The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells


Abstract
:1. Introduction
1.1. Acute Myeloid Leukemia
1.2. Leukemic Stem Cells
2. Malignant Cell Metabolism and Its Possible Clinical Importance in AML
2.1. Metabolism in Malignant Diseases
2.2. Metabolism in AML
3. The Phosphoinositide 3-Kinase (PI3K)-Akt-Mammalian Target of Rapamycin (mTOR) Pathway
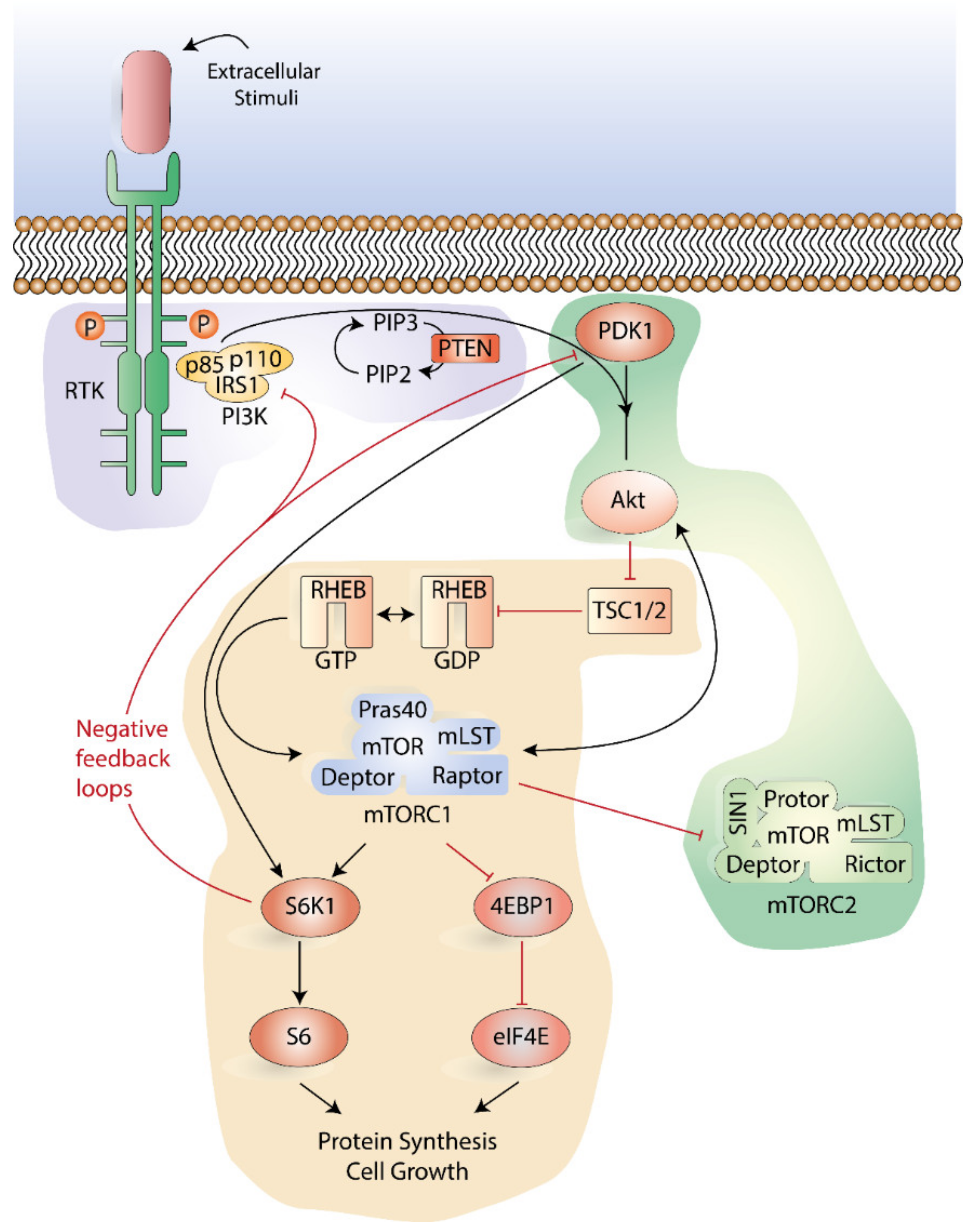
3.1. Function and Signaling of the PI3K-Akt-mTOR Pathway
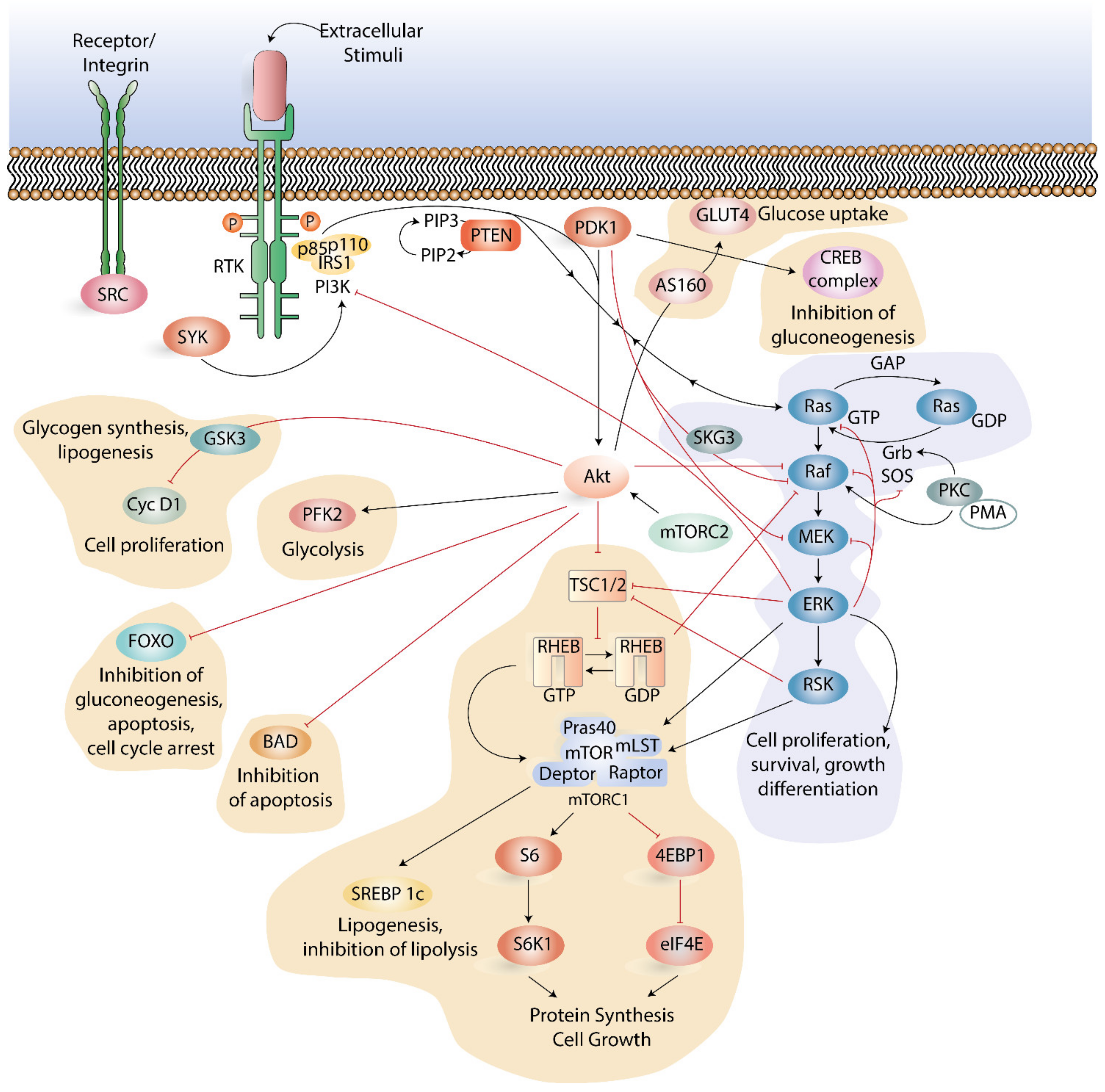
3.2. The Role of the PI3K-Akt-mTOR Pathway in Modulating Metabolism
3.3. PI3K-Akt-mTOR Signaling in AML
3.4. Crosstalk between PI3K-Akt-mTOR Pathway and other Signaling Pathways
4. Inhibition of the PI3K-Akt-mTOR Pathway in AML
5. Conclusions and Further Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4EBP1 | eIF4E -binding protein 1 |
| AML | acute myeloid leukemia |
| AMPK | AMP-activated protein kinase |
| APL | acute promyelocytic leukemia |
| BAD | BCL2 associated agonist of cell death |
| BCL2 | B-cell lymphoma-2 |
| CR | complete remission |
| CRi | complete remission with incomplete recovery |
| CREB | cAMP response element-binding protein |
| DFS | disease free survival |
| eIF4E | eukaryotic initiation factor-4E |
| ERK | extracellular signal-regulated kinase |
| FKBP38 | FK506-binding protein 38 |
| FLT3 | fms like tyrosine kinase 3 |
| GAB2 | GRB2 associated binding protein |
| GAP | GTPase-activating protein |
| GLUT1 | glucose transporter 1 |
| GPCR | G-protein-coupled receptor |
| GRB2 | growth factor receptor-bound protein 2 |
| GSK3 | glycogen synthase kinase 3 |
| HSC | hematopoietic stem cell |
| HSPC | hematopoietic stem/progenitor cell |
| IGF | insulin growth factor |
| IRS | insulin receptor substrates |
| IDH | isocitrate dehydrogenase |
| ITD | internal tandem duplication |
| LSC | leukemic stem cell |
| MEK | MAPK ERK kinase |
| mLST | mammalian lethal with SEC13 protein |
| mSIN1 | mammalian stress-activated protein kinase-interacting protein 1 |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mTOR complex 1 |
| mTORC2 | mTOR complex 2 |
| NPM1 | nucleophosmin 1 |
| OS | overall survival |
| PDK1 | phosphoinositide-dependent kinase-1 |
| PFK2 | phosphofructokinase-2 |
| PH | pleckstrin-homology |
| PI3K | phosphoinositide 3-kinase |
| PIKK | PI3K-related kinase |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PIP3 | phosphatidylinositol 3,4,5- trisphosphates |
| PKC | protein kinase C |
| PMA | Phorbol 12-myristate 13-acetate |
| PR | partial remission |
| Pras40 | proline-rich Akt substrate of 40 kDa |
| PTEN | protein deleted on chromosome 10 |
| RSK | Ribosomal S6 kinase |
| RHEB | Ras homologue enriched in brain (RHEB) |
| RTK | receptor tyrosine kinase |
| S6K | S6 kinase |
| SH2 | Src Homology 2 |
| SHIP1 | SH2 domain-containing inositol 5-phosphatases 1 |
| SHIP2 | SH2 domain-containing inositol 5-phosphatases 2 |
| SREBF1 | sterol regulatory element-binding transcription factor 1 |
| TCA | tricarboxylic acid |
| TKD | tyrosine kinase domain |
| TSC | tuberous sclerosis complex |
References
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Almond, L.M.; Charalampakis, M.; Ford, S.J.; Gourevitch, D.; Desai, A. Myeloid Sarcoma: Presentation, Diagnosis, and Treatment. Clin. Lymphoma Myeloma Leuk. 2017, 17, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Ganzel, C.; Douer, D. Extramedullary disease in APL: A real phenomenon to contend with or not? Best Pract. Res. Clin. Haematol. 2014, 27, 63–68. [Google Scholar] [CrossRef]
- Sanz, M.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coco, F.L.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic Acid and Arsenic Trioxide for Acute Promyelocytic Leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Coco, F.L.; Latagliata, R.; Breccia, M. Management of acute promyelocytic leukemia in the elderly. Mediterr. J. Hematol. Infect. Dis. 2013, 5, e2013045. [Google Scholar]
- De Kouchkovsky, I.; Abdul-Hay, M. ‘Acute myeloid leukemia: A comprehensive review and 2016 update’. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Chen, W.-L.; Wang, J.-H.; Zhao, A.-H.; Xu, X.; Wang, Y.-H.; Chen, T.-L.; Li, J.-M.; Mi, J.-Q.; Zhu, Y.-M.; Liu, Y.-F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef]
