Appraising the Genetic Architecture of Kernel Traits in Hexaploid Wheat Using GWAS

 ,
,
Abstract
:1. Introduction
2. Results
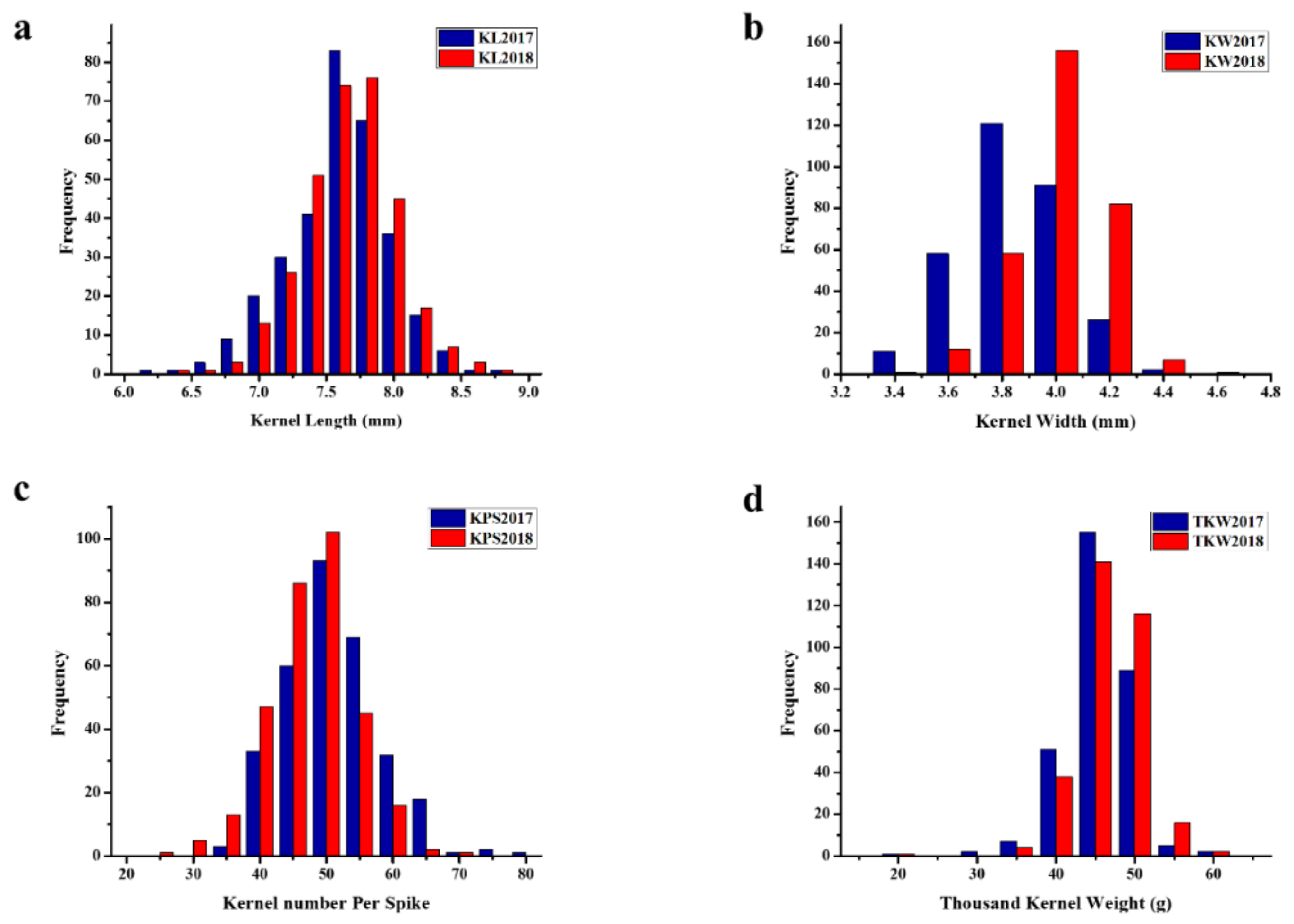
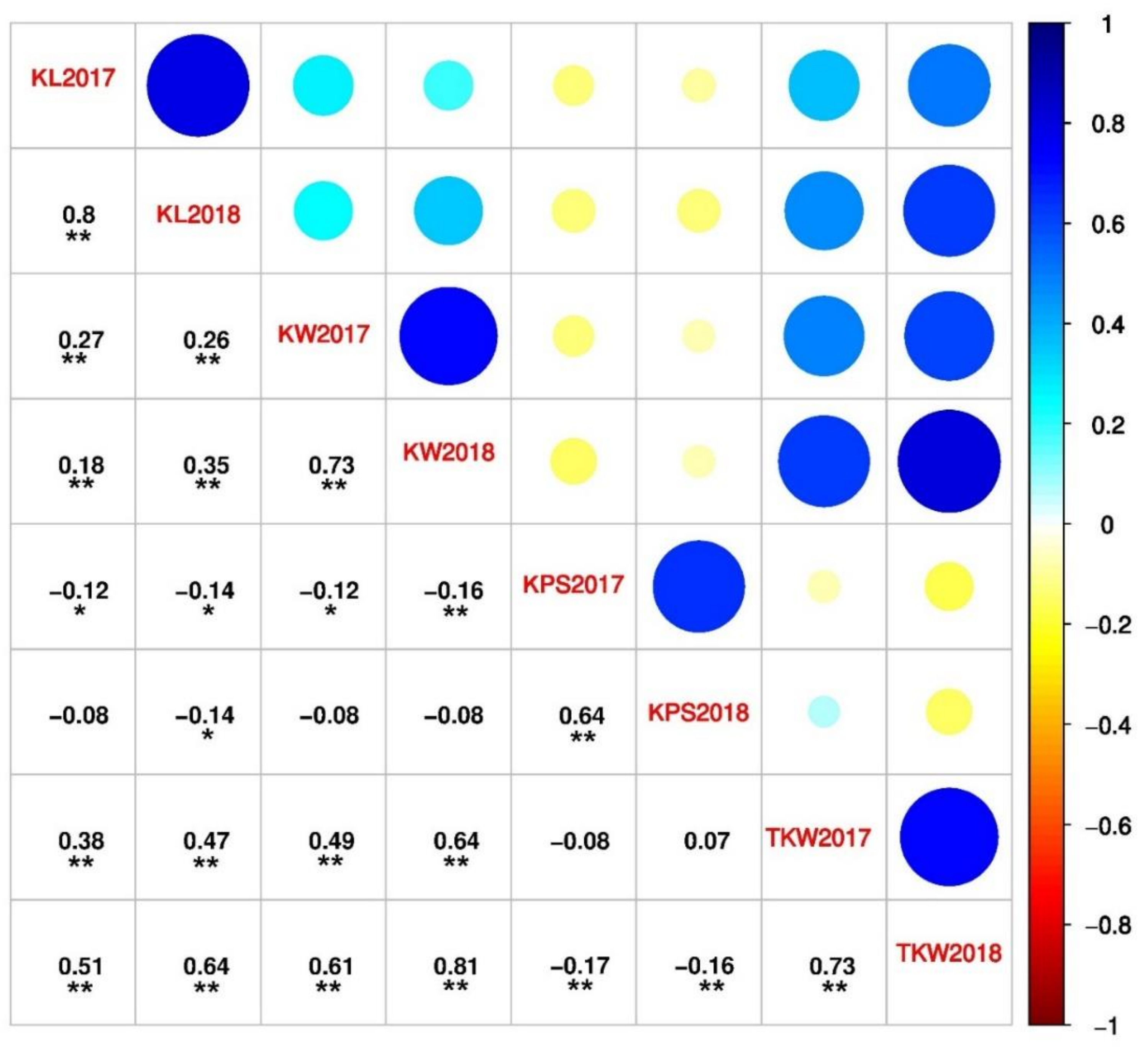
2.1. Phenotypic Evaluation, Correlation, and Heritability of Kernel Traits
2.2. Population Structure and LD Analysis
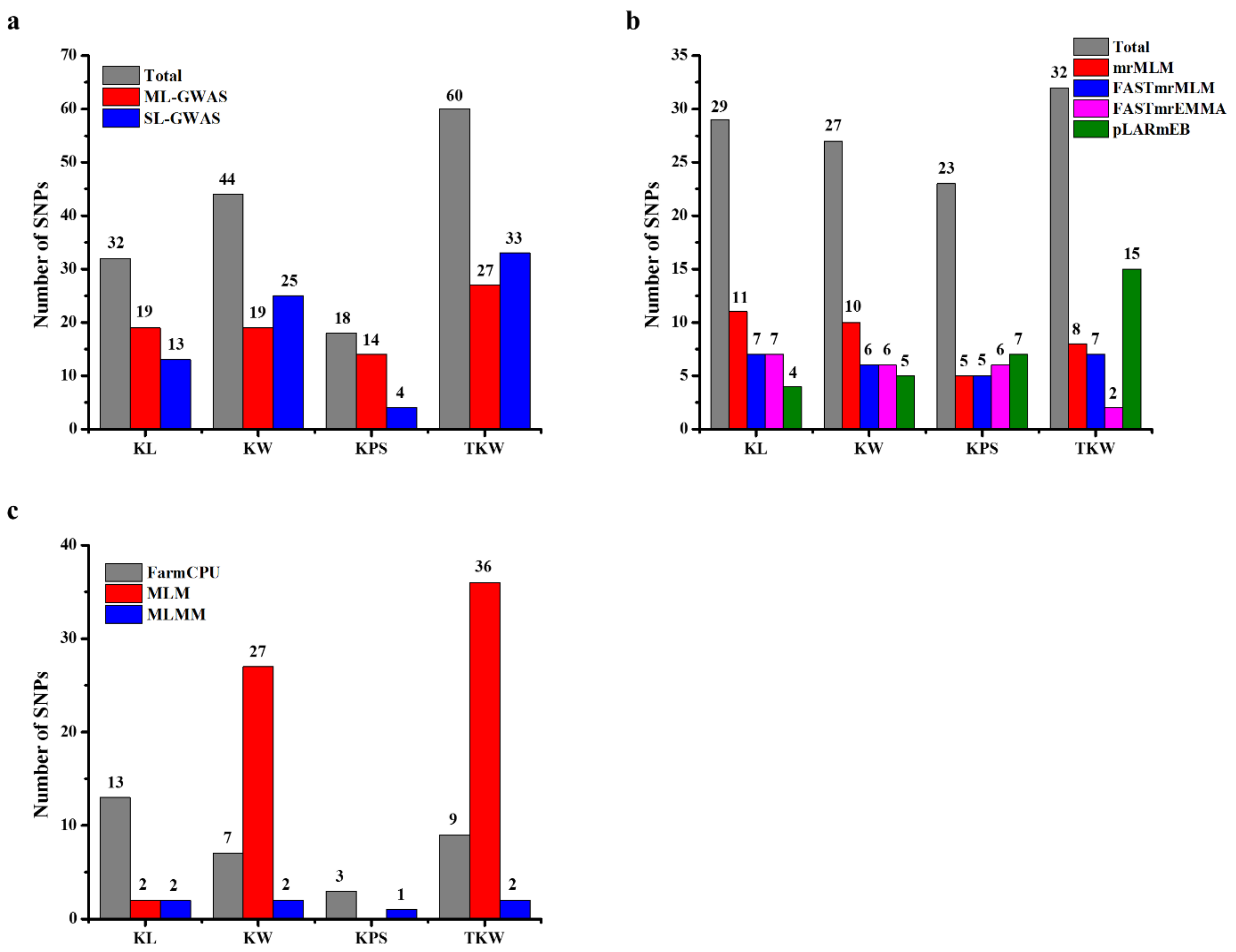
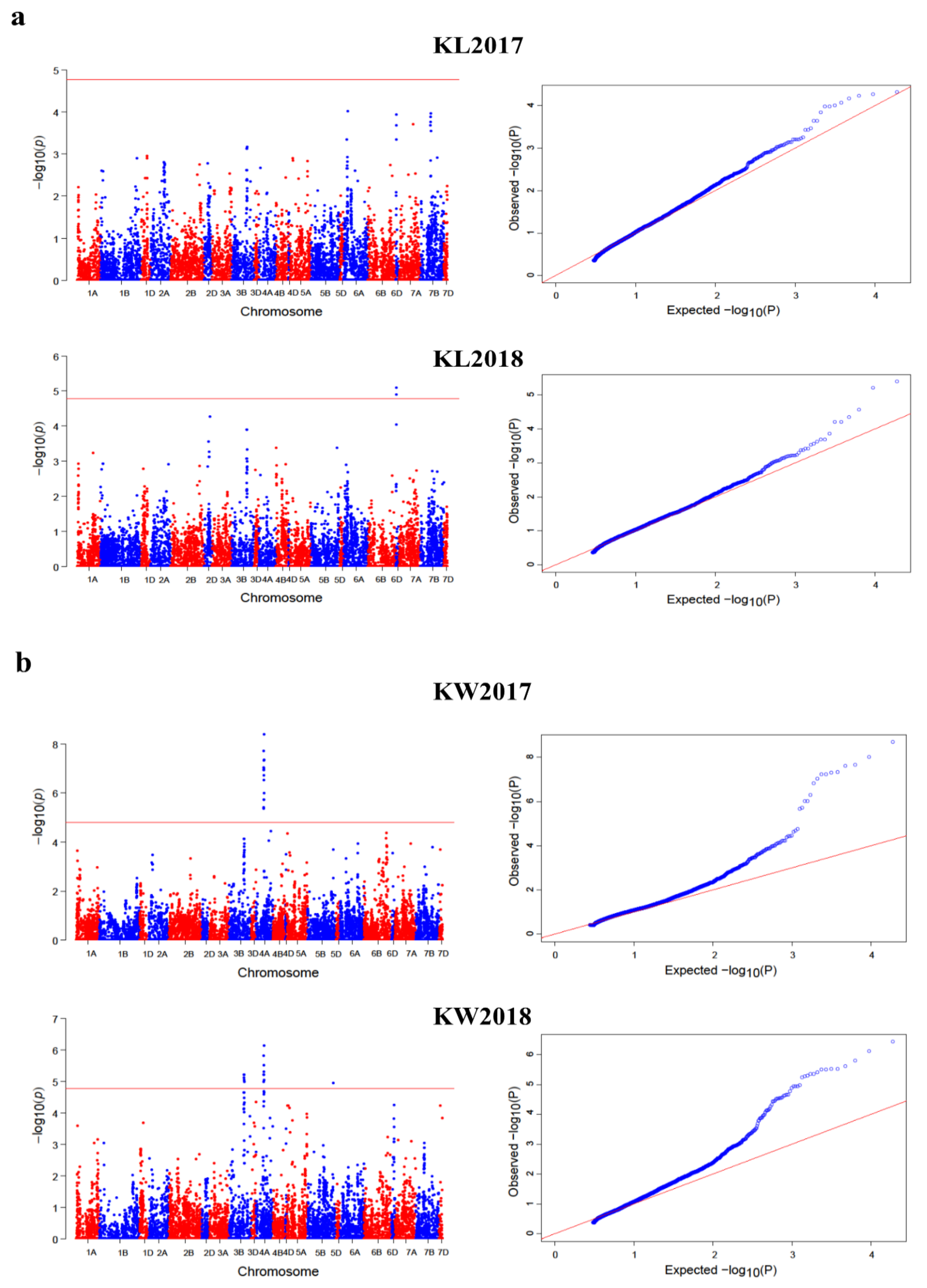
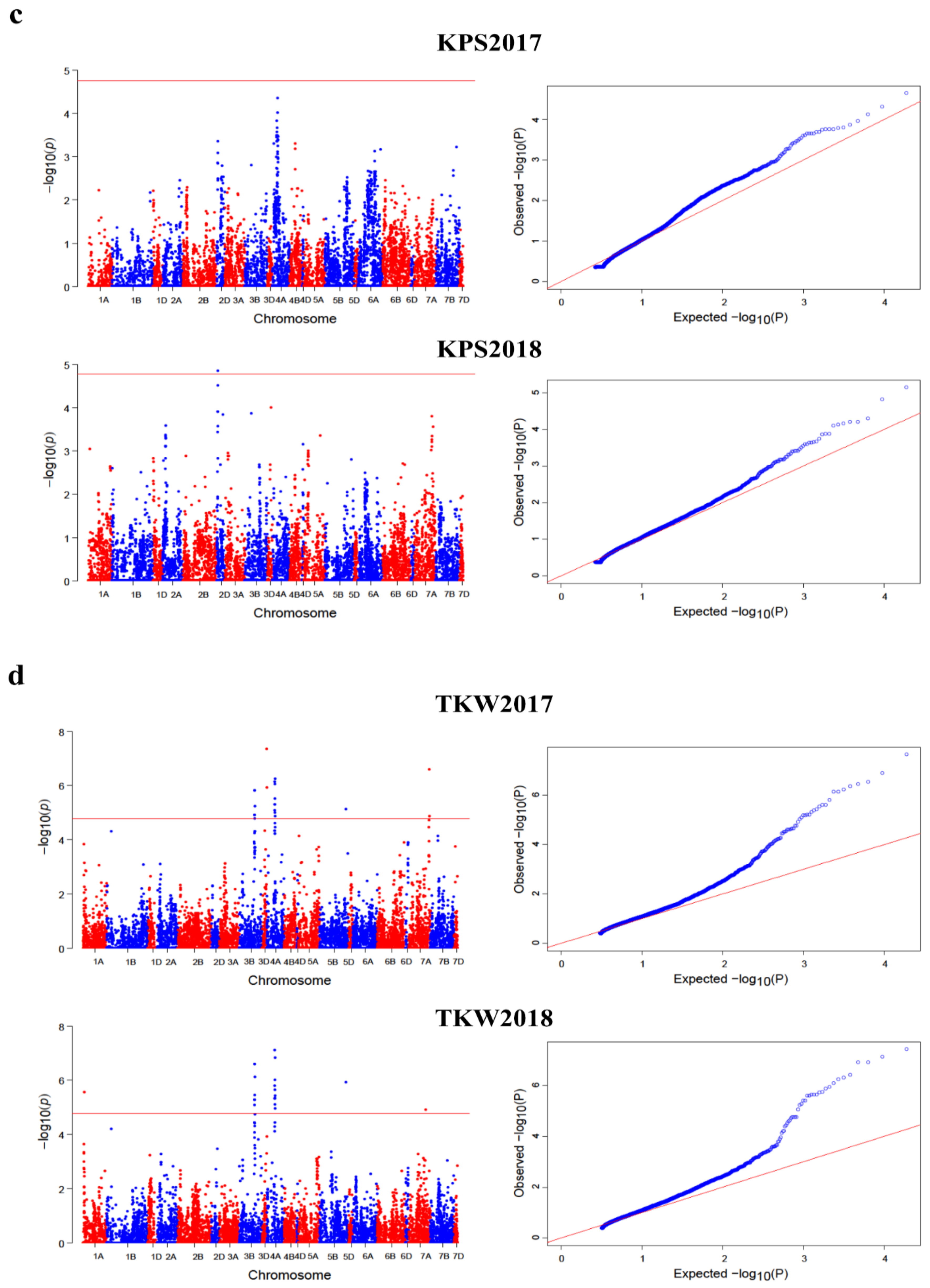
2.3. GWAS for Kernel Traits
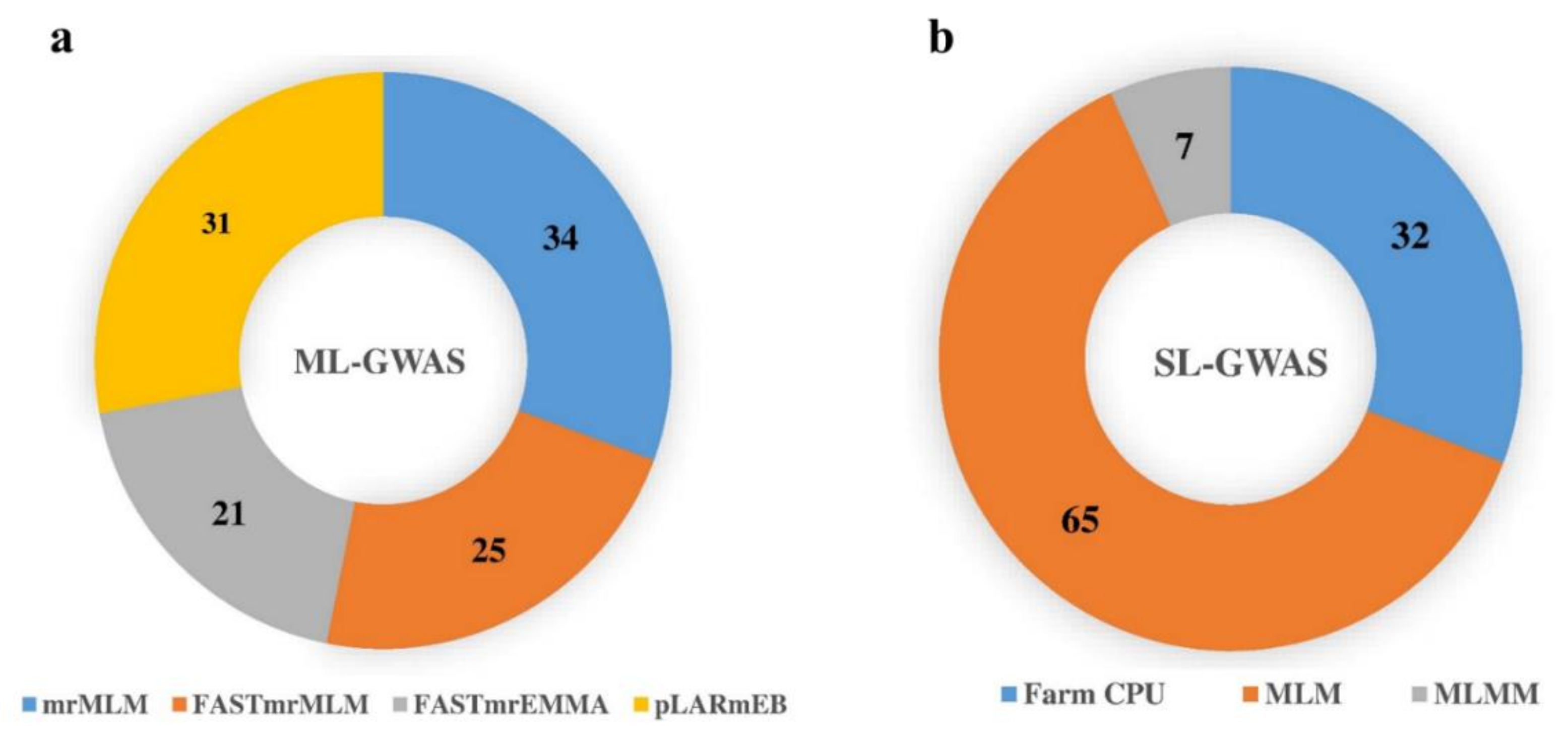
2.4. Loci Detected by Multi-Locus GWAS Methods
2.5. Loci Detected by Single-Locus GWAS Methods
2.6. SNPs Associated with More than One Trait
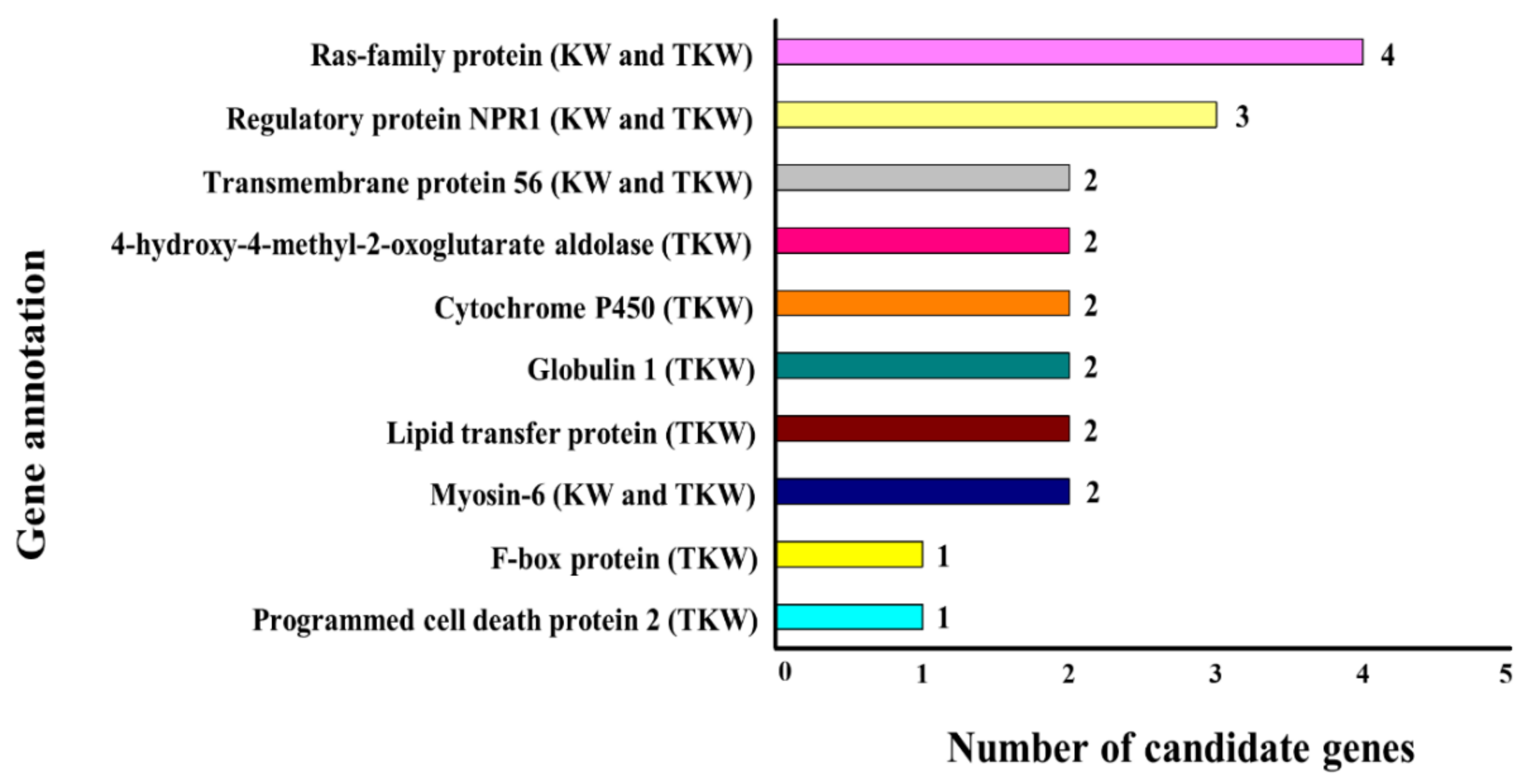
2.7. Mining of Candidate Genes
3. Discussion
3.1. Phenotypic Observations of Kernel Traits
3.2. GWAS Using High-Throughput Genotyping
3.3. Comparison of Present GWAS Results with Previous Studies
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotyping
4.3. Genotyping
4.4. Statistical Analysis
4.5. Population Structure and Kinship Analysis
4.6. Genome-Wide Association Studies
4.7. Candidate Gene Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GWAS | Genome-wide association study |
| ML | Multi-locus |
| SL | Single-locus |
| SNP | Single nucleotide polymorphism |
| QTL | Quantitative trait loci |
| KL | Kernel length |
| KW | Kernel width |
| KPS | Kernel per spike |
| TKW | Thousand kernel weight |
| MAF | Minor allele frequency |
| HWE | Hardy–Weinberg equilibrium |
| ANOVA | Analysis of variance |
| LD | Linkage disequilibrium |
| GAPIT | Genomic association and prediction integrated tool |
| MTA | Marker-trait associations |
| CV | Coefficient of variation |
References
- Muhammad, A.; Hao, H.; Xue, Y.; Alam, A.; Bai, S.; Hu, W.; Sajid, M.; Hu, Z.; Samad, R.A.; Li, Z.; et al. Survey of wheat straw stem characteristics for enhanced resistance to lodging. Cellulose 2020, 27, 2469–2484. [Google Scholar] [CrossRef]
- Zhang, D.; Zhou, Y.; Zhao, X.; Lv, L.; Zhang, C.; Li, J.; Sun, G.; Li, S.; Song, C. Development and utilization of introgression lines using synthetic octaploid wheat (aegilops tauschii× hexaploid wheat) as Donor. Front. Plant Sci. 2018, 9, 1113. [Google Scholar] [CrossRef] [PubMed]
- Bruinsma, J. The resources outlook: By how much do land, water and crop yields need to increase by 2050? In Looking Ahead in World Food and Agriculture: Perspectives to 2050; FAO: Rome, Italy, 2011; pp. 233–278. [Google Scholar]
- Quarrie, S.; Pekic Quarrie, S.; Radosevic, R.; Rancic, D.; Kaminska, A.; Barnes, J.; Leverington, M.; Ceoloni, C.; Dodig, D. Dissecting a wheat QTL for yield present in a range of environments: From the QTL to candidate genes. J. Exp. Bot. 2006, 57, 2627–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.Y.; Liu, D.C.; Guo, X.L.; Yang, W.L.; Sun, J.Z.; Wang, D.W.; Zhang, A. Genomic distribution of quantitative trait loci for yield and yield-related traits in common wheat. J. Integr. Plant Biol. 2010, 52, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Sorrells, M.E. Three-dimensional seed size and shape QTL in hexaploid wheat (Triticum aestivum L.) populations. Crop Sci. 2014, 54, 98–110. [Google Scholar] [CrossRef]
- Valluru, R.; Reynolds, M.P.; Salse, J. Genetic and molecular bases of yield-associated traits: A translational biology approach between rice and wheat. Theor. Appl. Genet. 2014, 127, 1463–1489. [Google Scholar] [CrossRef]
- Su, Z.; Hao, C.; Wang, L.; Dong, Y.; Zhang, X. Identification and development of a functional marker of TaGW2 associated with grain weight in bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2011, 122, 211–223. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, J.; Shi, C.; Chen, J.; Zheng, F.; Tian, J. Function of TaGW2-6A and its effect on grain weight in wheat (Triticum aestivum L.). Euphytica 2013, 192, 347–357. [Google Scholar] [CrossRef]
