Role of Aryl Hydrocarbon Receptor Activation and Autophagy in Psoriasis-Related Inflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
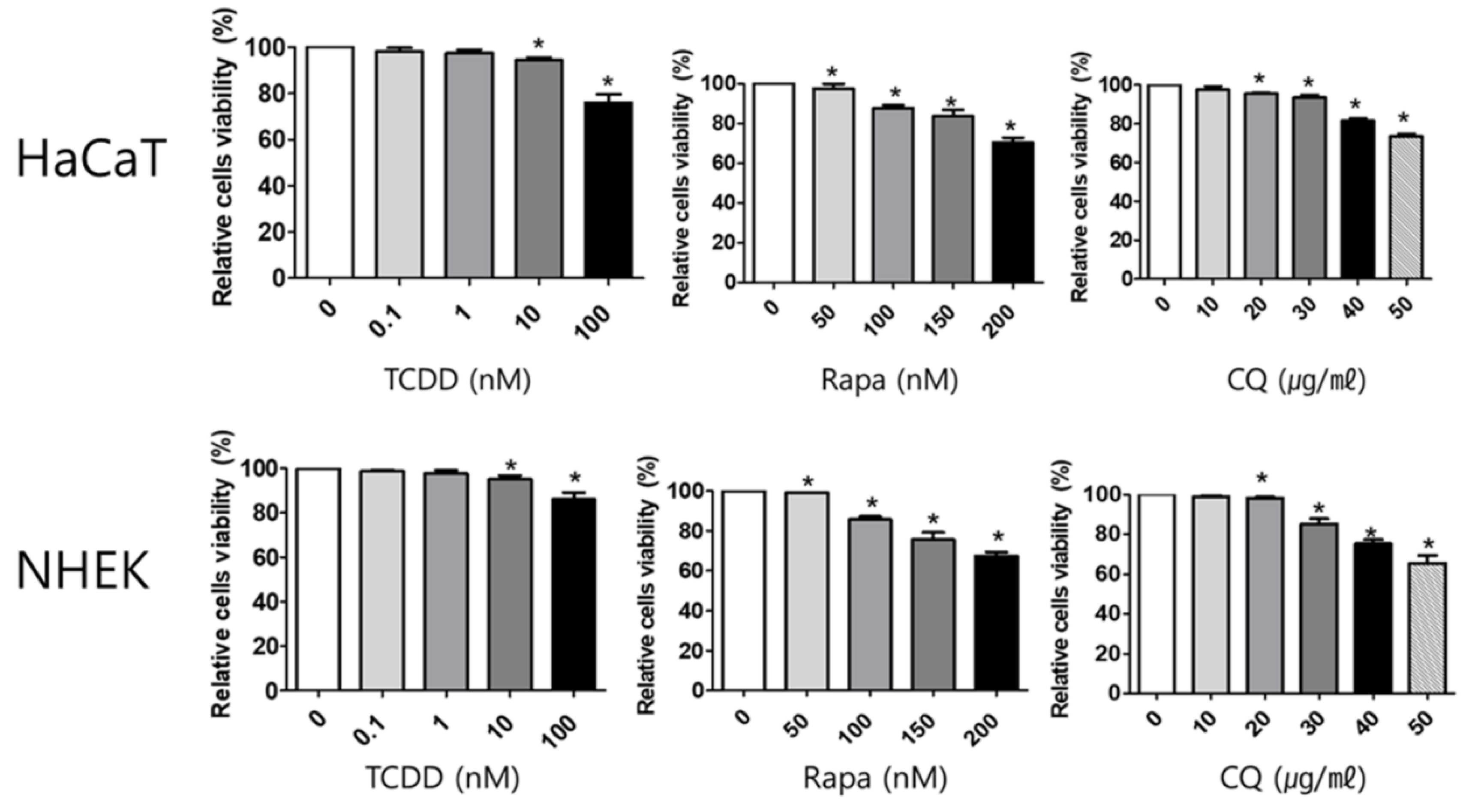
2.1. Cell Viability
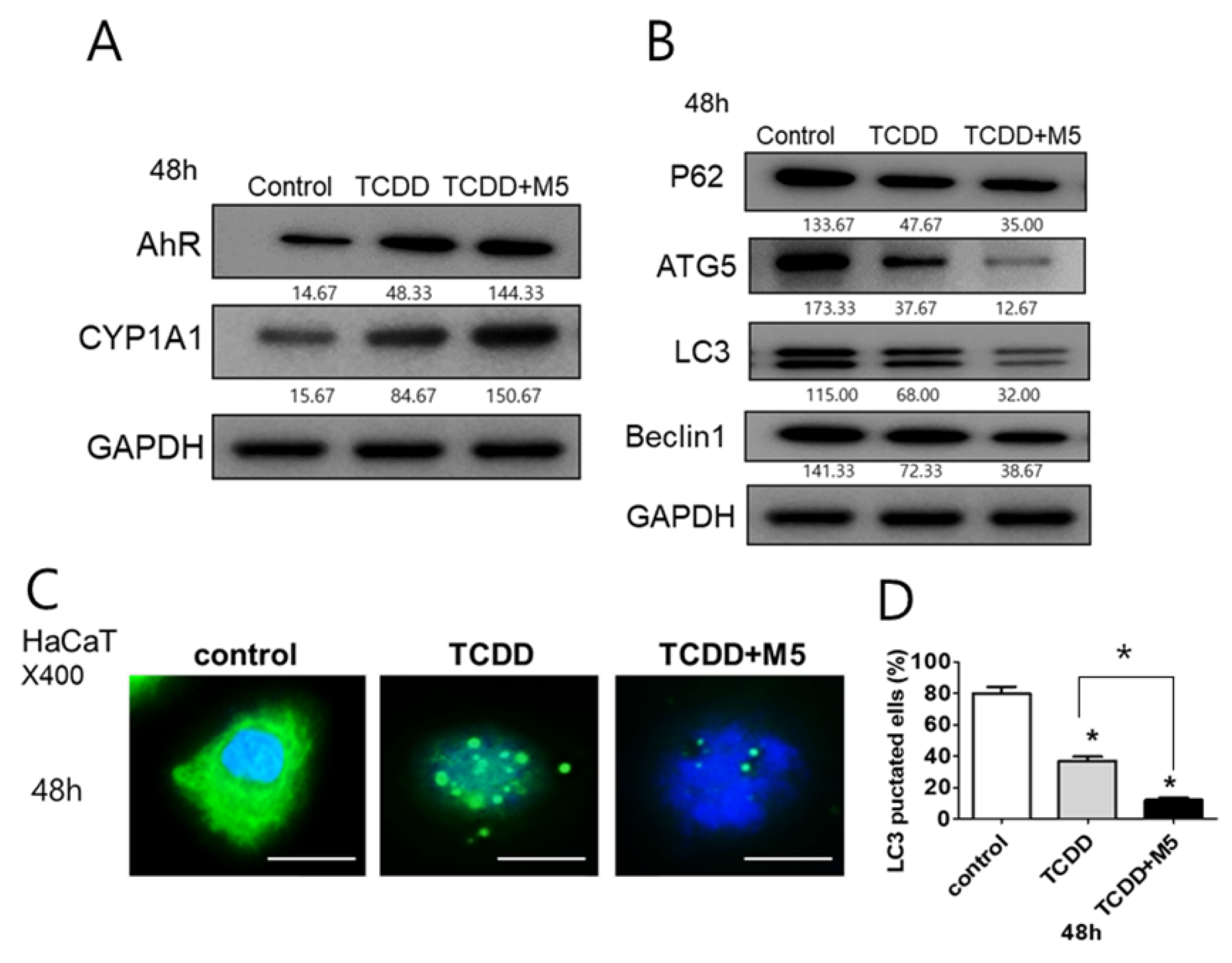
2.2. In M5-Stimulated HaCaT Cells, TCDD Treatment Significantly Upregulated AhR and CYP1A1 While Downregulating Autophagy-Related Factors and the Production of Autophagosomes
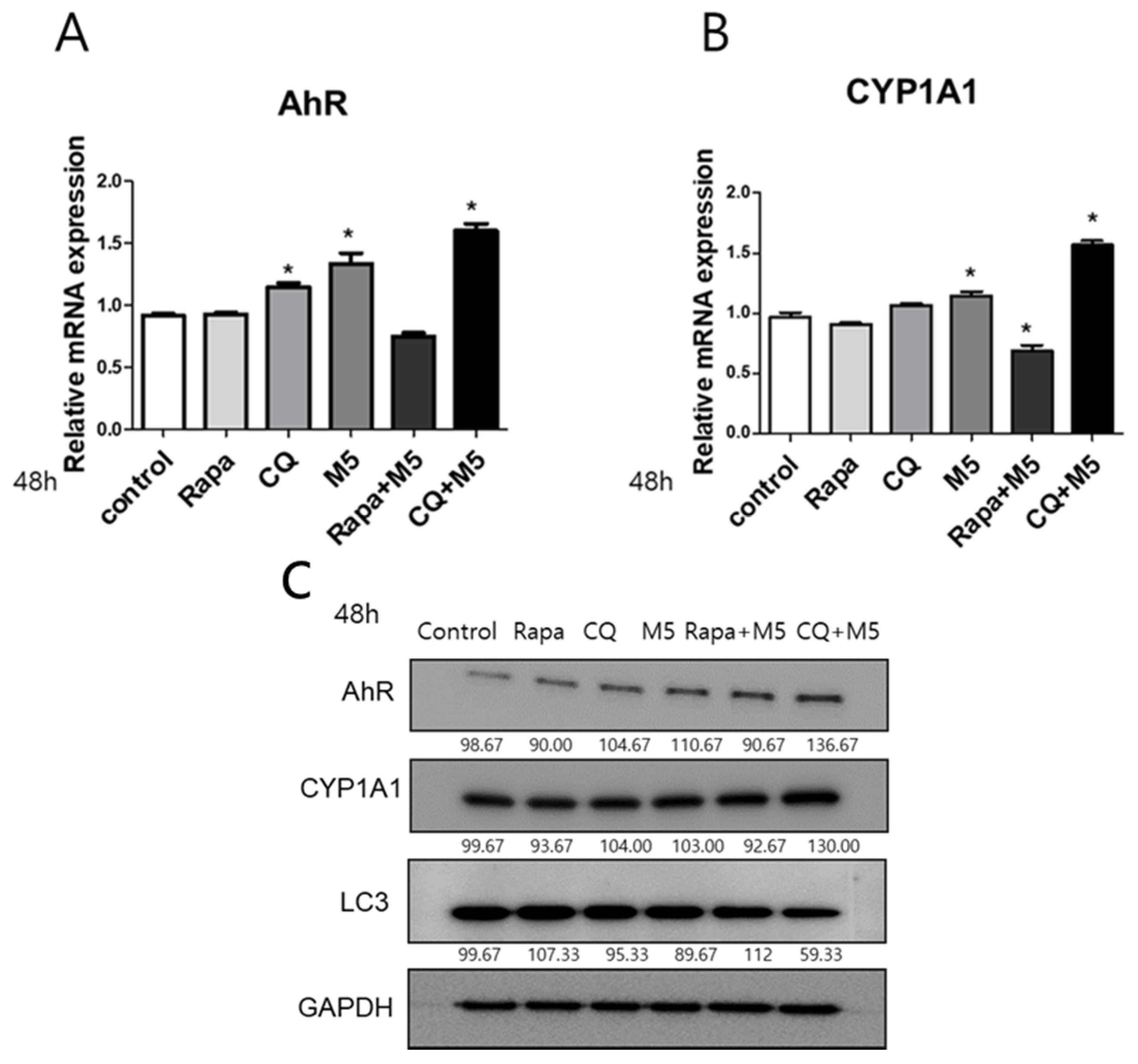
2.3. Autophagy Inhibition Upregulated AhR and CYP1A1 in M5-Stimulated HaCaT Cells
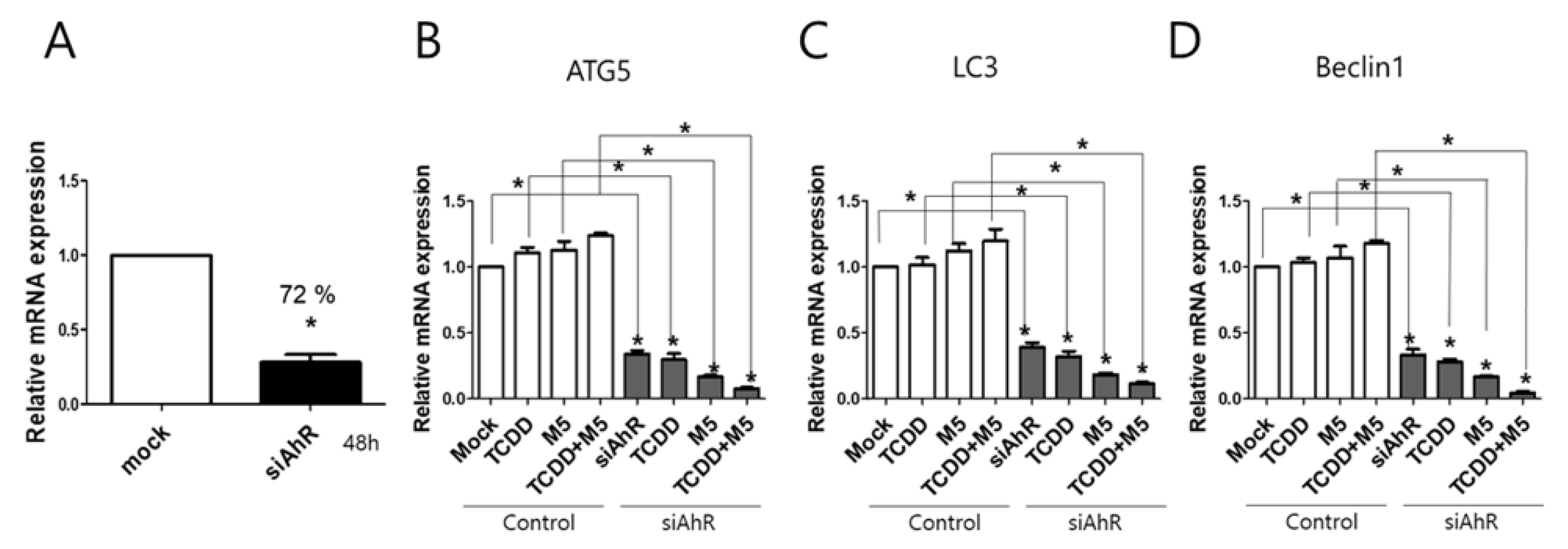
2.4. AhR Knockdown Significantly Attenuated Autophagy
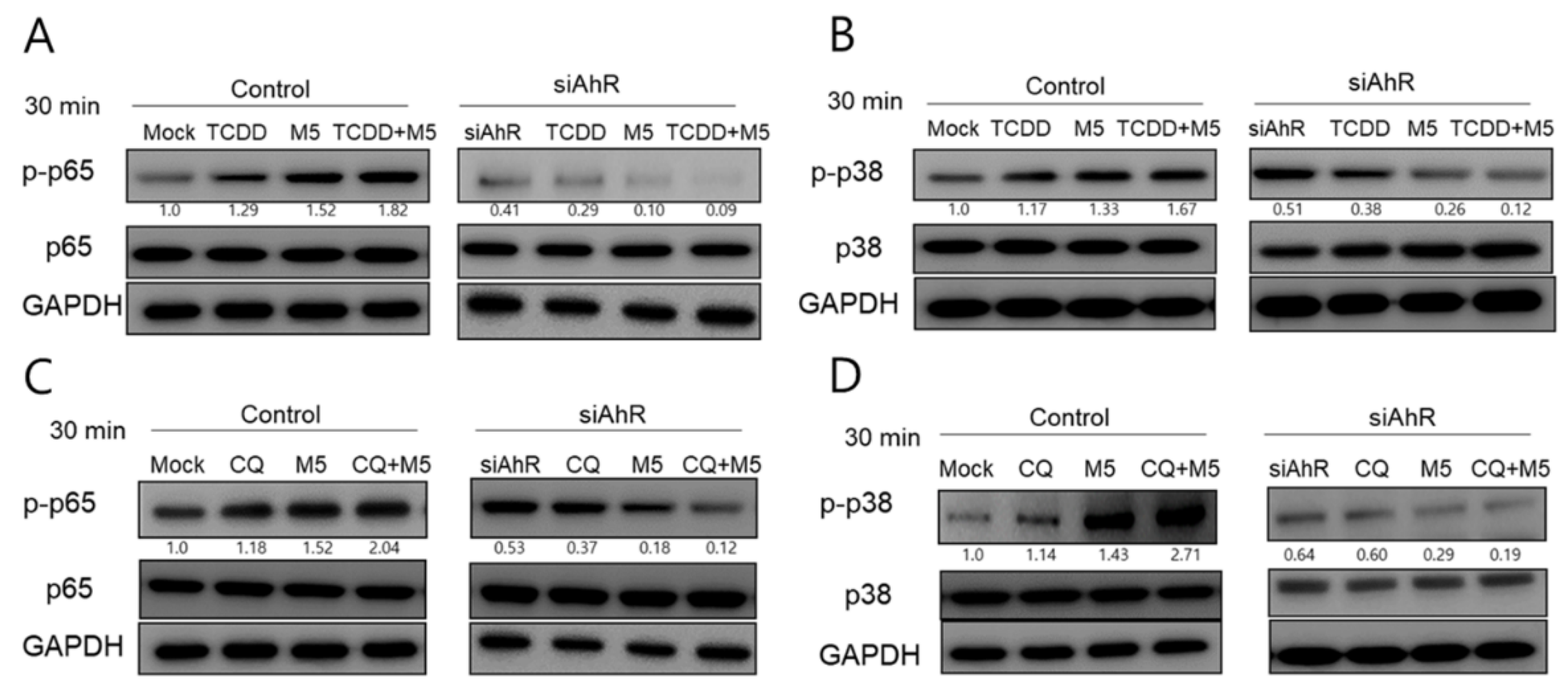
2.5. AhR Inhibition Blocked both TCDD- and Chloroquine-Induced Phosphorylation of p65NF-κB and p38MAPK in M5-Stimulated HaCaT Cells
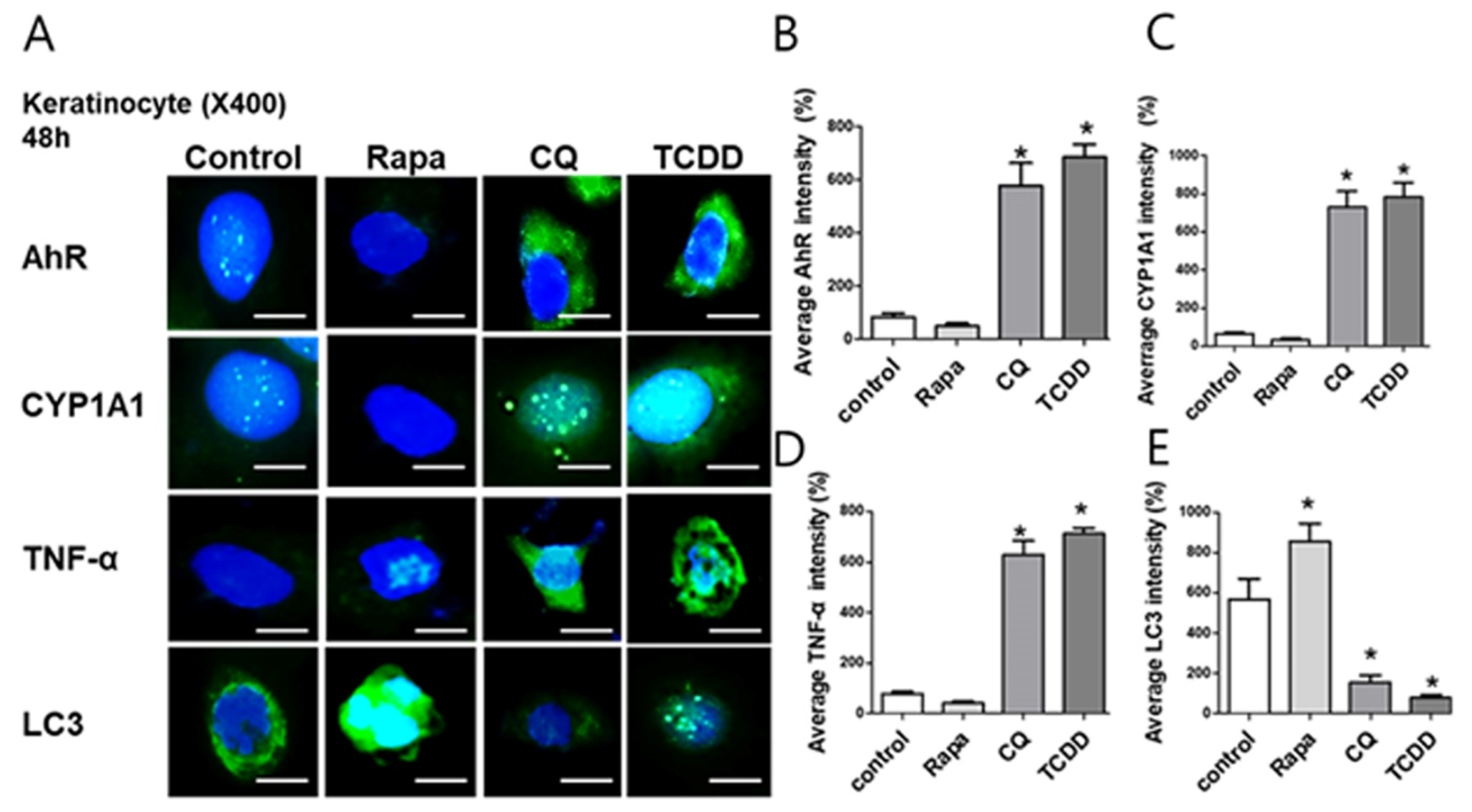
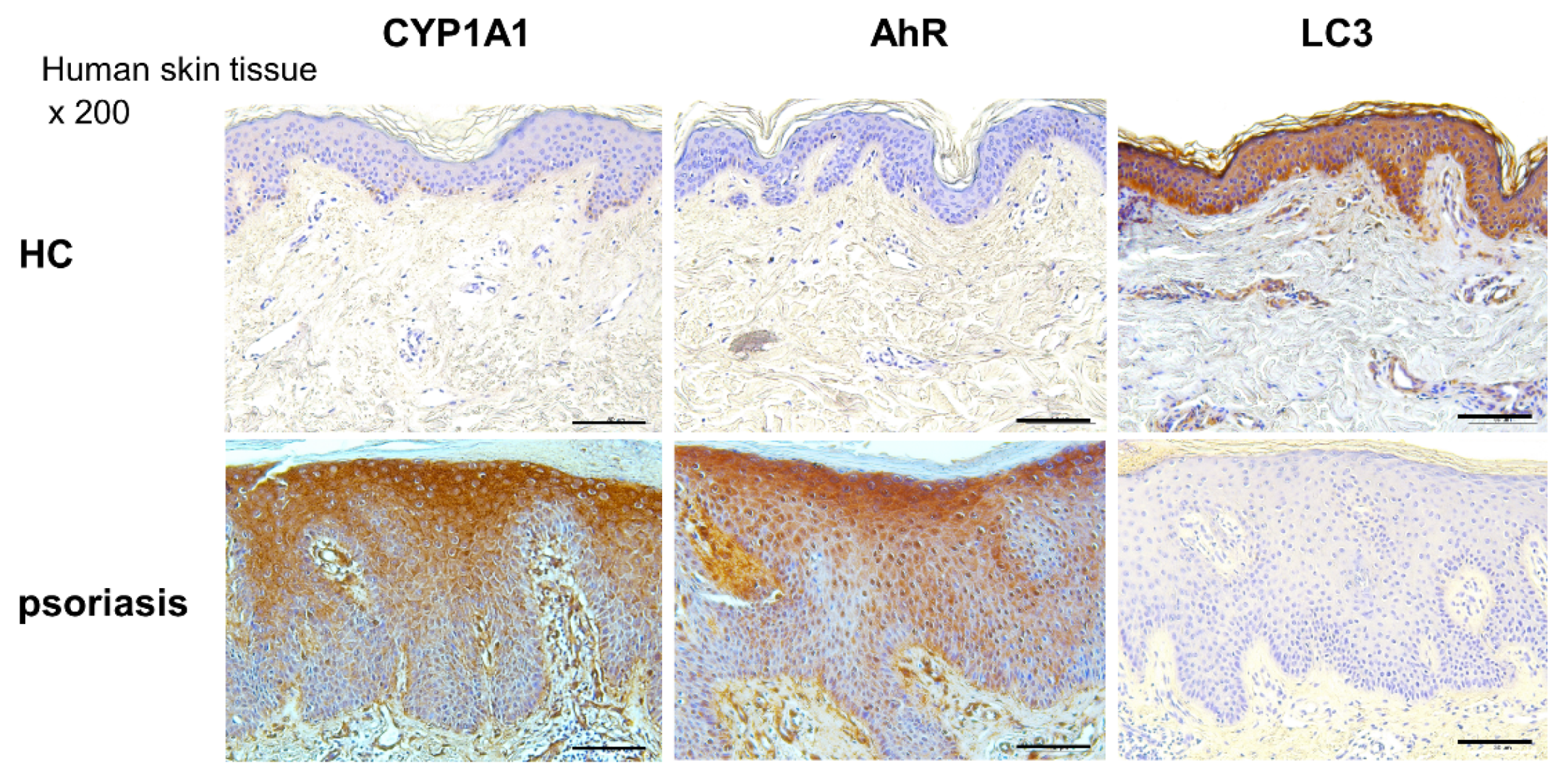
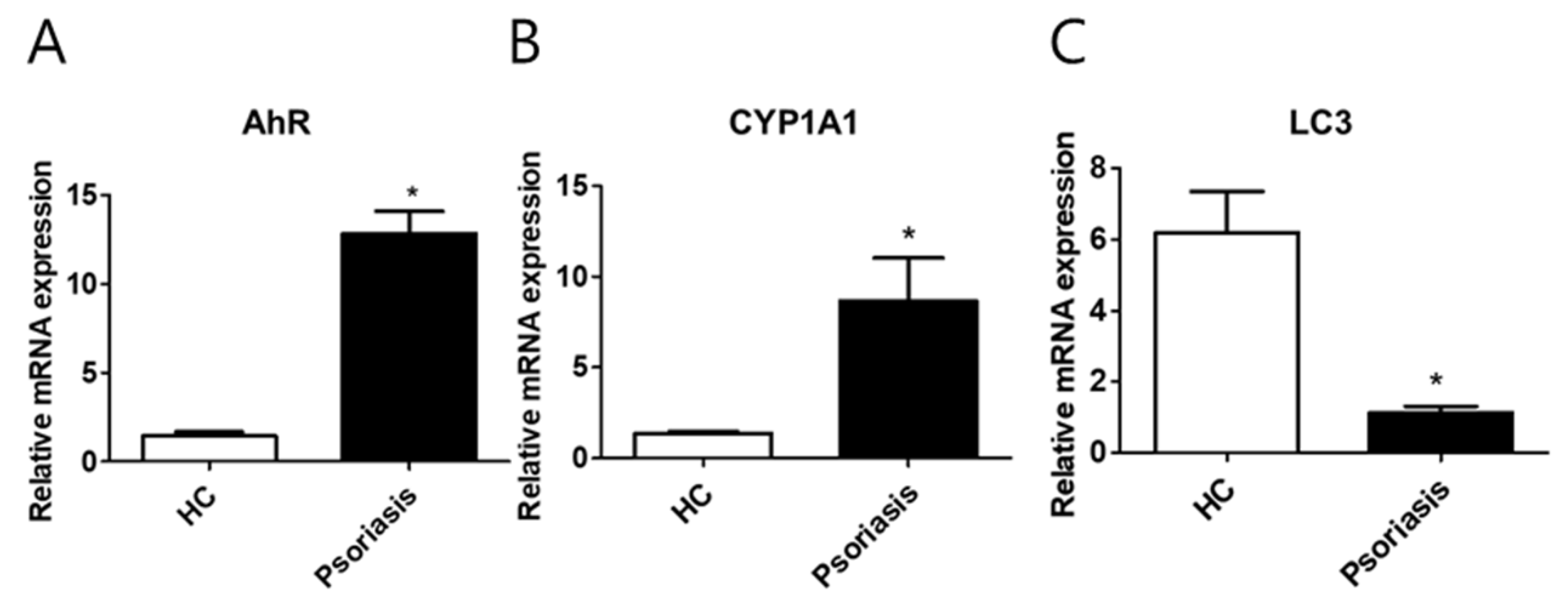
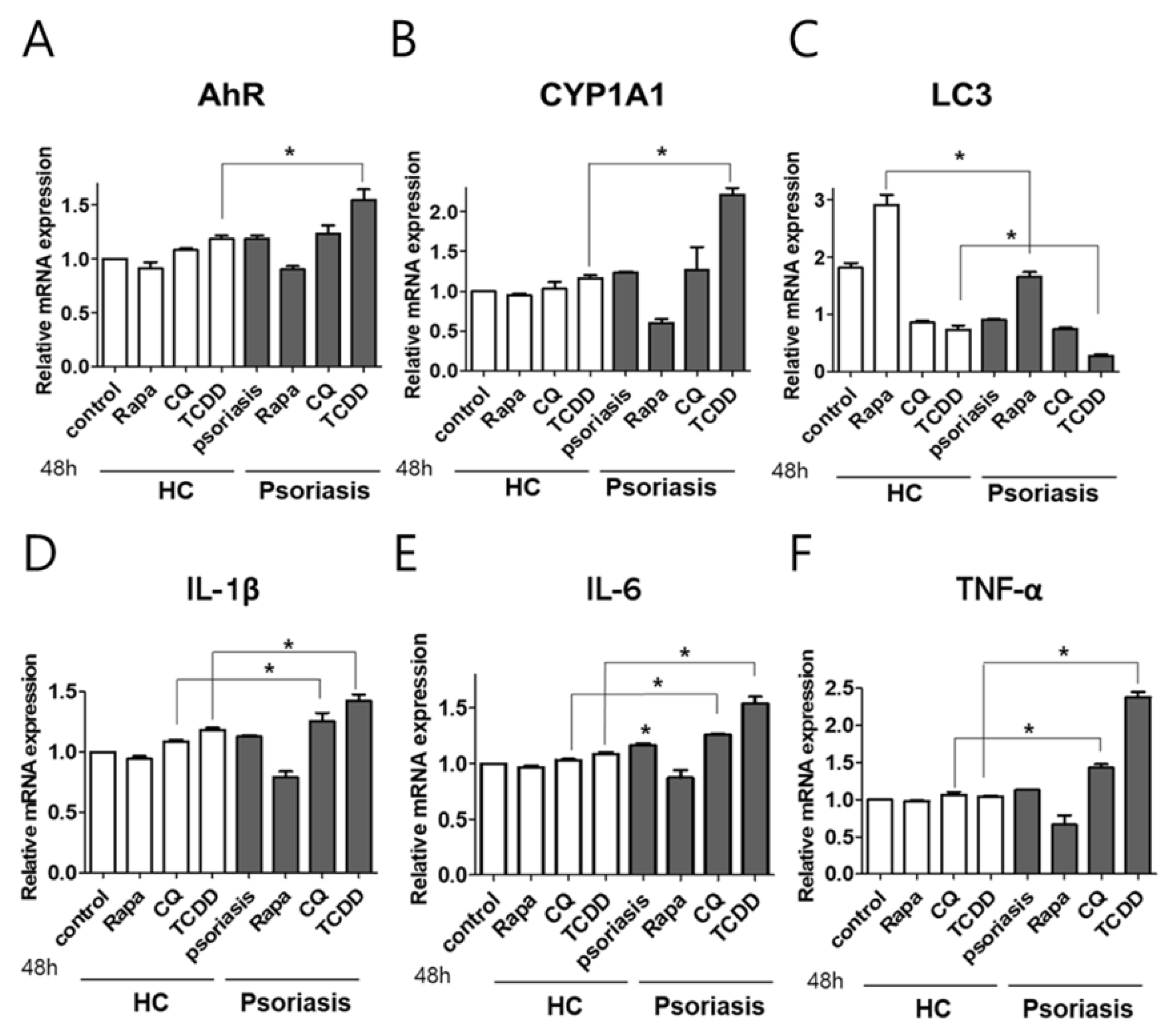
2.6. CYP1A1, LC3, and AhR Differential Expression in Human Lesional Psoriasis Skin
2.7. AhR or Autophagy Modulation in Human Psoriasis Skin Biopsies Enhanced the Production of Proinflammatory Cytokines
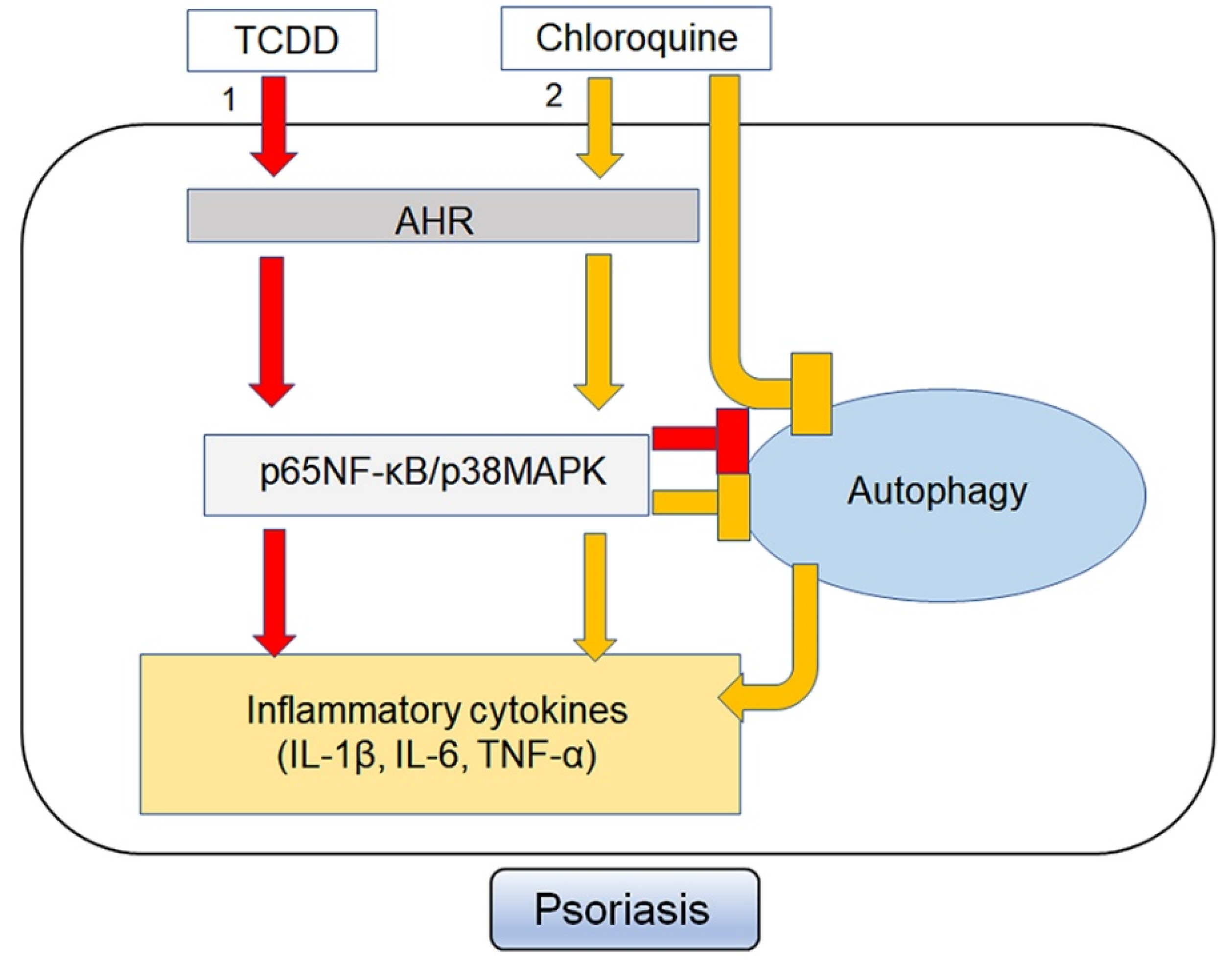
3. Discussion
4. Materials and Methods
4.1. Patients and Sample Collections
4.2. Cell Culture
4.3. Ex Vivo Culture
4.4. MTT Assay
4.5. Immunohistochemistry
4.6. Western Blot Analyses
4.7. Quantitative Reverse Transcriptase-PCR