- Kuo, T.-C.; Tian, T.-F.; Tseng, Y.J. 3Omics: A web-based systems biology tool for analysis, integration and visualization of human transcriptomic, proteomic and metabolomic data. BMC Syst. Biol. 2013, 7, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roboz, J.; Roboz, G.J. Mass spectrometry in leukemia research and treatment. Expert Rev. Hematol. 2015, 8, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.; Robertson, A.G.; Hoadley, K.A.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Shivarov, V.; Dolnik, A.; Lang, K.M.; Krönke, J.; Kuchenbauer, F.; Paschka, P.; Gaidzik, V.I.; Döhner, H.; Schlenk, R.F.; Döhner, K.; et al. MicroRNA expression-based outcome prediction in acute myeloid leukemia: Novel insights through cross-platform integrative analyses. Haematologica 2016, 101, e454–e456. [Google Scholar] [CrossRef] [Green Version]
- Reikvam, H.; Aasebø, E.; Brenner, A.; Bartaula-Brevik, S.; Grønningsæter, I.S.; Forthun, R.B.; Hovland, R.; Bruserud, Ø. High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype. J. Clin. Med. 2019, 8, 970. [Google Scholar] [CrossRef] [Green Version]
- Navada, S.C.; Steinmann, J.; Lübbert, M.; Silverman, L.R. Clinical development of demethylating agents in hematology. J. Clin. Investig. 2014, 124, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Szmigielska-Kaplon, A.; Robak, T. Hypomethylating agents in the treatment of myelodysplastic syndromes and myeloid leukemia. Curr. Cancer Drug Targets 2011, 11, 837–848. [Google Scholar] [CrossRef]
- Brandwein, J.M.; Zhu, N.; Kumar, R.; Leber, B.; Sabloff, M.; Sandhu, I.; Kassis, J.; Olney, H.J.; Elemary, M.; Schuh, A.C. Treatment of older patients with acute myeloid leukemia (AML): Revised Canadian consensus guidelines. Am. J. blood Res. 2017, 7, 30–40. [Google Scholar]
- Burnett, A.K.; Milligan, D.; Prentice, A.G.; Goldstone, A.H.; McMullin, M.F.; Hills, R.K.; Wheatley, K. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer 2007, 109, 1114–1124. [Google Scholar] [CrossRef]
- Adams, C.M.; Clark-Garvey, S.; Porcu, P.; Eischen, C.M. Targeting the Bcl-2 Family in B Cell Lymphoma. Front. Oncol. 2018, 8, 636. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Wei, A.H. How I treat acute myeloid leukemia in the era of new drugs. Blood 2020, 135, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguille, S.; Van Tendeloo, V.F.I.; Berneman, Z.N. Leukemia-associated antigens and their relevance to the immunotherapy of acute myeloid leukemia. Leukemia 2012, 26, 2186–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsykunova, G.; Reikvam, H.; Hovland, R.; Bruserud, Ø. The surface molecule signature of primary human acute myeloid leukemia (AML) cells is highly associated with NPM1 mutation status. Leukemia 2012, 26, 557–559. [Google Scholar] [CrossRef] [Green Version]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef]
- Hamburger, A.W.; Salmon, S.E. Primary bioassay of human tumor stem cells. Science 1977, 197, 461–463. [Google Scholar] [CrossRef]
- Wang, J.C.; Dick, J.E. Cancer stem cells: Lessons from leukemia. Trends Cell Biol. 2005, 15, 494–501. [Google Scholar] [CrossRef]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; Van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef]
- Bruserud, Ø.; Gjertsen, B.T.; Foss, B.; Huang, T.-S.; Tjønnfjord, G.; Ernst, P. New Strategies in the Treatment of Acute Myelogenous Leukemia (AML): In Vitro Culture of AML Cells-The Present Use in Experimental Studies and the Possible Importance for Future Therapeutic Approaches. Stem Cells 2001, 19, 1–11. [Google Scholar] [CrossRef]
- Hope, K.J.; Jin, L.; Dick, J.E. Human acute myeloid leukemia stem cells. Arch. Med Res. 2003, 34, 507–514. [Google Scholar] [CrossRef]