- Jaiswal, V.; Gahlaut, V.; Mathur, S.; Agarwal, P.; Khandelwal, M.K.; Khurana, J.P.; Tyagi, A.K.; Balyan, H.S.; Gupta, P.K. Identification of novel SNP in promoter sequence of TaGW2-6A associated with grain weight and other agronomic traits in wheat (Triticum aestivum L.). PLoS ONE 2015, 10, e0129400. [Google Scholar] [CrossRef] [Green Version]
- Heidari, B.; Sayed-Tabatabaei, B.E.; Saeidi, G.; Kearsey, M.; Suenaga, K. Mapping QTL for grain yield, yield components, and spike features in a doubled haploid population of bread wheat. Genome 2011, 54, 517–527. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J.; Li, R.; Deng, Z.; Zhang, K.; Liu, B.; Tian, J. Conditional QTL mapping of three yield components in common wheat (Triticum aestivum L.). Crop J. 2016, 4, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Hao, C.; Zhang, Y.; Zhang, B.; Cheng, X.; Qin, L.; Li, T.; Shi, W.; Chang, X.; Jing, R. Association and validation of yield-favored alleles in chinese cultivars of common wheat (Triticum aestivum L.). PLoS ONE 2015, 10, e0130029. [Google Scholar]
- Guo, J.; Zhang, Y.; Shi, W.; Zhang, B.; Zhang, J.; Xu, Y.; Cheng, X.; Cheng, K.; Zhang, X.; Hao, C. Association analysis of grain-setting rates in apical and basal spikelets in bread wheat (Triticum aestivum L.). Front. Plant Sci. 2015, 6, 1029. [Google Scholar] [CrossRef]
- Cuthbert, J.L.; Somers, D.J.; Brûlé-Babel, A.L.; Brown, P.D.; Crow, G.H. Molecular mapping of quantitative trait loci for yield and yield components in spring wheat (Triticum aestivum L.). Theor. Appl. Genet. 2008, 117, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Li, J.; Ding, A.; Zhao, C.; Wang, L.; Wang, X.; Li, S.; Bao, Y.; Li, X.; Feng, D. Conditional QTL mapping for plant height with respect to the length of the spike and internode in two mapping populations of wheat. Theor. Appl. Genet. 2011, 122, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Zhao, C.; Ding, A.; Li, J.; Wang, L.; Li, X.; Bao, Y.; Li, J.; Wang, H. Construction of an integrative linkage map and QTL mapping of grain yield-related traits using three related wheat RIL populations. Theor. Appl. Genet. 2014, 127, 659–675. [Google Scholar] [CrossRef]
- Edae, E.A.; Byrne, P.F.; Haley, S.D.; Lopes, M.S.; Reynolds, M.P. Genome-wide association mapping of yield and yield components of spring wheat under contrasting moisture regimes. Theor. Appl. Genet. 2014, 127, 791–807. [Google Scholar] [CrossRef]
- Azadi, A.; Mardi, M.; Hervan, E.M.; Mohammadi, S.A.; Moradi, F.; Tabatabaee, M.T.; Pirseyedi, S.M.; Ebrahimi, M.; Fayaz, F.; Kazemi, M. QTL mapping of yield and yield components under normal and salt-stress conditions in bread wheat (Triticum aestivum L.). Plant Mol. Biol. Rep. 2015, 33, 102–120. [Google Scholar] [CrossRef]
- Li, F.; Wen, W.; Liu, J.; Zhang, Y.; Cao, S.; He, Z.; Rasheed, A.; Jin, H.; Zhang, C.; Yan, J. Genetic architecture of grain yield in bread wheat based on genome-wide association studies. BMC Plant Biol. 2019, 19, 168. [Google Scholar] [CrossRef]
- Tsai, H.-Y.; Janss, L.L.; Andersen, J.R.; Orabi, J.; Jensen, J.D.; Jahoor, A.; Jensen, J. Genomic prediction and GWAS of yield, quality and disease-related traits in spring barley and winter wheat. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, A.M.; Barker, G.L.; Berry, S.T.; Coghill, J.A.; Gwilliam, R.; Kirby, S.; Robinson, P.; Brenchley, R.C.; D’Amore, R.; McKenzie, N. Transcript-specific, single-nucleotide polymorphism discovery and linkage analysis in hexaploid bread wheat (Triticum aestivum L.). Plant Biotechnol. J. 2011, 9, 1086–1099. [Google Scholar] [CrossRef] [Green Version]
- Cavanagh, C.R.; Chao, S.; Wang, S.; Huang, B.E.; Stephen, S.; Kiani, S.; Forrest, K.; Saintenac, C.; Brown-Guedira, G.L.; Akhunova, A. Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc. Natl. Acad. Sci. USA 2013, 110, 8057–8062. [Google Scholar] [CrossRef] [Green Version]
