CD36 and CD97 in Pancreatic Cancer versus Other Malignancies

 and
and
Abstract
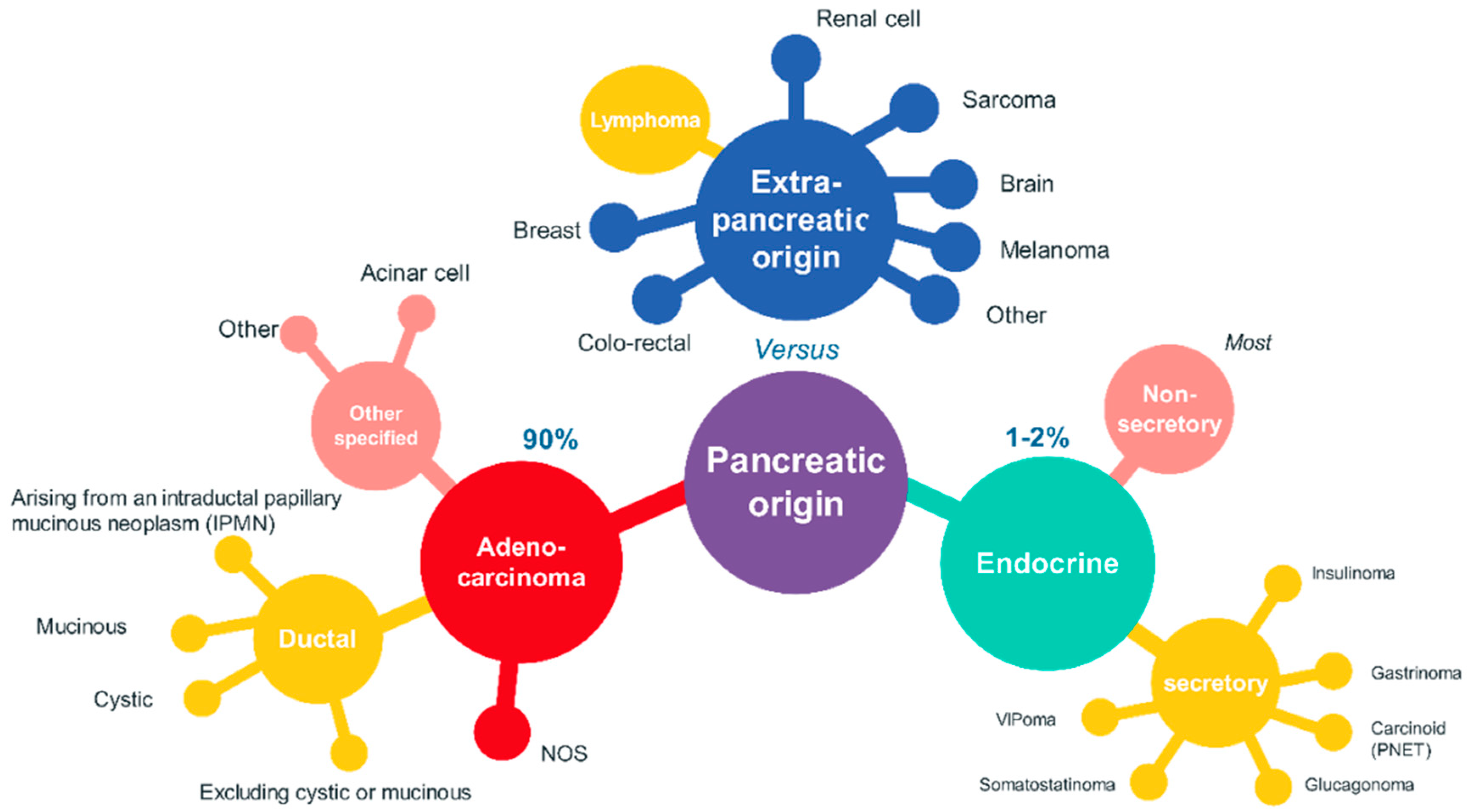
:1. Introduction
2. CD36 in Pancreatic Cancer vs. CD36 in Normal Tissues: Where Do We Stand?
2.1. CD36 in Normal Tissues
2.2. CD36 Promotes Tumor Metastasis in Pancreatic Cancer
2.3. CD36—A Mediator of the Engulfment of Pancreatic Tumor Microvesicles
2.4. CD36 Can Regulate Chemoresistance in Pancreatic Cancer
3. CD97 During Pancreatic Cancer vs. CD97 in Normal Tissue
3.1. Distribution and Functions of CD 97
3.2. Expression of CD97 in Pancreatic Cancer
4. Why Examine Concomitant Expression of CD36 and CD97s? (Why Bother with CD36 and CD97 in Pancreatic Cancer?)
5. Heterogeneity of Pancreatic-Cancer-Associated Fibroblasts
5.1. Tumor Microenvironment
5.2. Normal Fibroblast
5.3. Cancer-Associated Fibroblasts (CAFs)
5.4. Cancer-Associated Fibroblasts (CAFs) in PDAC
5.5. Secretome
5.6. Metabolism
5.7. Challenges to Studying Metabolic Interactions in the Tumor Microenvironment
5.8. Targeting CAFs Could Create New Therapeutic Avenues in Pancreatic Cancer Therapy
- -
- TGFβ and interleukin signaling—blocking antibodies inhibitors;
- -
- NFkB and TNFα signaling, to reduce perlecan secretion;
- -
- Cancer cells–ECM interaction: Hedgehog signaling through IPI-926 (sonidegib and vismodegib) or hyaluronic acid through enzymes (PEGPH20) blocking antibodies inhibitors
- -
- Immunosuppression in the tumor microenvironment [113].
5.9. Future Directions
6. Signaling Side: TGFβ, CD36, and CD97—Signaling in Pancreatic Cancer
6.1. Crosstalk with Other Pathways
6.2. Involvement of CD36 and CD 97 in Signaling Pathways
7. Favoring Quiescence (Cell Dormancy)—A Valid Therapeutic Strategy in Pancreatic Cancer?
8. Conclusions
- (a)
- “Therefore, reversal of activated fibroblasts to the quiescence state is an important area of investigation that may help the therapeutic management of a number of diseases including pancreatic cancer” [227].
- (b)
- “Thus, targeting the CAFs at this stage with molecules that can revert the back to “quiescent” state can be considered an attractive therapeutic strategy, as this will disrupt the tumor–stroma crosstalk and inhibit the tumor growth and progression” [228].