- Miyamoto, T.; Weissman, I.L.; Akashi, K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc. Natl. Acad. Sci. USA 2000, 97, 7521–7526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, A.; Chan, S.M.; Thomas, D.; Majeti, R. Biology and Clinical Relevance of Acute Myeloid Leukemia Stem Cells. Semin. Hematol. 2015, 52, 150–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taussig, D.C.; Vargaftig, J.; Miraki-Moud, F.; Griessinger, E.; Sharrock, K.; Luke, T.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34− fraction. Blood 2010, 115, 1976–1984. [Google Scholar] [CrossRef] [Green Version]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.J.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like Leukemia Stem Cells in Acute Myeloid Leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45, e45. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Herst, P.; Howman, R.A.; Neeson, P.J.; Berridge, M.; Ritchie, D.S. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. J. Leukoc. Biol. 2011, 89, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Fiegl, M.; McQueen, T.; Clise-Dwyer, K.; Andreeff, M. The warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res. 2008, 68, 5198–5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farge, T.; Saland, E.; De Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, B.C.; Fathi, A.T.; Dinardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2016, 31, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-H.; Chen, W.-L.; Li, J.-M.; Wu, S.-F.; Chen, T.-L.; Zhu, Y.-M.; Zhang, W.-N.; Li, Y.; Qiu, Y.-P.; Zhao, A.-H.; et al. Prognostic significance of 2-hydroxyglutarate levels in acute myeloid leukemia in China. Proc. Natl. Acad. Sci. USA 2013, 110, 17017–17022. [Google Scholar] [CrossRef] [Green Version]
- Kornblau, S.M.; Womble, M.; Qiu, Y.H.; Jackson, C.E.; Chen, W.; Konopleva, M.; Estey, E.H.; Andreeff, M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood 2006, 108, 2358–2365. [Google Scholar] [CrossRef] [Green Version]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Hennessy, B.T.; Smith, D.L.; Ram, P.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT Pathway for Cancer Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef]
- Sujobert, P.; Bardet, V.; Cornillet-Lefebvre, P.; Hayflick, J.S.; Prie, N.; Verdier, F.; Vanhaesebroeck, B.; Muller, O.; Pesce, F.; Ifrah, N.; et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 2005, 106, 1063–1066. [Google Scholar] [CrossRef] [Green Version]
- Piddock, R.E.; Bowles, K.M.; Rushworth, S.A. The Role of PI3K Isoforms in Regulating Bone Marrow Microenvironment Signaling Focusing on Acute Myeloid Leukemia and Multiple Myeloma. Cancers 2017, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Testa, J.R.; Moore, R.; LaRue, L. A portrait of AKT kinases: Human cancer and animal models depict a family with strong individualities. Cancer Biol. Ther. 2004, 3, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgering, B.; Coffer, P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, K.J.; Lang, H.; Wohlschlaeger, J.; Sotiropoulos, G.C.; Reis, H.; Schmid, K.W.; Baba, H.A. AKT and ERK1/2 signaling in intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2007, 13, 6470–6477. [Google Scholar] [CrossRef] [PubMed]
- Kunz, J.; Henriquez, R.; Schneider, U.; Deuter-Reinhard, M.; Movva, N.; Hall, M.N. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 1993, 73, 585–596. [Google Scholar] [CrossRef]
- Dowling, R.J.O.; Topisirovic, I.; Fonseca, B.D.; Sonenberg, N. Dissecting the role of mTOR: Lessons from mTOR inhibitors. Biochim. Biophys Acta 2010, 1804, 433–439. [Google Scholar] [CrossRef]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.N.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-E.; Chen, J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Frías, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 Is Necessary for Akt/PKB Phosphorylation, and Its Isoforms Define Three Distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.D.; Sonenberg, N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 2005, 433, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Kozlowski, M.T.; Weng, Q.-P.; Morrice, N.; Avruch, J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 1998, 8, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Ma, N.; Liu, A.; Shen, X.; Wang, Q.; Liu, Y.; Jiang, Y. Rheb Activates mTOR by Antagonizing Its Endogenous Inhibitor, FKBP38. Science 2007, 318, 977–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proud, C.G. Cell signaling. mTOR, Unleashed. Science 2007, 318, 926–927. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, Z.; Yin, J.; Quon, M.; Ye, J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J. Biol. Chem. 2008, 283, 35375–35382. [Google Scholar] [CrossRef] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb Binds and Regulates the mTOR Kinase. Curr. Biol. 2005, 15, 702–7133. [Google Scholar] [CrossRef] [Green Version]