- Winfield, M.O.; Allen, A.M.; Burridge, A.J.; Barker, G.L.; Benbow, H.R.; Wilkinson, P.A.; Coghill, J.; Waterfall, C.; Davassi, A.; Scopes, G. High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol. J. 2016, 14, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Zhang, N.; Fan, X.-l.; Zhang, W.; Zhao, C.-h.; Yang, L.-j.; Pan, R.-q.; Chen, M.; Han, J.; Zhao, X.-q. Utilization of a Wheat660K SNP array-derived high-density genetic map for high-resolution mapping of a major QTL for kernel number. Sci. Rep. 2017, 7, 1–12. [Google Scholar]
- Jin, H.; Wen, W.; Liu, J.; Zhai, S.; Zhang, Y.; Yan, J.; Liu, Z.; Xia, X.; He, Z. Genome-wide QTL mapping for wheat processing quality parameters in a Gaocheng 8901/Zhoumai 16 recombinant inbred line population. Front. Plant Sci. 2016, 7, 1032. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; He, Z.; Rasheed, A.; Wen, W.; Yan, J.; Zhang, P.; Wan, Y.; Zhang, Y.; Xie, C.; Xia, X. Genome-wide association mapping of black point reaction in common wheat (Triticum aestivum L.). BMC Plant Biol. 2017, 17, 220. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Zhang, F.; Yan, X.; Zhang, X.; Dong, Z.; Cui, D.; Chen, F. Genome-wide association study for 13 agronomic traits reveals distribution of superior alleles in bread wheat from the Yellow and Huai Valley of China. Plant Biotechnol. J. 2017, 15, 953–969. [Google Scholar] [CrossRef] [Green Version]
- Valluru, R.; Reynolds, M.P.; Davies, W.J.; Sukumaran, S. Phenotypic and genome-wide association analysis of spike ethylene in diverse wheat genotypes under heat stress. New Phytol. 2017, 214, 271–283. [Google Scholar] [CrossRef] [Green Version]
- Mu, J.; Wu, J.; Liu, S.; Dai, M.; Sun, D.; Huang, S.; Wang, Q.; Zeng, Q.; Yu, S.; Chen, L. Genome-Wide Linkage Mapping Reveals Stripe Rust Resistance in Common Wheat (Triticum aestivum) Xinong1376. Plant Dis. 2019, 103, 2742–2750. [Google Scholar] [CrossRef]
- Kthiri, D.; Loladze, A.; N’Diaye, A.; Nilsen, K.T.; Walkowiak, S.; Dreisigacker, S.; Ammar, K.; Pozniak, C.J. Mapping of genetic loci conferring resistance to leaf rust from three globally resistant durum wheat sources. Front. Plant Sci. 2019, 10, 1247. [Google Scholar] [CrossRef]
- Zhao, M.; Leng, Y.; Chao, S.; Xu, S.S.; Zhong, S. Molecular mapping of QTL for Fusarium head blight resistance introgressed into durum wheat. Theor. Appl. Genet. 2018, 131, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Assanga, S.O.; Fuentealba, M.; Zhang, G.; Tan, C.; Dhakal, S.; Rudd, J.C.; Ibrahim, A.M.; Xue, Q.; Haley, S.; Chen, J. Mapping of quantitative trait loci for grain yield and its components in a US popular winter wheat TAM 111 using 90K SNPs. PLoS ONE 2017, 12, e0189669. [Google Scholar] [CrossRef]
- Garcia, M.; Eckermann, P.; Haefele, S.; Satija, S.; Sznajder, B.; Timmins, A.; Baumann, U.; Wolters, P.; Mather, D.E.; Fleury, D. Genome-wide association mapping of grain yield in a diverse collection of spring wheat (Triticum aestivum L.) evaluated in southern Australia. PLoS ONE 2019, 14, e0211730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Wang, S.; Wei, W.; Xie, H.; Liu, K.; Zhang, C.; Wu, Z.; Jiang, H.; Cao, J.; Zhao, L. Genome-wide association study of pre-harvest sprouting tolerance using a 90K SNP array in common wheat (Triticum aestivum L.). Theor. Appl. Genet. 2019, 132, 2947–2963. [Google Scholar] [CrossRef]
- Mangini, G.; Gadaleta, A.; Colasuonno, P.; Marcotuli, I.; Signorile, A.M.; Simeone, R.; De Vita, P.; Mastrangelo, A.M.; Laidò, G.; Pecchioni, N. Genetic dissection of the relationships between grain yield components by genome-wide association mapping in a collection of tetraploid wheats. PLoS ONE 2018, 13, e0190162. [Google Scholar] [CrossRef]