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CD | cluster of differentiation |
| PDAC | pancreatic ductal adenocarcinoma |
| panIN | pancreatic intraepithelial neoplasia |
| SR-B2 | receptor class B type 2 |
| MVECs | microvascular endothelial cells |
| ER | endoplasmic reticulum |
| FAO | mitochondrial FA oxidation |
| CPT1 | carnitine palmitoyltransferase-1 |
| PI3K | phosphatidylinositol 3-kinase |
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| LCFAs | long-chain fatty acids |
| ox-LDL | oxidized low-density lipoprotein |
| ox-HDL | oxidized high-density lipoprotein |
| ox-PLs | oxidized phospholipids |
| FABPc | cytosolic FA binding protein |
| TSP | thrombospondins |
| AOPPs | advanced oxidation protein products |
| AGEs | advanced glycation end products |
| EMT | epithelial–mesenchymal transition |
| TSP-1 | thrombospondin-1 |
| TGFβ | transforming growth factor-β |
| EGFs | epidermal growth factors |
| PT | pancreatitis |
| CAF | cancer-associated fibroblast |
| ECM | extracellular matrix |
| PDGFRα | platelet-derived growth factor receptor-α |
| αSMA | α-smooth muscle actin |
| FAP | fibroblast activation protein |
| PSCs | pancreatic stellate cells |
References
- Croce, C.M. Oncogenes and cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, B.; Zeng, S.; Grutzmann, R.; Pilarsky, C. The Role of Exosomes in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, S.; Pottler, M.; Lan, B.; Grutzmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [Green Version]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Caravia, L.; Dudau, M.; Gherghiceanu, M.; Tanase, C.; Enciu, A.M. Could caveolae be acting as warnings of mitochondrial ageing? Mech. Ageing Dev. 2015, 146, 81–87. [Google Scholar] [CrossRef]
- Pistol-Tanase, C.; Raducan, E.; Dima, S.O.; Albulescu, L.; Alina, I.; Marius, P.; Cruceru, L.M.; Codorean, E.; Neagu, T.M.; Popescu, I. Assessment of soluble angiogenic markers in pancreatic cancer. Biomark. Med. 2008, 2, 447–455. [Google Scholar] [CrossRef] [Green Version]
- Tanase, C.P.; Neagu, M.; Albulescu, R.; Hinescu, M.E. Advances in pancreatic cancer detection. Adv. Clin. Chem. 2010, 51, 145–180. [Google Scholar] [CrossRef]
- Tanase, C.P.; Neagu, A.I.; Necula, L.G.; Mambet, C.; Enciu, A.M.; Calenic, B.; Cruceru, M.L.; Albulescu, R. Cancer stem cells: Involvement in pancreatic cancer pathogenesis and perspectives on cancer therapeutics. World J. Gastroenterol. 2014, 20, 10790–10801. [Google Scholar] [CrossRef]
- Albulescu, R.; Neagu, M.; Albulescu, L.; Tanase, C. Tissular and soluble miRNAs for diagnostic and therapy improvement in digestive tract cancers. Expert Rev. Mol. Diagn. 2011, 11, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Dima, S.O.; Tanase, C.; Albulescu, R.; Herlea, V.; Chivu-Economescu, M.; Purnichescu-Purtan, R.; Dumitrascu, T.; Duda, D.G.; Popescu, I. An exploratory study of inflammatory cytokines as prognostic biomarkers in patients with ductal pancreatic adenocarcinoma. Pancreas 2012, 41, 1001–1007. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, G.; You, L.; Yang, J.; Feng, M.; Qiu, J.; Zhao, F.; Liu, Y.; Cao, Z.; Zheng, L.; et al. Role of the microbiome in occurrence, development and treatment of pancreatic cancer. Mol. Cancer 2019, 18, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Dseagu, V.L.; Devesa, S.S.; Goggins, M.; Stolzenberg-Solomon, R. Pancreatic cancer incidence trends: Evidence from the Surveillance, Epidemiology and End Results (SEER) population-based data. Int. J. Epidemiol. 2018, 47, 427–439. [Google Scholar] [CrossRef]
- Haeberle, L.; Esposito, I. Pathology of pancreatic cancer. Transl. Gastroenterol. Hepatol. 2019, 4, 50. [Google Scholar] [CrossRef]
- Noushmehr, H.; D’Amico, E.; Farilla, L.; Hui, H.; Wawrowsky, K.A.; Mlynarski, W.; Doria, A.; Abumrad, N.A.; Perfetti, R. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic beta-cells and mediates fatty acid effects on insulin secretion. Diabetes 2005, 54, 472–481. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Zhou, L.; Shen, T.; Zhou, S.; Ding, G.; Cao, L. Down-expression of CD36 in pancreatic adenocarcinoma and its correlation with clinicopathological features and prognosis. J. Cancer 2018, 9, 578–583. [Google Scholar] [CrossRef] [Green Version]
- Lamaze, C.; Tardif, N.; Dewulf, M.; Vassilopoulos, S.; Blouin, C.M. The caveolae dress code: Structure and signaling. Curr. Opin. Cell Biol. 2017, 47, 117–125. [Google Scholar] [CrossRef]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef] [Green Version]
- Thorne, R.F.; Law, E.G.; Elith, C.A.; Ralston, K.J.; Bates, R.C.; Burns, G.F. The association between CD36 and Lyn protein tyrosine kinase is mediated by lipid. Biochem. Biophys. Res. Commun. 2006, 351, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Jain, S.S.; McFarlan, J.T.; Snook, L.A.; Chabowski, A.; Bonen, A. Exercise-and training-induced upregulation of skeletal muscle fatty acid oxidation are not solely dependent on mitochondrial machinery and biogenesis. J. Physiol. 2013, 591, 4415–4426. [Google Scholar] [CrossRef] [PubMed]
- Luiken, J.J.; Koonen, D.P.; Willems, J.; Zorzano, A.; Becker, C.; Fischer, Y.; Tandon, N.N.; Van Der Vusse, G.J.; Bonen, A.; Glatz, J.F. Insulin stimulates long-chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of FAT/CD36. Diabetes 2002, 51, 3113–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luiken, J.J.; Coort, S.L.; Willems, J.; Coumans, W.A.; Bonen, A.; van der Vusse, G.J.; Glatz, J.F. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes 2003, 52, 1627–1634. [Google Scholar] [CrossRef] [Green Version]