4.8. Transfection of siRNAs (Small Interfering RNA) Specific for AhR and ATG5
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Atopic dermatitis |
| AhR | Aryl hydrocarbon receptor |
| NHEKs | Normal human epidermal keratinocytes |
| TCDD | 2, 3, 7, 8-Tetrachlorodibenzo-P-Dioxin |
| MAPK | Mitogen-activated protein kinase |
| M5 | The mixture of five proinflammatory cytokines |
| DAPI | 4′,6-diamidino-2-phenylindole |
| NF-κB | Nuclear factor-kappaB |
| PM | Particulate matter |
| Rapa | Rapamycin |
| CQ | Chloroquine |
| FBS | Fetal bovine serum |
| FICZ | 6-formylindolo[3,2-b]carbazole |
| EDTA | Ethylenediaminetetraacetic acid |
| DMEM | Dulbecco’s modified eagle medium |
| ECL | Enhanced chemiluminescence |
| DMSO | Dimethyl sulfoxide |
| TBST | Tris-buffered saline 0.1% Tween 20 |
| PBS | Phosphate-buffered saline |
| FITC | Fluorescein-5-isothiocyanate |
| SDS-PAGE | Sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| IL-1 β | Interleukin-1 β |
| IL-6 | Interleukin-6 |
| TNF- α | Transforming growth factor- α |
References
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slominski, A.T.; Kim, T.K.; Janjetovic, Z.; Brożyna, A.A.; Żmijewski, M.A.; Xu, H.; Sutter, T.R.; Tuckey, R.C.; Jetten, A.M.; Crossman, D.K. Differential and Overlapping Effects of 20,23(OH)2D3 and 1,25(OH)₂D3 on Gene Expression in Human Epidermal Keratinocytes: Identification of AhR as an Alternative Receptor for 20,23(OH)₂D3. Int. J. Mol. Sci. 2018, 19, 3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Salguero, P.M.; Hilbert, D.M.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol. Appl. Pharmacol. 1996, 140, 173–179. [Google Scholar] [CrossRef]