- Gahlaut, V.; Jaiswal, V.; Singh, S.; Balyan, H.; Gupta, P. Multi-Locus Genome Wide Association Mapping for Yield and its contributing traits in Hexaploid Wheat under Different Water Regimes. Sci. Rep. 2019, 9, 1–15. [Google Scholar]
- Pradhan, S.; Babar, M.A.; Robbins, K.; Bai, G.; Mason, R.E.; Khan, J.; Shahi, D.; Avci, M.; Guo, J.; Hossain, M.M. Understanding the Genetic Basis of Spike Fertility to Improve Grain Number, Harvest Index, and Grain Yield in Wheat Under High Temperature Stress Environments. Front. Plant Sci. 2019, 10, 1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Li, J.; Cheng, Y.; Yao, F.; Long, L.; Wang, Y.; Wu, Y.; Li, J.; Wang, J.; Jiang, Q. Genome-wide association study reveals new loci for yield-related traits in Sichuan wheat germplasm under stripe rust stress. BMC Genom. 2019, 20, 640. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Hao, C.; Zhang, Y.; Cheng, J.; Zhang, Z.; Liu, J.; Yi, X.; Cheng, X.; Sun, D.; Xu, Y. A combined association mapping and linkage analysis of kernel number per spike in common wheat (Triticum aestivum L.). Front. Plant Sci. 2017, 8, 1412. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Tamba, C.L.; Ni, Y.-L.; Zhang, Y.-M. Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLoS Comput. Biol. 2017, 13, e1005357. [Google Scholar] [CrossRef]
- Segura, V.; Vilhjálmsson, B.J.; Platt, A.; Korte, A.; Seren, Ü.; Long, Q.; Nordborg, M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 2012, 44, 825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-B.; Feng, J.-Y.; Ren, W.-L.; Huang, B.; Zhou, L.; Wen, Y.-J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.-M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.-J.; Zhang, H.; Ni, Y.-L.; Huang, B.; Zhang, J.; Feng, J.-Y.; Wang, S.-B.; Dunwell, J.M.; Zhang, Y.-M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Feng, J.; Ni, Y.; Wen, Y.; Niu, Y.; Tamba, C.; Yue, C.; Song, Q.; Zhang, Y. pLARmEB: Integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-M.; Tamba, C.L. A fast mrMLM algorithm for multi-locus genome-wide association studies. bioRxiv 2018, 341784. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Mantovani, E.E.; Simsek, S.; Jain, S.; Elias, E.M.; Mergoum, M. Genome wide genetic dissection of wheat quality and yield related traits and their relationship with grain shape and size traits in an elite× non-adapted bread wheat cross. PLoS ONE 2019, 14, e0221826. [Google Scholar] [CrossRef] [PubMed]
- Abdipour, M.; Ebrahimi, M.; Izadi-Darbandi, A.; Mastrangelo, A.M.; Najafian, G.; Arshad, Y.; Mirniyam, G. Association between grain size and shape and quality traits, and path analysis of thousand grain weight in Iranian bread wheat landraces from different geographic regions. Not. Bot. Horti. Agrobot. Cluj Napoca 2016, 44, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.; Munkvold, J.; Sorrells, M. Comparison of digital image analysis using elliptic Fourier descriptors and major dimensions to phenotype seed shape in hexaploid wheat (Triticum aestivum L.). Euphytica 2013, 190, 99–116. [Google Scholar] [CrossRef]
- Gegas, V.C.; Nazari, A.; Griffiths, S.; Simmonds, J.; Fish, L.; Orford, S.; Sayers, L.; Doonan, J.H.; Snape, J.W. A genetic framework for grain size and shape variation in wheat. Plant Cell 2010, 22, 1046–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breseghello, F.; Sorrells, M.E. QTL analysis of kernel size and shape in two hexaploid wheat mapping populations. Field Crops Res. 2007, 101, 172–179. [Google Scholar] [CrossRef]