- Nieva, C.; Marro, M.; Santana-Codina, N.; Rao, S.; Petrov, D.; Sierra, A. The lipid phenotype of breast cancer cells characterized by Raman microspectroscopy: Towards a stratification of malignancy. PLoS ONE 2012, 7, e46456. [Google Scholar] [CrossRef]
- Iwao, Y.; Nakajou, K.; Nagai, R.; Kitamura, K.; Anraku, M.; Maruyama, T.; Otagiri, M. CD36 is one of important receptors promoting renal tubular injury by advanced oxidation protein products. Am. J. Physiol. Ren. Physiol. 2008, 295, F1871–F1880. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Li, W.; Silverstein, R.L. Advanced glycation end products induce a prothrombotic phenotype in mice via interaction with platelet CD36. Blood 2012, 119, 6136–6144. [Google Scholar] [CrossRef] [Green Version]
- Albert, M.L.; Pearce, S.F.; Francisco, L.M.; Sauter, B.; Roy, P.; Silverstein, R.L.; Bhardwaj, N. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J. Exp. Med. 1998, 188, 1359–1368. [Google Scholar] [CrossRef]
- Doens, D.; Valiente, P.A.; Mfuh, A.M.; Vo, A.X.T.; Tristan, A.; Carreno, L.; Quijada, M.; Nguyen, V.T.; Perry, G.; Larionov, O.V.; et al. Identification of Inhibitors of CD36-Amyloid Beta Binding as Potential Agents for Alzheimer’s Disease. Acs Chem. Neurosci. 2017, 8, 1232–1241. [Google Scholar] [CrossRef]
- Pennathur, S.; Pasichnyk, K.; Bahrami, N.M.; Zeng, L.; Febbraio, M.; Yamaguchi, I.; Okamura, D.M. The macrophage phagocytic receptor CD36 promotes fibrogenic pathways on removal of apoptotic cells during chronic kidney injury. Am. J. Pathol. 2015, 185, 2232–2245. [Google Scholar] [CrossRef] [Green Version]
- Ping, M.; Xiao, W.; Mo, L.; Xiao, X.; Song, S.; Tang, W.; Yang, X. Paeonol attenuates advanced oxidation protein product-induced oxidative stress injury in THP-1 macrophages. Pharmacology 2014, 93, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, N.; Nagai, R.; Ikemoto, M.; Arai, H.; Miyazaki, A.; Hakamata, H.; Horiuchi, S.; Nakayama, H. CD36, serves as a receptor for advanced glycation endproducts (AGE). J. Diabetes Complicat. 2002, 16, 56–59. [Google Scholar] [CrossRef]
- Yang, P.; Su, C.; Luo, X.; Zeng, H.; Zhao, L.; Wei, L.; Zhang, X.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. Dietary oleic acid-induced CD36 promotes cervical cancer cell growth and metastasis via up-regulation Src/ERK pathway. Cancer Lett. 2018, 438, 76–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enciu, A.M.; Radu, E.; Popescu, I.D.; Hinescu, M.E.; Ceafalan, L.C. Targeting CD36 as Biomarker for Metastasis Prognostic: How Far from Translation into Clinical Practice? Biomed Res. Int. 2018, 2018, 7801202. [Google Scholar] [CrossRef] [Green Version]
- Deng, M.; Cai, X.; Long, L.; Xie, L.; Ma, H.; Zhou, Y.; Liu, S.; Zeng, C. CD36 promotes the epithelial-mesenchymal transition and metastasis in cervical cancer by interacting with TGF-beta. J. Transl. Med. 2019, 17, 352. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Addai, J.; Tian, W.H.; Frolov, A.; Wheeler, T.M.; Thompson, T.C. Reduced infiltration of class A scavenger receptor positive antigen-presenting cells is associated with prostate cancer progression. Cancer Res. 2004, 64, 2076–2082. [Google Scholar] [CrossRef] [Green Version]
- Kubo, M.; Eguchi, H. ASO Author Reflections: Regulation of Chemoresistance in Pancreatic Ductal Adenocarcinoma by Scavenger Receptor CD36. Ann. Surg. Oncol. 2020, 27, 620–621. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Gotoh, K.; Eguchi, H.; Kobayashi, S.; Iwagami, Y.; Tomimaru, Y.; Akita, H.; Asaoka, T.; Noda, T.; Takeda, Y.; et al. Impact of CD36 on Chemoresistance in Pancreatic Ductal Adenocarcinoma. Ann. Surg. Oncol. 2020, 27, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Chan, C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci. Rep. 2016, 6, 18669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladanyi, A.; Mukherjee, A.; Kenny, H.A.; Johnson, A.; Mitra, A.K.; Sundaresan, S.; Nieman, K.M.; Pascual, G.; Benitah, S.A.; Montag, A.; et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018, 37, 2285–2301. [Google Scholar] [CrossRef]
- Pan, J.; Fan, Z.; Wang, Z.; Dai, Q.; Xiang, Z.; Yuan, F.; Yan, M.; Zhu, Z.; Liu, B.; Li, C. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3beta/beta-catenin pathway. J. Exp. Clin. Cancer Res. Cr. 2019, 38, 52. [Google Scholar] [CrossRef] [Green Version]
- Hale, J.S.; Otvos, B.; Sinyuk, M.; Alvarado, A.G.; Hitomi, M.; Stoltz, K.; Wu, Q.; Flavahan, W.; Levison, B.; Johansen, M.L.; et al. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells 2014, 32, 1746–1758. [Google Scholar] [CrossRef] [Green Version]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Guo, C.; Fisher, P.B.; Subjeck, J.R.; Wang, X.Y. Scavenger Receptors: Emerging Roles in Cancer Biology and Immunology. Adv. Cancer Res. 2015, 128, 309–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Kang, Y. Lipid Metabolism Fuels Cancer’s Spread. Cell Metab. 2017, 25, 228–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 2015, 5, 14752. [Google Scholar] [CrossRef] [Green Version]
- Celia-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef] [Green Version]
- Niculite, C.M.; Enciu, A.M.; Hinescu, M.E. CD 36: Focus on Epigenetic and Post-Transcriptional Regulation. Front. Genet. 2019, 10, 680. [Google Scholar] [CrossRef]