- Poland, A.; Knutson, J.C. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: Examination of the mechanism of toxicity. Annu Rev. Pharmacol Toxicol. 1982, 22, 517–554. [Google Scholar] [CrossRef]
- Schecter, A.; Cramer, P.; Boggess, K.; Stanley, J.; Papke, O.; Olson, J.; Silver, A.; Schmitz, M. Intake of dioxins and related compounds from food in the U.S. population. J. Toxicol. Environ. Health A 2001, 63, 1–18. [Google Scholar] [CrossRef]
- Schecter, A.; Birnbaum, L.; Ryan, J.J.; Constable, J.D. Dioxins: An overview. Environ. Res. 2006, 101, 419–428. [Google Scholar] [CrossRef]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef]
- Haarmann-Stemmann, T.; Abel, J.; Fritsche, E.; Krutmann, J. The AhR-Nrf2 pathway in keratinocytes: On the road to chemoprevention? J. Investig. Dermatol. 2012, 132, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Loertscher, J.A.; Lin, T.M.; Peterson, R.E.; Allen-Hoffmann, B.L. In utero exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin causes accelerated terminal differentiation in fetal mouse skin. Toxicol. Sci. 2002, 68, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Agostinis, P.; Garmyn, M.; Van Laethem, A. The Aryl hydrocarbon receptor: An illuminating effector of the UVB response. Sci. STKE 2007, 2007, pe49. [Google Scholar] [CrossRef]