- Xiao, Y.; He, S.; Yan, J.; Zhang, Y.; Zhang, Y.; Wu, Y.; Xia, X.; Tian, J.; Ji, W.; He, Z. Molecular mapping of quantitative trait loci for kernel morphology traits in a non-1BL. 1RS× 1BL. 1RS wheat cross. Crop Pasture Sci. 2011, 62, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, A.; Xia, X.; Ogbonnaya, F.; Mahmood, T.; Zhang, Z.; Mujeeb-Kazi, A.; He, Z. Genome-wide association for grain morphology in synthetic hexaploid wheats using digital imaging analysis. BMC Plant Biol. 2014, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Wang, Y.; Guo, Y.; Zhao, Y.; Kong, F.; Li, S. Characterization and mapping of QTLs on chromosome 2D for grain size and yield traits using a mutant line induced by EMS in wheat. Crop J. 2015, 3, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, J.; Scott, P.; Brinton, J.; Mestre, T.C.; Bush, M.; Del Blanco, A.; Dubcovsky, J.; Uauy, C. A splice acceptor site mutation in TaGW2-A1 increases thousand grain weight in tetraploid and hexaploid wheat through wider and longer grains. Theor. Appl. Genet. 2016, 129, 1099–1112. [Google Scholar] [CrossRef] [Green Version]
- Ramya, P.; Chaubal, A.; Kulkarni, K.; Gupta, L.; Kadoo, N.; Dhaliwal, H.; Chhuneja, P.; Lagu, M.; Gupt, V. QTL mapping of 1000-kernel weight, kernel length, and kernel width in bread wheat (Triticum aestivum L.). Appl. Genet. 2010, 51, 421–429. [Google Scholar] [CrossRef]
- Basile, S.M.L.; Ramírez, I.A.; Crescente, J.M.; Conde, M.B.; Demichelis, M.; Abbate, P.; Rogers, W.J.; Pontaroli, A.C.; Helguera, M.; Vanzetti, L.S. Haplotype block analysis of an Argentinean hexaploid wheat collection and GWAS for yield components and adaptation. BMC Plant Biol. 2019, 19, 553. [Google Scholar]
- Talini, R.F.; Brandolini, A.; Miculan, M.; Brunazzi, A.; Vaccino, P.; Pè, M.E.; Dell’Acqua, M. Genome wide association study of agronomic and quality traits in a world collection of the wild wheat relative Triticum urartu. Plant J. 2020, 102, 555–568. [Google Scholar] [CrossRef]
- Cheng, B.; Gao, X.; Cao, N.; Ding, Y.; Gao, Y.; Chen, T.; Xin, Z.; Zhang, L. Genome-wide association analysis of stripe rust resistance loci in wheat accessions from southwestern China. J. Appl. Genet. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hu, X.; Shi, T.; Yin, H.; Sun, D.; Yuanfeng, H.; Xia, X.; Luo, J.; Fernie, A.R.; He, Z. Metabolite-based genome-wide association study enables dissection of the flavonoid decoration pathway of wheat kernels. Plant Biotechnol. J. 2020, 10, 1111. [Google Scholar] [CrossRef]
- Ain, Q.-u.; Rasheed, A.; Anwar, A.; Mahmood, T.; Imtiaz, M.; He, Z.; Xia, X.; Quraishi, U.M. Genome-wide association for grain yield under rainfed conditions in historical wheat cultivars from Pakistan. Front. Plant Sci. 2015, 6, 743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Chen, D.; Alqudah, A.M.; Röder, M.S.; Ganal, M.W.; Schnurbusch, T. Genome-wide association analyses of 54 traits identified multiple loci for the determination of floret fertility in wheat. New Phytol. 2017, 214, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Barry, K.; Cao, F.; Zhou, M. Genome-wide association mapping for adult resistance to powdery mildew in common wheat. Mol. Biol. Rep. 2019, 1, 16. [Google Scholar] [CrossRef]
- Muqaddasi, Q.H.; Zhao, Y.; Rodemann, B.; Plieske, J.; Ganal, M.W.; Röder, M.S. Genome-wide association mapping and prediction of adult stage Septoria tritici blotch infection in European winter wheat via high-density marker arrays. Plant Genome 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, F.-P.; Zeng, Q.-D.; Wu, J.-H.; Wang, Q.-L.; Yang, Z.-J.; Liang, B.-P.; Kang, Z.-S.; Chen, X.-H.; Han, D.-J. QTL mapping and validation of adult plant resistance to stripe rust in Chinese wheat landrace Humai 15. Front. Plant Sci. 2018, 9, 968. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, G.; Leng, Y.; Wanjugi, H.; Xi, P.; Grosz, M.D.; Mergoum, M.; Zhong, S. Molecular mapping of Fusarium head blight resistance in the spring wheat line ND2710. Phytopathology 2018, 108, 972–979. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Assanga, S.O.; Dhakal, S.; Gu, X.; Tan, C.-T.; Yang, Y.; Rudd, J.; Hays, D.; Ibrahim, A.; Xue, Q. Validation of chromosomal locations of 90K array single nucleotide polymorphisms in US wheat. Crop Sci. 2016, 56, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Avni, R.; Nave, M.; Eilam, T.; Sela, H.; Alekperov, C.; Peleg, Z.; Dvorak, J.; Korol, A.; Distelfeld, A. Ultra-dense genetic map of durum wheat× wild emmer wheat developed using the 90K iSelect SNP genotyping assay. Mol. Breed. 2014, 34, 1549–1562. [Google Scholar] [CrossRef]