- Zhang, W.H.; Wang, W.Q.; Gao, H.L.; Xu, S.S.; Li, S.; Li, T.J.; Han, X.; Xu, H.X.; Li, H.; Jiang, W.; et al. Tumor-Infiltrating Neutrophils Predict Poor Survival of Non-Functional Pancreatic Neuroendocrine Tumor. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef]
- Sano, M.; Ijichi, H.; Takahashi, R.; Miyabayashi, K.; Fujiwara, H.; Yamada, T.; Kato, H.; Nakatsuka, T.; Tanaka, Y.; Tateishi, K.; et al. Blocking CXCLs-CXCR2 axis in tumor-stromal interactions contributes to survival in a mouse model of pancreatic ductal adenocarcinoma through reduced cell invasion/migration and a shift of immune-inflammatory microenvironment. Oncogenesis 2019, 8, 8. [Google Scholar] [CrossRef]
- Roufas, C.; Chasiotis, D.; Makris, A.; Efstathiades, C.; Dimopoulos, C.; Zaravinos, A. The Expression and Prognostic Impact of Immune Cytolytic Activity-Related Markers in Human Malignancies: A Comprehensive Meta-analysis. Front. Oncol. 2018, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Xin, Z.; Hu, W.; Jiang, S.; Yang, Z.; Yan, X.; Li, X.; Yang, Y.; Chen, F. Forkhead box O proteins: Crucial regulators of cancer EMT. Semin. Cancer Biol. 2018, 50, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y. CD36 tango in cancer: Signaling pathways and functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef] [PubMed]
- Karamitopoulou, E. Role of epithelial-mesenchymal transition in pancreatic ductal adenocarcinoma: Is tumor budding the missing link? Front. Oncol. 2013, 3, 221. [Google Scholar] [CrossRef] [Green Version]
- Miyashita, H.; Watanabe, H.; Ohe, H.; Itakura, Y.; Ohnishi, K.; Hayami, H.; Watanabe, M. [An application of 2D-Doppler color flow mapping to the prostate]. Nihon Hinyokika Gakkai Zasshi. Jpn. J. Urol. 1988, 79, 235–238. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, R.T.; Veronese, N.; Nottegar, A.; Malleo, G.; Smith, L.; Demurtas, J.; Cheng, L.; Wood, L.D.; Silvestris, N.; Salvia, R.; et al. Prognostic Role of High-Grade Tumor Budding in Pancreatic Ductal Adenocarcinoma: A Systematic Review and Meta-Analysis with a Focus on Epithelial to Mesenchymal Transition. Cancers 2019, 11, 113. [Google Scholar] [CrossRef] [Green Version]
- Galvan, J.A.; Zlobec, I.; Wartenberg, M.; Lugli, A.; Gloor, B.; Perren, A.; Karamitopoulou, E. Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br. J. Cancer 2015, 112, 1944–1950. [Google Scholar] [CrossRef] [Green Version]
- Chu, L.Y.; Ramakrishnan, D.P.; Silverstein, R.L. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 2013, 122, 1822–1832. [Google Scholar] [CrossRef] [Green Version]
- Murphy-Ullrich, J.E.; Poczatek, M. Activation of latent TGF-beta by thrombospondin-1: Mechanisms and physiology. Cytokine Growth Factor Rev. 2000, 11, 59–69. [Google Scholar] [CrossRef]
- Simonian, S.J.; Stuart, F.P.; Hill, J.L.; Mahajan, S.K. Conversion of a Scribner shunt to an arteriovenous fistula for chronic dialysis. Surgery 1977, 82, 448–451. [Google Scholar]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef]
- Tkach, M.; Thery, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soung, Y.H.; Ford, S.; Zhang, V.; Chung, J. Exosomes in Cancer Diagnostics. Cancers 2017, 9, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Lam, E.W.; Sun, Y. Extracellular vesicles in the tumor microenvironment: Old stories, but new tales. Mol. Cancer 2019, 18, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Pfeiler, S.; Thakur, M.; Grunauer, P.; Megens, R.T.A.; Joshi, U.; Coletti, R.; Samara, V.; Muller-Stoy, G.; Ishikawa-Ankerhold, H.; Stark, K.; et al. CD36-triggered cell invasion and persistent tissue colonization by tumor microvesicles during metastasis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 1860–1872. [Google Scholar] [CrossRef] [Green Version]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Iwagami, Y.; Eguchi, H.; Nagano, H.; Akita, H.; Hama, N.; Wada, H.; Kawamoto, K.; Kobayashi, S.; Tomokuni, A.; Tomimaru, Y.; et al. miR-320c regulates gemcitabine-resistance in pancreatic cancer via SMARCC1. Br. J. Cancer 2013, 109, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Pang, B.; Xu, X.; Lu, Y.; Jin, H.; Yang, R.; Jiang, C.; Shao, D.; Liu, Y.; Shi, J. Prediction of new targets and mechanisms for quercetin in the treatment of pancreatic cancer, colon cancer, and rectal cancer. Food Funct. 2019, 10, 5339–5349. [Google Scholar] [CrossRef]
- Eichler, W.; Aust, G.; Hamann, D. Characterization of an early activation-dependent antigen on lymphocytes defined by the monoclonal antibody BL-Ac(F2). Scand. J. Immunol. 1994, 39, 111–115. [Google Scholar] [CrossRef]
- Safaee, M.; Clark, A.J.; Ivan, M.E.; Oh, M.C.; Bloch, O.; Sun, M.Z.; Oh, T.; Parsa, A.T. CD97 is a multifunctional leukocyte receptor with distinct roles in human cancers (Review). Int. J. Oncol. 2013, 43, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ward, Y.; Tian, L.; Lake, R.; Guedez, L.; Stetler-Stevenson, W.G.; Kelly, K. CD97, an adhesion receptor on inflammatory cells, stimulates angiogenesis through binding integrin counterreceptors on endothelial cells. Blood 2005, 105, 2836–2844. [Google Scholar] [CrossRef] [PubMed]
- Ward, Y.; Lake, R.; Martin, P.L.; Killian, K.; Salerno, P.; Wang, T.; Meltzer, P.; Merino, M.; Cheng, S.Y.; Santoro, M.; et al. CD97 amplifies LPA receptor signaling and promotes thyroid cancer progression in a mouse model. Oncogene 2013, 32, 2726–2738. [Google Scholar] [CrossRef] [Green Version]