- Jux, B.; Kadow, S.; Esser, C. Langerhans cell maturation and contact hypersensitivity are impaired in aryl hydrocarbon receptor-null mice. J. Immunol. 2009, 182, 6709–6717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doria, A.; Gatto, M.; Punzi, L. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845. [Google Scholar]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.Y.; Zhang, X.Y.; Wang, X.F.; Sun, B.C. Autophagy enhances the aggressiveness of human colorectal cancer cells and their ability to adapt to apoptotic stimulus. Cancer Biol. Med. 2012, 9, 105–110. [Google Scholar]
- Gupta, R.; Debbaneh, M.G.; Liao, W. Genetic Epidemiology of Psoriasis. Curr. Dermatol. Rep. 2014, 3, 61–78. [Google Scholar] [CrossRef]
- Guilloteau, K.; Paris, I.; Pedretti, N.; Boniface, K.; Juchaux, F.; Huguier, V.; Guillet, G.; Bernard, F.X.; Lecron, J.C.; Morel, F. Skin Inflammation Induced by the Synergistic Action of IL-17A, IL-22, Oncostatin M, IL-1 α, and TNF- α Recapitulates Some Features of Psoriasis. J. Immunol. 2010, 184, 5263–5270. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Jagannath, C.; Liu, X.D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Funatake, C.J.; Marshall, N.B.; Steppan, L.B.; Mourich, D.V.; Kerkvliet, N.I. Cutting edge: Activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+ CD25+ cells with characteristics of regulatory T cells. J. Immunol. 2005, 175, 4184–4188. [Google Scholar] [CrossRef] [Green Version]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef] [Green Version]