- Parmar, N.; Tamanoi, F. Rheb G-Proteins and the Activation of mTORC1. Enzymes 2010, 27, 39–56. [Google Scholar]
- Reikvam, H.; Nepstad, I.; Bruserud, O.; Hatfield, K.J. Pharmacological targeting of the PI3K/mTOR pathway alters the release of angioregulatory mediators both from primary human acute myeloid leukemia cells and their neighboring stromal cells. Oncotarget 2013, 4, 830–843. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.; Thompson, C.B. Signaling in Control of Cell Growth and Metabolism. Cold Spring Harb. Perspect. Biol. 2012, 4, a006783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braccini, L.; Ciraolo, E.; Martini, M.; Pirali, T.; Germena, G.; Rolfo, K.; Hirsch, E. PI3K keeps the balance between metabolism and cancer. Adv. Biol. Regul. 2012, 52, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulain, L.; Sujobert, P.; Zylbersztejn, F.; Barreau, S.; Stuani, L.; Lambert, M.; Palama, T.; Chesnais, V.; Birsen, R.; Vergez, F.; et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia 2017, 31, 2326–2335. [Google Scholar] [CrossRef]
- Elstrom, R.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Tian, Y.; Yu, Z.; Shi, D.; Wang, J.; Zhang, C.; Peng, R.; Chen, X.; Liu, C.; Chen, Y.; et al. Targeting PDK1 with dichloroacetophenone to inhibit acute myeloid leukemia (AML) cell growth. Oncotarget 2016, 7, 1395–1407. [Google Scholar] [CrossRef] [Green Version]
- Pereira, O.; Teixeira, A.; Sampaio-Marques, B.; Castro, I.; Girao, H.; Ludovico, P. Signalling mechanisms that regulate metabolic profile and autophagy of acute myeloid leukaemia cells. J. Cell. Mol. Med. 2018, 22, 4807–4817. [Google Scholar] [CrossRef]
- Nepstad, I.; Hatfield, K.; Tvedt, T.A.; Reikvam, H.; Bruserud, Ø. Clonal Heterogeneity Reflected by PI3K-AKT-mTOR Signaling in Human Acute Myeloid Leukemia Cells and Its Association with Adverse Prognosis. Cancers (Basel) 2018, 10, 332. [Google Scholar] [CrossRef] [Green Version]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Aasebø, E.; Hernandez-Valladares, M.; Hagen, K.M.; Rye, K.P.; Berven, F.S.; Selheim, F.; Reikvam, H.; et al. Effects of insulin and pathway inhibitors on the PI3K-Akt-mTOR phosphorylation profile in acute myeloid leukemia cells. Signal Transduct. Target. Ther. 2019, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Kornblau, S.M.; Tibes, R.; Qiu, Y.H.; Chen, W.; Kantarjian, H.M.; Andreeff, M.; Coombes, K.R.; Mills, G.B. Functional proteomic profiling of AML predicts response and survival. Blood 2009, 113, 154–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, Y.H.; Eom, J.I.; Cheong, J.-W.; Maeng, H.O.; Kim, J.Y.; Jeung, H.K.; Lee, S.T.; Lee, M.H.; Hahn, J.S.; Ko, Y.W. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: Its significance as a prognostic variable. Leukemia 2003, 17, 995–997. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Drakos, E.; Grammatikakis, I.; Schlette, E.J.; Li, J.; Leventaki, V.; Staikou-Drakopoulou, E.; Patsouris, E.; Panayiotidis, P.; Medeiros, L.J.; et al. mTOR signaling is activated by FLT3 kinase and promotes survival of FLT3-mutated acute myeloid leukemia cells. Mol. Cancer 2010, 9, 292. [Google Scholar] [CrossRef] [Green Version]
- Nepstad, I.; Hatfield, K.; Aasebø, E.; Hernandez-Valladares, M.; Brenner, A.; Bartaula-Brevik, S.; Berven, F.S.; Selheim, F.; Skavland, J.; Gjertsen, B.T.; et al. Two acute myeloid leukemia patient subsets are identified based on the constitutive PI3K-Akt-mTOR signaling of their leukemic cells; a functional, proteomic, and transcriptomic comparison. Expert Opin. Ther. Targets 2018, 22, 639–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepstad, I.; Reikvam, H.; Brenner, A.; Bruserud, O.; Hatfield, K. Resistance to the Antiproliferative In Vitro Effect of PI3K-Akt-mTOR Inhibition in Primary Human Acute Myeloid Leukemia Cells Is Associated