- Han, S.L.; Xu, C.; Wu, X.L.; Li, J.L.; Liu, Z.; Zeng, Q.Q. The impact of expressions of CD97 and its ligand CD55 at the invasion front on prognosis of rectal adenocarcinoma. Int. J. Colorectal Dis. 2010, 25, 695–702. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Wu, H.; Jiao, Y.; Zheng, J. Expression and prognostic value of CD97 and its ligand CD55 in pancreatic cancer. Oncol. Lett. 2015, 9, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Trojanowicz, B.; Ye, L.; Li, C.; Zhang, L.; Li, X.; Li, G.; Zheng, Y.; Chen, L. The invasion and metastasis promotion role of CD97 small isoform in gastric carcinoma. PLoS ONE 2012, 7, e39989. [Google Scholar] [CrossRef]
- Somasundaram, A.; Ardanowski, N.; Opalak, C.F.; Fillmore, H.L.; Chidambaram, A.; Broaddus, W.C. Wilms tumor 1 gene, CD97, and the emerging biogenetic profile of glioblastoma. Neurosurg. Focus 2014, 37, E14. [Google Scholar] [CrossRef] [Green Version]
- Ward, Y.; Lake, R.; Yin, J.J.; Heger, C.D.; Raffeld, M.; Goldsmith, P.K.; Merino, M.; Kelly, K. LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res. 2011, 71, 7301–7311. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Xu, X.; Tang, J.; Zhang, W.; Zhangyuan, G.; Ji, J.; Deng, L.; Lu, S.; Zhuo, H.; Sun, B. CD97 Promotes Tumor Aggressiveness Through the Traditional G Protein-Coupled Receptor-Mediated Signaling in Hepatocellular Carcinoma. Hepatology 2018, 68, 1865–1878. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, K.; Reynolds, J.C.; Paik, C.H.; Sood, V.K.; Maloney, P.J.; Larson, S.M.; Reba, R.C. Immunoreactivity affects the biodistribution and tumor targeting of radiolabeled anti-P97 Fab fragment. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 1990, 31, 202–210. [Google Scholar]
- Jaspars, L.H.; Vos, W.; Aust, G.; Van Lier, R.A.; Hamann, J. Tissue distribution of the human CD97 EGF-TM7 receptor. Tissue Antigens 2001, 57, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Kwakkenbos, M.J.; Pouwels, W.; Matmati, M.; Stacey, M.; Lin, H.H.; Gordon, S.; van Lier, R.A.; Hamann, J. Expression of the largest CD97 and EMR2 isoforms on leukocytes facilitates a specific interaction with chondroitin sulfate on B cells. J. Leukoc. Biol. 2005, 77, 112–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwakkenbos, M.J.; Matmati, M.; Madsen, O.; Pouwels, W.; Wang, Y.; Bontrop, R.E.; Heidt, P.J.; Hoek, R.M.; Hamann, J. An unusual mode of concerted evolution of the EGF-TM7 receptor chimera EMR2. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 2582–2584. [Google Scholar] [CrossRef] [Green Version]
- Veninga, H.; Becker, S.; Hoek, R.M.; Wobus, M.; Wandel, E.; van der Kaa, J.; van der Valk, M.; de Vos, A.F.; Haase, H.; Owens, B.; et al. Analysis of CD97 expression and manipulation: Antibody treatment but not gene targeting curtails granulocyte migration. J. Immunol. 2008, 181, 6574–6583. [Google Scholar] [CrossRef] [Green Version]
- Yeon Won, H.; Hwan Mun, S.; Shin, B.; Lee, S.K. Contradictory Role of CD97 in Basal and Tumor Necrosis Factor-Induced Osteoclastogenesis In Vivo. Arthritis Rheumatol. 2016, 68, 1301–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjong, W.Y.; Lin, H.H. The RGD motif is involved in CD97/ADGRE5-promoted cell adhesion and viability of HT1080 cells. Sci. Rep. 2019, 9, 1517. [Google Scholar] [CrossRef] [Green Version]
- Gray, J.X.; Haino, M.; Roth, M.J.; Maguire, J.E.; Jensen, P.N.; Yarme, A.; Stetler-Stevenson, M.A.; Siebenlist, U.; Kelly, K. CD97 is a processed, seven-transmembrane, heterodimeric receptor associated with inflammation. J. Immunol. 1996, 157, 5438–5447. [Google Scholar]
- Kop, E.N.; Adriaansen, J.; Smeets, T.J.; Vervoordeldonk, M.J.; van Lier, R.A.; Hamann, J.; Tak, P.P. CD97 neutralisation increases resistance to collagen-induced arthritis in mice. Arthritis Res. Ther. 2006, 8, R155. [Google Scholar] [CrossRef] [Green Version]
- Aust, G.; Steinert, M.; Schutz, A.; Boltze, C.; Wahlbuhl, M.; Hamann, J.; Wobus, M. CD97, but not its closely related EGF-TM7 family member EMR2, is expressed on gastric, pancreatic, and esophageal carcinomas. Am. J. Clin. Pathol. 2002, 118, 699–707. [Google Scholar] [CrossRef]
- Boltze, C.; Schneider-Stock, R.; Aust, G.; Mawrin, C.; Dralle, H.; Roessner, A.; Hoang-Vu, C. CD97, CD95 and Fas-L clearly discriminate between chronic pancreatitis and pancreatic ductal adenocarcinoma in perioperative evaluation of cryocut sections. Pathol. Int. 2002, 52, 83–88. [Google Scholar] [CrossRef]
- Steinert, M.; Wobus, M.; Boltze, C.; Schutz, A.; Wahlbuhl, M.; Hamann, J.; Aust, G. Expression and regulation of CD97 in colorectal carcinoma cell lines and tumor tissues. Am. J. Pathol. 2002, 161, 1657–1667. [Google Scholar] [CrossRef] [Green Version]
- Hamann, J.; Wishaupt, J.O.; van Lier, R.A.; Smeets, T.J.; Breedveld, F.C.; Tak, P.P. Expression of the activation antigen CD97 and its ligand CD55 in rheumatoid synovial tissue. Arthritis Rheum. 1999, 42, 650–658. [Google Scholar] [CrossRef]
- Mikesch, J.H.; Schier, K.; Roetger, A.; Simon, R.; Buerger, H.; Brandt, B. The expression and action of decay-accelerating factor (CD55) in human malignancies and cancer therapy. Cell. Oncol. Off. J. Int. Soc. Cell. Oncol. 2006, 28, 223–232. [Google Scholar] [CrossRef]