- Sheoran, S.; Jaiswal, S.; Kumar, D.; Raghav, N.; Sharma, R.; Pawar, S.; Paul, S.; Iquebal, M.A.; RAI, A.; Kumar, D. Uncovering genomic regions associated with 36 agro-morphological traits in Indian spring wheat using GWAS. Front. Plant Sci. 2019, 10, 527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, S.; Li, W.; Liu, S.; Li, X.; Fang, Y.; Zhang, J.; Wang, Y.; Xu, S.; Zhang, J. Identification of QTNs controlling seed protein content in soybean using multi-locus genome-wide association studies. Front. Plant Sci. 2018, 9, 1690. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Ma, Q.; Li, M.; Hao, F.; Wang, C. Multi-locus genome-wide association studies of fiber-quality related traits in Chinese early-maturity upland cotton. Front. Plant Sci. 2018, 9, 1169. [Google Scholar] [CrossRef] [Green Version]
- Arora, S.; Singh, N.; Kaur, S.; Bains, N.S.; Uauy, C.; Poland, J.; Chhuneja, P. Genome-wide association study of grain architecture in wild wheat Aegilops tauschii. Front. Plant Sci. 2017, 8, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamil, M.; Ali, A.; Gul, A.; Ghafoor, A.; Napar, A.A.; Ibrahim, A.M.; Naveed, N.H.; Yasin, N.A.; Mujeeb-Kazi, A. Genome-wide association studies of seven agronomic traits under two sowing conditions in bread wheat. BMC Plant Biol. 2019, 19, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-X.; Zhu, Y.-L.; Zhang, D.-X.; Shao, H.; Liu, P.; Hu, J.-B.; Zhang, H.; Zhang, H.-P.; Chang, C.; Lu, J. Genome-wide association study for grain yield and related traits in elite wheat varieties and advanced lines using SNP markers. PLoS ONE 2017, 12, e0188662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Xu, Z.; Fan, X.; Zhou, Q.; Cao, J.; Wang, F.; Ji, G.; Yang, L.; Feng, B.; Wang, T. A genome-wide association study of wheat spike related traits in China. Front. Plant Sci. 2018, 9, 1584. [Google Scholar] [CrossRef]
- Hou, J.; Jiang, Q.; Hao, C.; Wang, Y.; Zhang, H.; Zhang, X. Global selection on sucrose synthase haplotypes during a century of wheat breeding. Plant Physiol. 2014, 164, 1918–1929. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Wang, Q.; Li, Z.; Cheng, H.; Li, Z.; Liu, X.; Song, W.; Appels, R.; Zhao, H. Expression of Ta CYP 78A3, a gene encoding cytochrome P450 CYP 78A3 protein in wheat (Triticum aestivum L.), affects seed size. Plant J. 2015, 83, 312–325. [Google Scholar] [CrossRef]
- Gao, F.; Wen, W.; Liu, J.; Rasheed, A.; Yin, G.; Xia, X.; Wu, X.; He, Z. Genome-wide linkage mapping of QTL for yield components, plant height and yield-related physiological traits in the Chinese wheat cross Zhou 8425B/Chinese Spring. Front. Plant Sci. 2015, 6, 1099. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Zhao, G.; Huang, D.; Jia, J. Candidate loci involved in domestication and improvement detected by a published 90K wheat SNP array. Sci. Rep. 2017, 7, 1–13. [Google Scholar]
- Yang, R.-C.; Peng, F.Y.; Hu, Z. Inferring defense-related gene families in Arabidopsis and wheat. BMC Genom. 2017, 18, 980. [Google Scholar] [CrossRef] [Green Version]