with Altered Cell Metabolism. Int. J. Mol. Sci. 2018, 19, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grønningsæter, I.; Fredly, H.; Gjertsen, B.T.; Hatfield, K.; Bruserud, Ø. Systemic Metabolomic Profiling of Acute Myeloid Leukemia Patients before and During Disease-Stabilizing Treatment Based on All-Trans Retinoic Acid, Valproic Acid, and Low-Dose Chemotherapy. Cells 2019, 8, 1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandts, C.H.; Sargin, B.; Rode, M.; Biermann, C.; Lindtner, B.; Schwäble, J.; Buerger, H.; Müller-Tidow, C.; Choudhary, C.; McMahon, M.; et al. Constitutive Activation of Akt by Flt3 Internal Tandem Duplications Is Necessary for Increased Survival, Proliferation, and Myeloid Transformation. Cancer Res. 2005, 65, 9643–9650. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, D.; Nogami, A.; Okada, K.; Akiyama, H.; Umezawa, Y.; Miura, O. FLT3-ITD Activates RSK1 to Enhance Proliferation and Survival of AML Cells by Activating mTORC1 and eIF4B Cooperatively with PIM or PI3K and by Inhibiting Bad and BIM. Cancers 2019, 11, 1827. [Google Scholar] [CrossRef] [Green Version]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Levis, M. FLT3 mutations in acute myeloid leukemia: What is the best approach in 2013? Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 220–226. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Waterfield, M. Signaling by Distinct Classes of Phosphoinositide 3-Kinases. Exp. Cell Res. 1999, 253, 239–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, Y.; Ohnishi, H.; Kitanaka, A.; Ishida, T.; Tanaka, T. Constitutive activation of PI3K is involved in the spontaneous proliferation of primary acute myeloid leukemia cells: Direct evidence of PI3K activation. Leukemia 2004, 18, 1438–1440. [Google Scholar] [CrossRef] [PubMed]
- Billottet, C.; Grandage, V.L.; Gale, R.E.; Quattropani, A.; Rommel, C.; Vanhaesebroeck, B.; Khwaja, A. A selective inhibitor of the p110delta isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene 2006, 25, 6648–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Simpson, S.-E.; Scialla, T.J.; Bagg, A.; Carroll, M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood 2003, 102, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Tamburini, J.; Elie, C.; Bardet, V.; Chapuis, N.; Park, S.; Broët, P.; Cornillet-Lefebvre, P.; Lioure, B.; Ugo, V.; Blanchet, O.; et al. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood 2007, 110, 1025–1028. [Google Scholar] [CrossRef] [Green Version]
- Gallay, N.; Dos Santos, C.; Cuzin, L.; Bousquet, M.; Gouy, V.S.; Chaussade, C.; Attal, M.; Payrastre, B.; Demur, C.; Récher, C. The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 2009, 23, 1029–1038. [Google Scholar] [CrossRef] [Green Version]
- Récher, C.; Dos Santos, C.; Demur, C.; Payrastre, B. mTOR, A New Therapeutic Target in Acute Myeloid Leukemia. Cell Cycle 2005, 4, 1540–1549. [Google Scholar]
- Tamburini, J.; Chapuis, N.; Bardet, V.; Park, S.; Sujobert, P.; Willems, L.; Ifrah, N.; Dreyfus, F.; Mayeux, P.; Lacombe, C.; et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: Rationale for therapeutic inhibition of both pathways. Blood 2008, 111, 379–382. [Google Scholar] [CrossRef] [Green Version]
- Chapuis, N.; Tamburini, J.; Cornillet-Lefebvre, P.; Gillot, L.; Bardet, V.; Willems, L.; Park, S.; Green, A.S.; Ifrah, N.; Dreyfus, F.; et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: Therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica 2010, 95, 415–423. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Chambard, J.C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Kodaki, T.; Woscholski, R.; Hallberg, B.; Downward, J.; Parker, P.J. The activation of phosphatidylinositol 3-kinase by Ras. Curr. Biol. 1994, 4, 798–806. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-Oh Kinase as a Direct Target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Suire, S.; Hawkins, P.; Stephens, L. Activation of phosphoinositide 3-kinase gamma by Ras. Curr. Biol. 2002, 12, 1068–1075. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Carriere, A.; Romeo, Y.; Acosta-Jaquez, H.A.; Moreau, J.; Bonneil, E.; Thibault, P.; Fingar, D.C.; Roux, P.P. ERK1/2 Phosphorylate Raptor to Promote Ras-dependent Activation of mTOR Complex 1 (mTORC1). J. Biol. Chem. 2011, 286, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Carnevale, J.; Ross, L.; Puissant, A.; Banerji, V.; Stone, R.M.; DeAngelo, D.J.; Ross, K.N.; Stegmaier, K. SYK regulates mTOR signaling in AML. Leukemia 2013, 27, 2118–2128. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Cox, D. Syk Regulates Multiple Signaling Pathways Leading to CX3CL1 Chemotaxis in Macrophages. J. Biol. Chem. 2011, 286, 14762–14769. [Google Scholar] [CrossRef] [Green Version]