- Vogl, U.M.; Ohler, L.; Rasic, M.; Frischer, J.M.; Modak, M.; Stockl, J. Evaluation of Prognostic Immune Signatures in Patients with Breast, Colorectal and Pancreatic Cancer Receiving Chemotherapy. Anticancer Res. 2017, 37, 1947–1955. [Google Scholar] [CrossRef] [Green Version]
- Heinzelmann, K.; Lehmann, M.; Gerckens, M.; Noskovicova, N.; Frankenberger, M.; Lindner, M.; Hatz, R.; Behr, J.; Hilgendorff, A.; Konigshoff, M.; et al. Cell-surface phenotyping identifies CD36 and CD97 as novel markers of fibroblast quiescence in lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L682–L696. [Google Scholar] [CrossRef]
- Schosserer, M.; Grillari, J.; Breitenbach, M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front. Oncol. 2017, 7, 278. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.A. The evolving concept of cell identity in the single cell era. Development 2019, 146, dev.169748. [Google Scholar] [CrossRef] [Green Version]
- Nurmik, M.; Ullmann, P.; Rodriguez, F.; Haan, S.; Letellier, E. In search of definitions: Cancer-associated fibroblasts and their markers. Int. J. Cancer 2020, 146, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Codrici, E.; Enciu, A.M.; Popescu, I.D.; Mihai, S.; Tanase, C. Glioma Stem Cells and Their Microenvironments: Providers of Challenging Therapeutic Targets. Stem Cells Int. 2016, 2016, 5728438. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.N.; Heiden, M.G.V. Metabolism in the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2020, 4, 17–40. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopa, K.B.; Kusiak, A.A.; Szopa, M.D.; Ferdek, P.E.; Jakubowska, M.A. Pancreatic Cancer and Its Microenvironment-Recent Advances and Current Controversies. Int. J. Mol. Sci. 2020, 21, 3218. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.; et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Watari, N.; Hotta, Y.; Mabuchi, Y. Morphological studies on a vitamin A-storing cell and its complex with macrophage observed in mouse pancreatic tissues following excess vitamin A administration. Okajimas Folia Anat. Jpn. 1982, 58, 837–858. [Google Scholar] [CrossRef] [Green Version]
- Wehr, A.Y.; Furth, E.E.; Sangar, V.; Blair, I.A.; Yu, K.H. Analysis of the human pancreatic stellate cell secreted proteome. Pancreas 2011, 40, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cell: Physiologic role, role in fibrosis and cancer. Curr. Opin. Gastroenterol. 2015, 31, 416–423. [Google Scholar] [CrossRef]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta 2009, 1791, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Neesse, A.; Frese, K.K.; Bapiro, T.E.; Nakagawa, T.; Sternlicht, M.D.; Seeley, T.W.; Pilarsky, C.; Jodrell, D.I.; Spong, S.M.; Tuveson, D.A. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12325–12330. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Strell, C.; Paulsson, J.; Jin, S.B.; Tobin, N.P.; Mezheyeuski, A.; Roswall, P.; Mutgan, C.; Mitsios, N.; Johansson, H.; Wickberg, S.M.; et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J. Natl. Cancer Inst. 2019, 111, 983–995. [Google Scholar] [CrossRef]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Hofman, P.; Tartare-Deckert, S.; Feral, C.C.; Meneguzzi, G.; Gaggioli, C. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef] [Green Version]
- Avery, D.; Govindaraju, P.; Jacob, M.; Todd, L.; Monslow, J.; Pure, E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 67, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Bassi, D.E.; Klein-Szanto, A.J.; Cukierman, E. Stroma-derived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. Am. J. Pathol. 2005, 167, 475–488. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, N.; Ranftl, R.; Chicherova, I.; Slaven, N.D.; Moeendarbary, E.; Farrugia, A.J.; Lam, M.; Semiannikova, M.; Westergaard, M.C.W.; Tchou, J.; et al. Dickkopf-3 links HSF1 and YAP/TAZ signalling to control aggressive behaviours in cancer-associated fibroblasts. Nat. Commun. 2019, 10, 130. [Google Scholar] [CrossRef]
- Fordyce, C.; Fessenden, T.; Pickering, C.; Jung, J.; Singla, V.; Berman, H.; Tlsty, T. DNA damage drives an activin a-dependent induction of cyclooxygenase-2 in premalignant cells and lesions. Cancer Prev. Res. (Phila) 2010, 3, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 282–295. [Google Scholar] [CrossRef]
- Okumura, T.; Ohuchida, K.; Kibe, S.; Iwamoto, C.; Ando, Y.; Takesue, S.; Nakayama, H.; Abe, T.; Endo, S.; Koikawa, K.; et al. Adipose tissue-derived stromal cells are sources of cancer-associated fibroblasts and enhance tumor progression by dense collagen matrix. Int. J. Cancer 2019, 144, 1401–1413. [Google Scholar] [CrossRef]
- DeFilippis, R.A.; Chang, H.; Dumont, N.; Rabban, J.T.; Chen, Y.Y.; Fontenay, G.V.; Berman, H.K.; Gauthier, M.L.; Zhao, J.; Hu, D.; et al. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discov. 2012, 2, 826–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFilippis, R.A.; Fordyce, C.; Patten, K.; Chang, H.; Zhao, J.; Fontenay, G.V.; Kerlikowske, K.; Parvin, B.; Tlsty, T.D. Stress signaling from human mammary epithelial cells contributes to phenotypes of mammographic density. Cancer Res. 2014, 74, 5032–5044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 2019, 19, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somerville, T.D.; Biffi, G.; Dassler-Plenker, J.; Hur, S.K.; He, X.Y.; Vance, K.E.; Miyabayashi, K.; Xu, Y.; Maia-Silva, D.; Klingbeil, O.; et al. Squamous trans-differentiation of pancreatic cancer cells promotes stromal inflammation. ELife 2020, 9, e53381. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.S.; Keshari, K.R. Cancer Metabolism. In Imaging and Metabolism; Springer International Publishing: Berlin/Heidelberg, Germany, 2018. [Google Scholar] [CrossRef]