- Zanke, C.D.; Ling, J.; Plieske, J.; Kollers, S.; Ebmeyer, E.; Korzun, V.; Argillier, O.; Stiewe, G.; Hinze, M.; Neumann, F. Analysis of main effect QTL for thousand grain weight in European winter wheat (Triticum aestivum L.) by genome-wide association mapping. Front. Plant Sci. 2015, 6, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Zhao, L.; Ren, Y.; Dong, Z.; Cui, D.; Chen, F. Genome-wide association study revealed that the TaGW8 gene was associated with kernel size in Chinese bread wheat. Sci. Rep. 2019, 9, 1–10. [Google Scholar]
- Consortium, I.W.G.S. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar]
- Bluhm, B.; Zhao, X.; Flaherty, J.; Xu, J.-R.; Dunkle, L. RAS2 regulates growth and pathogenesis in Fusarium graminearum. Mol. Plant Microbe Interact. 2007, 20, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Ramsdale, M. Programmed cell death in pathogenic fungi. BBA-Mol. Cell Res. 2008, 1783, 1369–1380. [Google Scholar] [CrossRef] [Green Version]
- Backer, R.; Naidoo, S.; van den Berg, N. The nonexpressor of pathogenesis-related genes 1 (NPR1) and related Family: Mechanistic insights in plant disease resistance. Front. Plant Sci. 2019, 10, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diethelm, M.; Schmolke, M.; Groth, J.; Friedt, W.; Schweizer, G.; Hartl, L. Association of allelic variation in two NPR1-like genes with Fusarium head blight resistance in wheat. Mol. Breed. 2014, 34, 31–43. [Google Scholar] [CrossRef]
- Walter, S.; Brennan, J.M.; Arunachalam, C.; Ansari, K.I.; Hu, X.; Khan, M.R.; Trognitz, F.; Trognitz, B.; Leonard, G.; Egan, D. Components of the gene network associated with genotype-dependent response of wheat to the Fusarium mycotoxin deoxynivalenol. Func. Integr. Genom. 2008, 8, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Doohan, F. Transcript profiling of the phytotoxic response of wheat to the Fusarium mycotoxin deoxynivalenol. Mycotoxin Res. 2011, 27, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kesari, P.; Dhindwal, S.; Choudhary, A.K.; Katiki, M.; Verma, A.; Ambatipudi, K.; Tomar, S.; Sharma, A.K.; Mishra, G. A novel function for globulin in sequestering plant hormone: Crystal structure of Wrightia tinctoria 11S globulin in complex with auxin. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanabata, T.; Shibaya, T.; Hori, K.; Ebana, K.; Yano, M. SmartGrain: High-throughput phenotyping software for measuring seed shape through image analysis. Plant Physiol. 2012, 160, 1871–1880. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y.; Nguyen, A.T.; Yoshioka, M.; Iehisa, J.C.; Takumi, S. Identification of quantitative trait loci controlling grain size and shape in the D genome of synthetic hexaploid wheat lines. Breed. Sci. 2013, 63, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Liu, M.; Yan, Y.; Qing, C.; Zhang, X.; Zhang, Y.; Long, Y.; Wang, L.; Pan, L.; Zou, C. Genetic dissection of maize embryonic callus regenerative capacity using multi-locus genome-wide association studies. Front. Plant Sci. 2018, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Ren, W.-L.; Wen, Y.-J.; Dunwell, J.M.; Zhang, Y.-M. pKWmEB: Integration of Kruskal–Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study. Heredity 2018, 120, 208–218. [Google Scholar] [CrossRef]
- Li, H.; Peng, Z.; Yang, X.; Wang, W.; Fu, J.; Wang, J.; Han, Y.; Chai, Y.; Guo, T.; Yang, N. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 2013, 45, 43. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2004, 21, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Thrash, A.; Tang, J.D.; DeOrnellis, M.; Peterson, D.G.; Warburton, M.L. PAST: The pathway association studies tool to infer biological meaning from GWAS datasets. Plants 2020, 9, 58. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Methods (1–4) | SNPs | Chr | Position (bp) | LOD > 3 | r2 (%) | Repeated in Years |
|---|---|---|---|---|---|---|---|
| KL | 1,2,3 | RAC875_29540_391 | 1A | 264 | 4.44–6.42 | 3.41–4.84 | 2018 |
| 2,3 | BS00089524_51 | 1B | 637 | 3.07–5.99 | 4.64–6.57 | 2018 | |