- Quintana, F.J.; Sherr, D.H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 2013, 65, 1148–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Lin, H.; Yu, J.; Yu, J.; Hu, Z. Autophagy and the immune response. Adv. Exp. Med. Biol. 2019, 1206, 595–634. [Google Scholar] [PubMed]
- Carew, J.S.; Medina, E.C.; Esquivel, J.A.; Mahalingam, D.; Swords, R.; Kelly, K.; Zhang, H.; Huang, P.; Mita, A.C.; Mita, M.M.; et al. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell. Mol. Med. 2010, 14, 2448–2459. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Subhawong, T.; Albert, J.M.; Kim, K.W.; Geng, L.; Sekhar, K.R.; Gi, Y.J.; Lu, B. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer. Res. 2006, 66, 10040–10047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.J.; Boyer, J.A.; Muku, G.E.; Murray, I.A.; Gowda, K.; Desai, D.; Amin, S.G.; Glick, A.B.; Perdew, G.H. Editor’s Highlight: Ah Receptor Activation Potentiates Neutrophil Chemoattractant (C-X-C Motif) Ligand 5 Expression in Keratinocytes and Skin. Toxicol. Sci. 2017, 160, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Nograles, K.E.; Zaba, L.C.; Guttman-Yassky, E.; Fuentes-Duculan, J.; Suarez-Farinas, M.; Cardinale, I.; Khatcherian, A.; Gonzalez, J.; Pierson, K.C.; White, T.R.; et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br. J. Dermatol. 