- Nizri, E.; Bar-David, S.; Aizic, A.; Sternbach, N.; Lahat, G.; Wolf, I.; Klausner, J. Desmoplasia in Lymph Node Metastasis of Pancreatic Adenocarcinoma Reveals Activation of Cancer-Associated Fibroblasts Pattern and T-helper 2 Immune Cell Infiltration. Pancreas 2019, 48, 367–373. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.; Sainson, R.C. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin. Cancer Biol. 2014, 25, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyan, S.W.; Kuo, W.H.; Huang, C.K.; Pan, C.C.; Shew, J.Y.; Chang, K.J.; Lee, E.Y.; Lee, W.H. Breast cancer cells induce cancer-associated fibroblasts to secrete hepatocyte growth factor to enhance breast tumorigenesis. PLoS ONE 2011, 6, e15313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGFbeta1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Matsuo, Y.; Ding, Q.; Desaki, R.; Maemura, K.; Mataki, Y.; Shinchi, H.; Natsugoe, S.; Takao, S. Hypoxia inducible factor-1 alpha plays a pivotal role in hepatic metastasis of pancreatic cancer: An immunohistochemical study. J. Hepato Biliary Pancreat. Sci. 2014, 21, 105–112. [Google Scholar] [CrossRef]
- Samain, R.; Sanz-Moreno, V. Cancer-associated fibroblasts: Activin A adds another string to their bow. Embo Mol. Med. 2020, 12, e12102. [Google Scholar] [CrossRef]
- Sun, Y.; Fan, X.; Zhang, Q.; Shi, X.; Xu, G.; Zou, C. Cancer-associated fibroblasts secrete FGF-1 to promote ovarian proliferation, migration, and invasion through the activation of FGF-1/FGFR4 signaling. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317712592. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Models Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [Green Version]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and Cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J. Exp. Clin. Cancer Res. CR 2019, 38, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Yan, M.; Wang, X.; Xu, Q.; Zhu, X.; Shi, J.; Li, Z.; Zhang, J.; Chen, W. Cancer-associated Fibroblast-derived IL-6 Promotes Head and Neck Cancer Progression via the Osteopontin-NF-kappa B Signaling Pathway. Theranostics 2018, 8, 921–940. [Google Scholar] [CrossRef] [PubMed]
- Sanford-Crane, H.; Abrego, J.; Sherman, M.H. Fibroblasts as Modulators of Local and Systemic Cancer Metabolism. Cancers 2019, 11, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, T.; Oldham, W.M.; Grasset, E.M.; Bourget, I.; Boulter, E.; Pisano, S.; Hofman, P.; Bellvert, F.; Meneguzzi, G.; Bulavin, D.V.; et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019, 29, 124–140.e10. [Google Scholar] [CrossRef] [Green Version]
- Valencia, T.; Kim, J.Y.; Abu-Baker, S.; Moscat-Pardos, J.; Ahn, C.S.; Reina-Campos, M.; Duran, A.; Castilla, E.A.; Metallo, C.M.; Diaz-Meco, M.T.; et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 2014, 26, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Balaji, U.; Freinkman, E.; McCue, P.; Witkiewicz, A.K. Unique metabolic features of pancreatic cancer stroma: Relevance to the tumor compartment, prognosis, and invasive potential. Oncotarget 2016, 7, 78396–78411. [Google Scholar] [CrossRef] [Green Version]
- Francescone, R.; Vendramini-Costa, D.B.; Franco-Barraza, J.; Wagner, J.; Muir, A.; Lau, A.N.; Gabitova, L.; Pazina, T.; Gupta, S.; Luong, T.; et al. The NetrinG1/NGL-1 Axis promotes pancreatic tumorigenesis through cancer associated fibroblast driven nutritional support and immunosuppression. BioRxiv 2019, 330209. [Google Scholar] [CrossRef] [Green Version]
- Shan, T.; Lu, H.; Ji, H.; Li, Y.; Guo, J.; Chen, X.; Wu, T. Loss of stromal caveolin-1 expression: A novel tumor microenvironment biomarker that can predict poor clinical outcomes for pancreatic cancer. PLoS ONE 2014, 9, e97239. [Google Scholar] [CrossRef]
- Codrici, E.; Albulescu, L.; Popescu, I.D.; Mihai, S.; Enciu, A.M.; Albulescu, R.; Tanase, C.; Hinescu, M.E. Caveolin-1-Knockout Mouse as a Model of Inflammatory Diseases. J. Immunol. Res. 2018, 2018, 2498576. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nadauld, L.; Ootani, A.; Corney, D.C.; Pai, R.K.; Gevaert, O.; Cantrell, M.A.; Rack, P.G.; Neal, J.T.; Chan, C.W.; et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat. Med. 2014, 20, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, A.J.; Castellanos, J.A.; Nagathihalli, N.S.; Merchant, N.B.; Skala, M.C. Optical Imaging of Drug-Induced Metabolism Changes in Murine and Human Pancreatic Cancer Organoids Reveals Heterogeneous Drug Response. Pancreas 2016, 45, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.W.; Lane, A.N.; Higashi, R.M. Stable Isotope Resolved Metabolomics Studies in Ex Vivo TIssue Slices. Bio-Protocol 2016, 6, e1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology 2011, 141, 1486–1497. [Google Scholar] [CrossRef]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://reactome.org/PathwayBrowser/#/R-HSA-2644603&PATH=R-HSA-1643685,R-HSA-5663202&DTAB=AN&ANALYSIS=MjAyMDA2MTcyMTMxMjlfMzk3 (accessed on 29 July 2020).