| 1,2,4 | Kukri_07961_503 | 6D | 3,491,885 | 6.14–7.50 | 3.63–7.65 | 2017, 2018 | |
| 1,2 | Kukri_c8827_217 | 7A | 3,732,575 | 3.56–3.74 | 2.70–4.40 | 2017 | |
| 1,4 | Tdurum_contig102328_129 | 7B | 4,030,937 | 3.94–6.58 | 5.36–10.51 | 2017 | |
| 1,3 | Kukri_c64387_218 | 7B | 4,049,359 | 3.22–4.07 | 2.58–4.84 | 2018 | |
| 1,2 | Tdurum_contig15734_221 | 7B | 4,127,623 | 3.60–4.59 | 2.64–4.33 | 2018 | |
| 1 | Ku_008899_90 | 3D | 1,664,604 | 3.64–3.86 | 5.22–5.40 | 2017, 2018 | |
| KW | 1,2 | RAC875_0643_1548 | 2A | 105,090 | 3.10–3.31 | 1.10–3.99 | 2017 |
| 2,3,4 | tplb0034e07_1581 | 2A | 79,588 | 3.86–4.29 | 1.28–1.94 | 2018 | |
| 1,2 | Kukri_c48194_641 | 4A | 1,892,065 | 3.35–6.32 | 4.59–6.97 | 2017 | |
| 1,4 | Ra_02239_504 | 6A | 2,962,548 | 3.95–4.59 | 1.88–4.77 | 2018 | |
| 2,3 | Tdurum_contig15734_221 | 7B | 4,127,623 | 3.40–4.33 | 2.92–3.65 | 2017 | |
| 2,3 | D_contig07330_330 | 7D | 4,292,747 | 3.01–6.28 | 2.50–6.00 | 2017 | |
| 1 | BS00021738_51 | 4A | 1,804,501 | 4.98–7.30 | 14.34–16.13 | 2017, 2018 | |
| KPS | 2,3,4 | BS00074341_51 | 1A | 109 | 4.18–6.18 | 2.90–3.96 | 2018 |
| 1,4 | Tdurum_contig54559_211 | 4B | 2,056,791 | 3.27–4.03 | 2.16–4.47 | 2017 | |
| 2,4 | Kukri_08268_79 | 5A | 2,172,100 | 3.26–3.51 | 2.39–3.87 | 2018 | |
| 2,3 | GENE_3726_78 | 6B | 3,355,233 | 3.28–3.68 | 2.32–2.40 | 2018 | |
| 1,2,3,4 | BobWhite_049_3064 | 7A | 3,802,457 | 3.26–9.02 | 2.83–8.15 | 2018 | |
| 2,4 | wsnp_Ku_rep_004159_90704469 | 7A | 3,806,941 | 5.07–5.36 | 3.41–4.07 | 2018 | |
| TKW | 2,4 | wsnp_Ex_c33778_42210283 | 4A | 1,954,125 | 5.99–7.90 | 1.21–6.84 | 2017 |
| 1,4 | RAC875_rep_c83934_91 | 7A | 3,886,333 | 3.76–10.77 | 2.27–5.15 | 2017 | |
| 2,4 | wsnp_Ku_0552_3060297 | 7A | 3,779,727 | 3.83–4.82 | 2.24–3.50 | 2018 | |
| 1,2 | Kukri_rep_c97425_164 | 7A | 3,791,938 | 3.19–3.34 | 1.86–3.34 | 2018 | |
| 2,4 | D_contig07330_330 | 7D | 4,292,747 | 3.43–3.54 | 0.49–3.10 | 2017 |
| Trait | Methods | SNP | Chr | Position (bp) | Repeated in Years |
|---|---|---|---|---|---|
| KL | pLARmEB, MLMM | Kukri_07961_503 | 6D | 3,491,885 | 2017, 2018 |
| KL | mrMLM, FarmCPU | Kukri_c8827_217 | 7A | 3,732,575 | 2017 |
| KL | pLARmEB, FarmCPU | Tdurum_contig102328_129 | 7B | 4,030,937 | 2017 |
| KW | mrMLM, MLMM | BS00021738_51 | 4A | 1,804,501 | 2017, 2018 |
| KW | FASTmrMLM, FarmCPU | Tdurum_contig15734_221 | 7B | 4,127,623 | 2017 |
| KPS | mrMLM, MLMM | wsnp_Ra_rep_007017_90667618 | 4A | 1,774,526 | 2017 |
| KPS | mrMLM, FarmCPU | BobWhite_049_3064 | 7A | 3,802,457 | 2018 |
| TKW | FASTmrEMMA, FarmCPU | BobWhite_09733_301 | 1B | 868 | 2017 |
| TKW | mrMLM, MLMM | BS00110286_51 | 3D | 1,727,487 | 2017 |
| TKW | mrMLM, MLMM | Ex_07338_401 | 4A | 1,800,847 | 2017, 2018 |
| TKW | mrMLM, MLM | Kukri_rep_c97425_164 | 7A | 3,791,938 | 2018 |
| TKW | pLARmEB, MLM | RAC875_rep_c83934_91 | 7A | 3,886,333 | 2017 |
| TKW | FASTmrEMMA, MLM | wsnp_Ex_c4068_7351806 | 4A | 1,803,131 | 2017, 2018 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhammad, A.; Hu, W.; Li, Z.; Li, J.; Xie, G.; Wang, J.; Wang, L. Appraising the Genetic Architecture of Kernel Traits in Hexaploid Wheat Using GWAS. Int. J. Mol. Sci. 2020, 21, 5649. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165649
Muhammad A, Hu W, Li Z, Li J, Xie G, Wang J, Wang L. Appraising the Genetic Architecture of Kernel Traits in Hexaploid Wheat Using GWAS. International Journal of Molecular Sciences. 2020; 21(16):5649. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165649
Chicago/Turabian StyleMuhammad, Ali, Weicheng Hu, Zhaoyang Li, Jianguo Li, Guosheng Xie, Jibin Wang, and Lingqiang Wang. 2020. "Appraising the Genetic Architecture of Kernel Traits in Hexaploid Wheat Using GWAS" International Journal of Molecular Sciences 21, no. 16: 5649. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165649