- Bartaula-Brevik, S.; Brattås, M.K.L.; Tvedt, T.H.A.; Reikvam, H.; Bruserud, O. Splenic tyrosine kinase (SYK) inhibitors and their possible use in acute myeloid leukemia. Expert Opin. Investig. Drugs 2018, 27, 377–387. [Google Scholar] [CrossRef]
- Krisenko, M.O.; Geahlen, R. Calling in SYK: SYK’s dual role as a tumor promoter and tumor suppressor in cancer. Biochim. Biophys. Acta 2015, 1853, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puissant, A.; Fenouille, N.; Alexe, G.; Pikman, Y.; Bassil, C.F.; Mehta, S.; Du, J.; Kazi, J.U.; Luciano, F.; Rönnstrand, L.; et al. SYK is a critical regulator of FLT3 in acute myeloid leukemia. Cancer Cell 2014, 25, 226–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R. Central role of PI3K–SYK interaction in fibrinogen-induced lamellipodia and filopodia formation in platelets. FEBS Open Bio 2016, 6, 1285–1296. [Google Scholar] [CrossRef]
- Reikvam, H.; Nepstad, I.; Tamburini, J. Predicting effects of kinase inhibitor in therapy for myeloid malignancies – the challenges in capturing disease heterogeneity. Expert Opin. Investig. Drugs 2013, 22, 1365–1370. [Google Scholar] [CrossRef] [Green Version]
- Fransecky, L.; Mochmann, L.H.; Baldus, C.D. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol. Cell. Ther. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Chapuis, N.; Tamburini, J.; Bardet, V.; Cornillet-Lefebvre, P.; Willems, L.; Green, A.; Mayeux, P.; Lacombe, C.; Bouscary, D. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica 2010, 95, 819–828. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Thompson, J.E.; Carroll, M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood 2005, 106, 4261–4268. [Google Scholar] [CrossRef] [Green Version]
- Perl, A.E.; Kasner, M.T.; Tsai, D.E.; Vogl, D.T.; Loren, A.W.; Schuster, S.J.; Porter, D.L.; Stadtmauer, E.A.; Goldstein, S.C.; Frey, N.V.; et al. A Phase I Study of the Mammalian Target of Rapamycin Inhibitor Sirolimus and MEC Chemotherapy in Relapsed and Refractory Acute Myelogenous Leukemia. Clin. Cancer Res. 2009, 15, 6732–6739. [Google Scholar] [CrossRef] [Green Version]
- Yee, K.W.; Zeng, Z.; Konopleva, M.; Verstovsek, S.; Ravandi, F.; Ferrajoli, A.; Thomas, D.; Wierda, W.; Apostolidou, E.; Albitar, M.; et al. Phase I/II Study of the Mammalian Target of Rapamycin Inhibitor Everolimus (RAD001) in Patients with Relapsed or Refractory Hematologic Malignancies. Clin. Cancer Res. 2006, 12, 5165–5173. [Google Scholar] [CrossRef] [Green Version]
- Rizzieri, D.A.; Feldman, E.; DiPersio, J.F.; Gabrail, N.; Stock, W.; Strair, R.; Rivera, V.M.; Albitar, M.; Bedrosian, C.L.; Giles, F.J. A Phase 2 Clinical Trial of Deforolimus (AP23573, MK-8669), a Novel Mammalian Target of Rapamycin Inhibitor, in Patients with Relapsed or Refractory Hematologic Malignancies. Clin. Cancer Res. 2008, 14, 2756–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, A.; Mayerhofer, M.; Herndlhofer, S.; Knoebl, P.; Sillaber, C.; Sperr, W.R.; Jaeger, U.; Valent, P. Evaluation of in vivo antineoplastic effects of rapamycin in patients with chemotherapy-refractory AML. Eur. J. Intern. Med. 2009, 20, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chapuis, N.; Marcoux, F.S.; Récher, C.; Prebet, T.; Chevallier, P.; Cahn, J.-Y.; Leguay, T.; Bories, P.; Witz, F.; et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia 2013, 27, 1479–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadori, S.; Stasi, R.; Martelli, A.M.; Venditti, A.; Meloni, G.; Pane, F.; Martinelli, G.; Lunghi, M.; Pagano, L.; Cilloni, D.; et al. Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients with acute myeloid leukaemia: Results of a phase II GIMEMA study (AML-1107). Br. J. Haematol. 2012, 156, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.A.; Breuleux, M. Optimal targeting of the mTORC1 kinase in human cancer. Curr. Opin. Cell Biol. 2009, 21, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.A.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a Dual PI3K/mTOR Inhibitor, Prevents PI3K Signaling and Inhibits the Growth of Cancer Cells with Activating PI3K Mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar] [CrossRef] [Green Version]