2008, 159, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- Brembilla, N.C.; Ramirez, J.M.; Chicheportiche, R.; Sorg, O.; Saurat, J.H.; Chizzolini, C. In vivo dioxin favors interleukin-22 production by human CD4+ T cells in an aryl hydrocarbon receptor (AhR)-dependent manner. PLoS ONE 2011, 6, e18741. [Google Scholar]
- Lowes, M.A.; Russell, C.B.; Martin, D.A.; Towne, J.E.; Krueger, J.G. The IL-23/T17 pathogenic axis in psoriasis is amplified by keratinocyte responses. Trends. Immunol. 2013, 34, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Sukseree, S.; Eckhart, L.; Tschachler, E.; Watanapokasin, R. Autophagy in epithelial homeostasis and defense. Front. Biosci. 2013, 5, 1000–1010. [Google Scholar]
- Douroudis, K.; Kingo, K.; Traks, T.; Reimann, E.; Raud, K.; Ratsep, R.; Mössner, R.; Silm, H.; Vasar, E.; Kõks, S. Polymorphisms in the ATG16L1 gene are associated with psoriasis vulgaris. Acta Derm. Venereol. 2012, 92, 85–87. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell. Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.M.; Shin, D.M.; Yuk, J.M.; Shi, G.; Choi, D.K.; Lee, S.H.; Huang, S.M.; Kim, J.M.; Kim, C.D.; Lee, J.H.; et al. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J. Immunol. 2011, 186, 1248–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, P.; Saini, N. PI3K/AKT/mTOR activation and autophagy inhibition plays a key role in increased cholesterol during IL-17A mediated inflammatory response in psoriasis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1795–1803. [Google Scholar] [CrossRef]
