature | 14 | PWAR5 | Literature | |
| 7 | LINC00315 | lncRNADisease_P | 15 | LINC00479 | lncRNADisease_P | |
| 8 | GABPB1-AS1 | Unconfirmed | ||||
| Colon cancer | 1 | NPSR1-AS1 | GEO | 9 | LINC00477 | lncRNADisease_P |
| 2 | MEG3 | Lnc2Cancer, lncRNADisease | 10 | PARD6G-AS1 | lncRNADisease_P | |
| 3 | H19 | Lnc2Cancer, lncRNADisease | 11 | OIP5-AS1 | lncRNADisease_P | |
| 4 | CCAT2 | Lnc2Cancer, lncRNADisease | 12 | LINC01184 | lncRNADisease_P | |
| 5 | HOTAIR | Lnc2Cancer, lncRNADisease | 13 | CARMN | lncRNADisease_P | |
| 6 | CCAT1 | Lnc2Cancer, lncRNADisease | 14 | MEG8 | lncRNADisease_P | |
| 7 | MALAT1 | Lnc2Cancer, lncRNADisease | 15 | GABPB1-AS | lncRNADisease_P | |
| 8 | GATA3-AS1 | lncRNADisease_P | ||||
| Osteosarcoma | 1 | HOTAIR | Lnc2Cancer, lncRNADisease | 9 | MEG8 | lncRNADisease_P |
| 2 | LINC00673 | Lnc2Cancer, lncRNADisease | 10 | GNAS-AS1 | lncRNADisease_P | |
| 3 | MIR17HG | lncRNADisease_P | 11 | PTCSC2 | lncRNADisease_P | |
| 4 | HULC | Lnc2Cancer, lncRNADisease_P | 12 | LINC00319 | Unconfirmed | |
| 5 | TUSC7 | Lnc2Cancer, lncRNADisease | 13 | GABPB1-AS1 | Unconfirmed | |
| 6 | HOTTIP | Lnc2Cancer, lncRNADisease | 14 | LINC00473 | Lnc2Cancer, lncRNADisease_P | |
| 7 | MEG3 | Lnc2Cancer, lncRNADisease | 15 | VLDLR-AS1 | lncRNADisease | |
| 8 | BANCR | Lnc2Cancer, lncRNADisease |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xuan, P.; Jia, L.; Zhang, T.; Sheng, N.; Li, X.; Li, J. LDAPred: A Method Based on Information Flow Propagation and a Convolutional Neural Network for the Prediction of Disease-Associated lncRNAs. Int. J. Mol. Sci. 2019, 20, 4458. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184458
Xuan P, Jia L, Zhang T, Sheng N, Li X, Li J. LDAPred: A Method Based on Information Flow Propagation and a Convolutional Neural Network for the Prediction of Disease-Associated lncRNAs. International Journal of Molecular Sciences. 2019; 20(18):4458. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184458
Chicago/Turabian StyleXuan, Ping, Lan Jia, Tiangang Zhang, Nan Sheng, Xiaokun Li, and Jinbao Li. 2019. "LDAPred: A Method Based on Information Flow Propagation and a Convolutional Neural Network for the Prediction of Disease-Associated lncRNAs" International Journal of Molecular Sciences 20, no. 18: 4458. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20184458