- Wunderle, L.; Badura, S.; Lang, F.; Wolf, A.; Schleyer, E.; Serve, H.; Goekbuget, N.; Pfeifer, H.; Bug, G.; Ottmann, O.G. Safety and Efficacy Of BEZ235, a Dual PI3-Kinase /mTOR Inhibitor, In Adult Patients With Relapsed Or Refractory Acute Leukemia: Results Of a Phase I Study. Blood 2013, 122, 2675. [Google Scholar] [CrossRef]
- Raynaud, F.; Eccles, S.; Clarke, P.A.; Hayes, A.; Nutley, B.; Alix, S.; Henley, A.; Di-Stefano, F.; Ahmad, Z.; Guillard, S.; et al. Pharmacologic Characterization of a Potent Inhibitor of Class I Phosphatidylinositide 3-Kinases. Cancer Res. 2007, 67, 5840–5850. [Google Scholar] [CrossRef] [Green Version]
- Pongas, G.; Fojo, T. BEZ235: When Promising Science Meets Clinical Reality. Oncologist 2016, 21, 1033–1034. [Google Scholar] [CrossRef] [Green Version]
- Gojo, I.; Perl, A.; Luger, S.; Baer, M.R.; Norsworthy, K.J.; Bauer, K.S.; Tidwell, M.; Fleckinger, S.; Carroll, M.; Sausville, E.A. Phase I study of UCN-01 and perifosine in patients with relapsed and refractory acute leukemias and high-risk myelodysplastic syndrome. Investig. New Drugs 2013, 31, 1217–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampath, D.; Malik, A.; Plunkett, W.; Nowak, B.; Williams, B.; Burton, M.; Verstovsek, S.; Faderl, S.; Garcia-Manero, G.; List, A.F.; et al. Phase I clinical, pharmacokinetic, and pharmacodynamic study of the Akt-inhibitor triciribine phosphate monohydrate in patients with advanced hematologic malignancies. Leuk. Res. 2013, 37, 1461–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herschbein, L.; Liesveld, J.L. Dueling for dual inhibition: Means to enhance effectiveness of PI3K/Akt/mTOR inhibitors in AML. Blood Rev. 2017, 32, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.; Nimer, S. Recent advances in the understanding and treatment of acute myeloid leukemia. F1000Research 2018, 7, 1196. [Google Scholar] [CrossRef] [Green Version]
- Geiger, T.L.; Rubnitz, J.E. New approaches for the immunotherapy of acute myeloid leukemia. Discov. Med. 2015, 19, 275–284. [Google Scholar]
- Liu, D.; Mamorska-Dyga, A. Syk inhibitors in clinical development for hematological malignancies. J. Hematol. Oncol. 2017, 10, 145. [Google Scholar] [CrossRef] [Green Version]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [Green Version]
- Maiti, A.; Naqvi, K.; Kadia, T.; Borthakur, G.; Takahashi, K.; Bose, P.; Daver, N.; Patel, A.; Alvarado, Y.; Ohanian, M.; et al. Phase II Trial of MEK Inhibitor Binimetinib (MEK162) in RAS-mutant Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2019, 19, 142–148.e1. [Google Scholar] [CrossRef]
- Bose, P.; Grant, S. Rational Combinations of Targeted Agents in AML. J. Clin. Med. 2015, 4, 634–664. [Google Scholar] [CrossRef]
- Reikvam, H.; Tamburini, J.; Skrede, S.; Holdhus, R.; Poulain, L.; Ersvaer, E.; Hatfield, K.; Bruserud, Ø.; Ersvær, E. Antileukaemic effect of PI3K-mTOR inhibitors in acute myeloid leukaemia-gene expression profiles reveal CDC25B expression as determinate of pharmacological effect. Br. J. Haematol. 2014, 164, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filì, C.; Candoni, A.; Zannier, M.E.; Olivieri, J.; Imbergamo, S.; Caizzi, M.; Nadali, G.; Di Bona, E.; Ermacora, A.; Gottardi, M.; et al. Efficacy and toxicity of Decitabine in patients with acute myeloid leukemia (AML): A multicenter real-world experience. Leuk. Res. 2019, 76, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Dinardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uy, G.L.; Rettig, M.P.; Stone, R.M.; Konopleva, M.Y.; Andreeff, M.; McFarland, K.; Shannon, W.; Fletcher, T.R.; Reineck, T.; Eades, W.; et al. A phase 1/2 study of chemosensitization with plerixafor plus G-CSF in relapsed or refractory acute myeloid leukemia. Blood Cancer J. 2017, 7, e542. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Dinardo, C.D.; Stein, E.M.; De Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib inIDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Stein, E.M.; Dinardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study | mTOR Small-Molecule Inhibitor | Patients | Treatment | Summary of Results | Toxicity/ Major Side Affects |