- Di Meglio, P.; Duarte, J.H.; Ahlfors, H.; Owens, N.D.; Li, Y.; Villanova, F.; Tosi, I.; Hirota, K.; Nestle, F.O.; Mrowietz, U.; et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity 2014, 40, 989–1001. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.S.; Lee, J.E.; Myung, C.H.; Park, J.I.; Jo, C.S.; Hwang, J.S. Particulate Matter-Induced Aryl Hydrocarbon Receptor Regulates Autophagy in Keratinocytes. Biomol. Ther. 2019, 570–576. [Google Scholar] [CrossRef]
- Duarte, J.H.; Di Meglio, P.; Hirota, K.; Ahlfors, H.; Stockinger, B. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLoS. One. 2013, 8, e79819. [Google Scholar] [CrossRef] [Green Version]
- Tauchi, M.; Hida, A.; Negishi, T.; Katsuoka, F.; Noda, S.; Mimura, J.; Hosoya, T.; Yanaka, A.; Aburatani, H.; Fujii-Kuriyama, Y.; et al. Constitutive expression of aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol. Cell Biol. 2005, 25, 9360–9368. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, K.A.; Elferink, C.J. Timing is everything: Consequences of transient and sustained AhR activity. Biochem. Pharmacol. 2009, 77, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Wincent, E.; Amini, N.; Luecke, S.; Glatt, H.; Bergman, J.; Crescenzi, C.; Rannug, A.; Rannug, U. The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J. Biol. Chem. 2009, 284, 2690–2696. [Google Scholar] [CrossRef] [Green Version]