|---|---|---|---|---|---|
| Rizzieri et al [122] | Ridaforolimus (also known as AP23573, MK-8669, or Deforolimus) | 55 patients, 23 patients with AML and three with other myeloid malignancies | Ridaforolimus 12.5 mg intravenous infusion for 5 days every 2 weeks | No complete remissions (CR) or partial remissions ( PR) Stable disease for a minority of patients | Mouth sores Fatigue Nausea Thrombocytopenia |
| Perl et al [120] | Sirolimus (also known as Rapamycin) | 29 patients with refractory or relapsed AML | Sirolimus in a 12 mg loading dose on day 1 followed by 4 mg/d on days 2 to 7, in parallel with chemotherapy. | CR or PR in 6 (22%) of the 27 patients who completed chemotherapy | Marrow aplasia Multi organ failure |
| Park et al. [124] | Everolimus (also known as RAD001) | 28 AML patients below 65 years of age in first relapse. | Everolimus in increasing doses from 10 to 70 mg, administrated orally on days 1 and 7 in combination with conventional 3 + 7 daunorubicin + cytarabine induction therapy. | CR in 68% of patients. Subsequent intensification with allogeneic stem cell transplantation in 29% of patients | Gastrointestinal Respiratory |
| Amadori et al. [125] | Temsirolimus (also known as CCI-779) | 53 patients with primary refractory or first relapse AML | Clofarabine 20 mg/m2 on days 1–5 and temsirolimus 25 mg on days 1, 8, and 15 If CR or CRi- monthly temsirolimus maintenance therapy | CR in 8% of patients CRi in 13% of patients Median DFS 3.5 months. Median OS 4 months (9.1 months for responders) | Infectious complications Febrile neutropenia Transaminitis |
| Targets | Potential Agents | Potential Advantages in Combination with PI3K-Akt-mTOR Inhibitors | Key References |
|---|---|---|---|
| DNA methylation | Azacitidine, decitabine | Potential synergism through the increase of Akt suppression and the promotion of mTOR inhibitor expression such as PTEN | [141,142] |
| BCL-2 | Venetoclax | Potential to inhibit AML cell growth | [143] |
| SYK | Fostamatinib | As SYK cross-reacts with the PI3K-Akt-mTOR, it may be a more broadly applicable therapeutic strategy | [136] |
| MEK | Binimetinib | Inhibition of both Ras-Raf-MEK-ERK and PI3K-Akt-mTOR pathways and their crosstalk can decrease signaling activity in both pathways, especially in RAS mutated cases | [138] |
| CXCR4/CXC12 | Plerixafor | CXCR4 antagonist can lead to sensitization for both conventional chemotherapy and signaling cascade inhibitors | [144] |
| FLT3 | Midostaurin, gilteritinib | Dual inhibition of FLT3 activation and downstream intracellular targets may potentially have synergistic effects, especially in FLT3 mutated cases | [145,146] |
| CD33 | Gemtuzumab ozogamicin | Inhibition of extracellular binding and signaling can potentiate the effect of PI3K-Akt-mTOR inhibition | [147] |
| IDH1 | Ivosidenib | Potentiates the alterations in metabolism associated with PI3K-Akt-mTOR, especially in IDH1 mutated cases | [148] |
| IDH2 | Enasidenib | Potentiates the alterations in metabolism associated with PI3K-Akt-mTOR, especially in IDH2 mutated cases | [149] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082907
Nepstad I, Hatfield KJ, Grønningsæter IS, Reikvam H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. International Journal of Molecular Sciences. 2020; 21(8):2907. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082907
Chicago/Turabian StyleNepstad, Ina, Kimberley Joanne Hatfield, Ida Sofie Grønningsæter, and Håkon Reikvam. 2020. "The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells" International Journal of Molecular Sciences 21, no. 8: 2907. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082907