- Haarmann-Stemmann, T.; Esser, C.; Krutmann, J. The Janus-Faced Role of Aryl Hydrocarbon Receptor Signaling in the Skin: Consequences for Prevention and Treatment of Skin Disorders. J. Investig. Dermatol. 2015, 135, 2572–2576. [Google Scholar] [CrossRef] [Green Version]
- Stobbe-Maicherski, N.; Wolff, S.; Wolff, C.; Abel, J.; Sydlik, U.; Frauenstein, K.; Haarmann-Stemmann, T. The interleukin-6-type cytokine oncostatin M induces aryl hydrocarbon receptor expression in a STAT3-dependent manner in human HepG2 hepatoma cells. FEBS J. 2013, 280, 6681–6690. [Google Scholar] [CrossRef] [PubMed]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.R.; Kang, S.Y.; Kim, H.O.; Park, C.W.; Chung, B.Y. Role of Aryl Hydrocarbon Receptor Activation and Autophagy in Psoriasis-Related Inflammation. Int. J. Mol. Sci. 2020, 21, 2195. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062195
Kim HR, Kang SY, Kim HO, Park CW, Chung BY. Role of Aryl Hydrocarbon Receptor Activation and Autophagy in Psoriasis-Related Inflammation. International Journal of Molecular Sciences. 2020; 21(6):2195. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062195
Chicago/Turabian StyleKim, Hye Ran, Seok Young Kang, Hye One Kim, Chun Wook Park, and Bo Young Chung. 2020. "Role of Aryl Hydrocarbon Receptor Activation and Autophagy in Psoriasis-Related Inflammation" International Journal of Molecular Sciences 21, no. 6: 2195. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062195