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-beta targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-beta/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Ellenrieder, V.; Fernandez Zapico, M.E.; Urrutia, R. TGFbeta-mediated signaling and transcriptional regulation in pancreatic development and cancer. Curr. Opin. Gastroenterol. 2001, 17, 434–440. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef]
- Hunt, K.K.; Fleming, J.B.; Abramian, A.; Zhang, L.; Evans, D.B.; Chiao, P.J. Overexpression of the tumor suppressor gene Smad4/DPC4 induces p21waf1 expression and growth inhibition in human carcinoma cells. Cancer Res. 1998, 58, 5656–5661. [Google Scholar]
- Grau, A.M.; Zhang, L.; Wang, W.; Ruan, S.; Evans, D.B.; Abbruzzese, J.L.; Zhang, W.; Chiao, P.J. Induction of p21waf1 expression and growth inhibition by transforming growth factor beta involve the tumor suppressor gene DPC4 in human pancreatic adenocarcinoma cells. Cancer Res. 1997, 57, 3929–3934. [Google Scholar]
- Lecanda, J.; Ganapathy, V.; D’Aquino-Ardalan, C.; Evans, B.; Cadacio, C.; Ayala, A.; Gold, L.I. TGFbeta prevents proteasomal degradation of the cyclin-dependent kinase inhibitor p27kip1 for cell cycle arrest. Cell Cycle 2009, 8, 742–756. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, I.; Imoto, M.; Adjei, P.N.; Gores, G.J.; Subramaniam, M.; Spelsberg, T.C.; Urrutia, R. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J. Clin. Investig. 1997, 99, 2365–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Marin-Garcia, P.; Ping, P.; Stein, L.; D’Eustachio, P.; Hermjakob, H. Reactome diagram viewer: Data structures and strategies to boost performance. Bioinformatics 2018, 34, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Seymour, A.B.; Hoque, A.T.; Schutte, M.; da Costa, L.T.; Redston, M.S.; Caldas, C.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; et al. Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res. 1995, 55, 4670–4675. [Google Scholar]
- Wilentz, R.E.; Iacobuzio-Donahue, C.A.; Argani, P.; McCarthy, D.M.; Parsons, J.L.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: Evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000, 60, 2002–2006. [Google Scholar]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar]
- Kawaguchi, Y.; Cooper, B.; Gannon, M.; Ray, M.; MacDonald, R.J.; Wright, C.V. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet. 2002, 32, 128–134. [Google Scholar] [CrossRef]
- Bardeesy, N.; Cheng, K.H.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Ehdaie, B.; Ohara, N.; Yoshino, T.; Deng, C.X. Synergistic action of Smad4 and Pten in suppressing pancreatic ductal adenocarcinoma formation in mice. Oncogene 2010, 29, 674–686. [Google Scholar] [CrossRef] [Green Version]
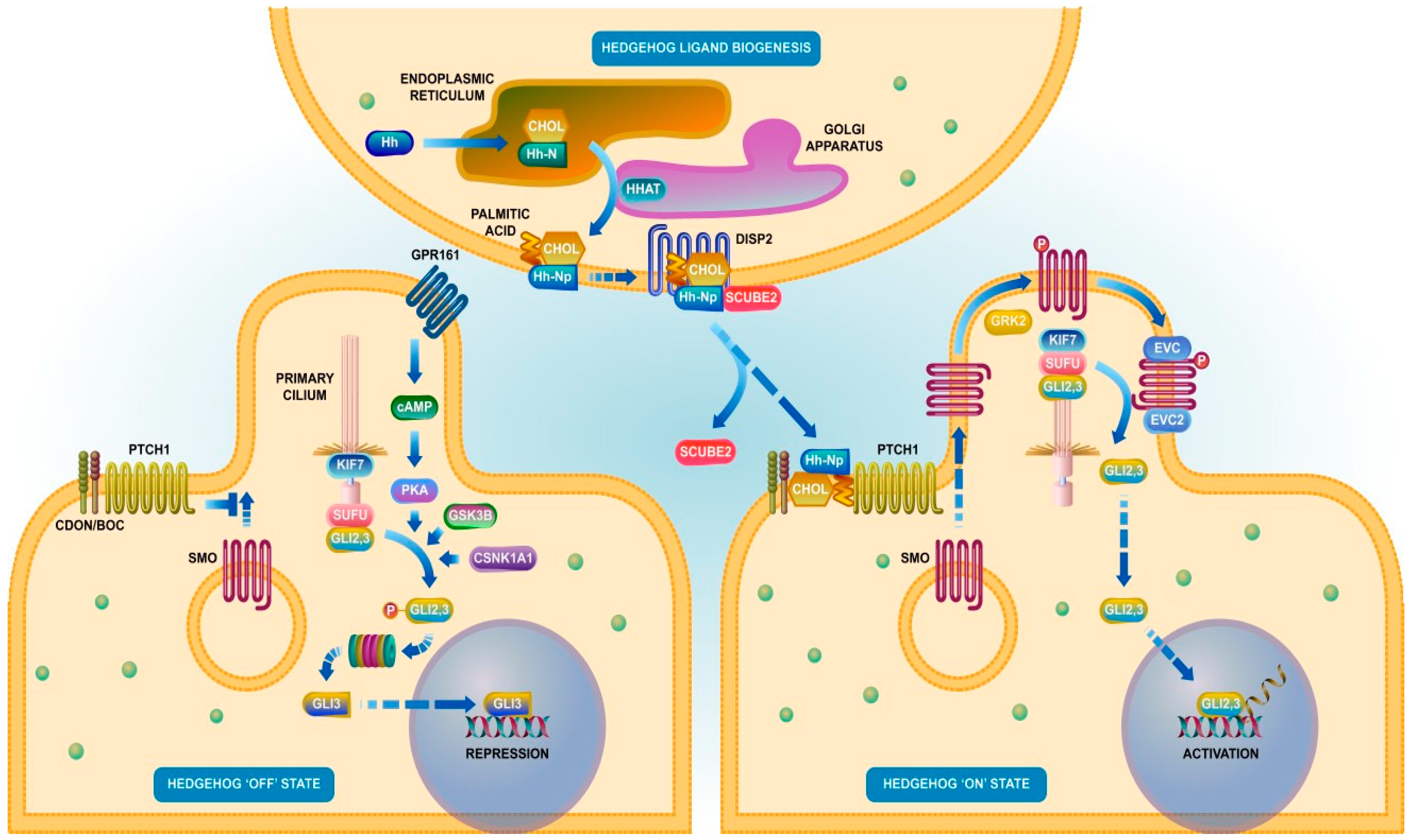
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Revi. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Chang, C.F.; Lin, S.S. Sonic Hedgehog Signaling in Organogenesis, Tumors, and Tumor Microenvironments. Int. J. Mol. Sci. 2020, 21, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Dong, J.; Li, Q.; Jin, Y.; Chen, B.; Zhou, M. Hedgehog Signaling in Pancreatic Fibrosis and Cancer. Medicine 2016, 95, e2996. [Google Scholar] [CrossRef]
- Gu, J.; Saiyin, H.; Fu, D.; Li, J. Stroma—A Double-Edged Sword in Pancreatic Cancer: A Lesson From Targeting Stroma in Pancreatic Cancer With Hedgehog Signaling Inhibitors. Pancreas 2018, 47, 382–389. [Google Scholar] [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [Green Version]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [Green Version]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernandez-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Hwang, R.F.; Moore, T.T.; Hattersley, M.M.; Scarpitti, M.; Yang, B.; Devereaux, E.; Ramachandran, V.; Arumugam, T.; Ji, B.; Logsdon, C.D.; et al. Inhibition of the hedgehog pathway targets the tumor-associated stroma in pancreatic cancer. Mol. Cancer Res. Mcr. 2012, 10, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Steinway, S.N.; Zanudo, J.G.; Ding, W.; Rountree, C.B.; Feith, D.J.; Loughran, T.P., Jr.; Albert, R. Network modeling of TGFbeta signaling in hepatocellular carcinoma epithelial-to-mesenchymal transition reveals joint sonic hedgehog and Wnt pathway activation. Cancer Res. 2014, 74, 5963–5977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inamoto, S.; Itatani, Y.; Yamamoto, T.; Minamiguchi, S.; Hirai, H.; Iwamoto, M.; Hasegawa, S.; Taketo, M.M.; Sakai, Y.; Kawada, K. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid-Derived Suppressor Cells through the CCL15-CCR1 Chemokine Axis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 492–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albo, D.; Berger, D.H.; Vogel, J.; Tuszynski, G.P. Thrombospondin-1 and transforming growth factor beta-1 upregulate plasminogen activator inhibitor type 1 in pancreatic cancer. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 1999, 3, 411–417. [Google Scholar] [CrossRef]
- Fang, Y.; Shen, Z.Y.; Zhan, Y.Z.; Feng, X.C.; Chen, K.L.; Li, Y.S.; Deng, H.J.; Pan, S.M.; Wu, D.H.; Ding, Y. CD36 inhibits beta-catenin/c-myc-mediated glycolysis through ubiquitination of GPC4 to repress colorectal tumorigenesis. Nat. Commun. 2019, 10, 3981. [Google Scholar] [CrossRef] [Green Version]
- Huiping, C.; Kristjansdottir, S.; Jonasson, J.G.; Magnusson, J.; Egilsson, V.; Ingvarsson, S. Alterations of E-cadherin and beta-catenin in gastric cancer. BMC Cancer 2001, 1, 16. [Google Scholar] [CrossRef] [Green Version]
- Joo, Y.E.; Rew, J.S.; Kim, H.S.; Choi, S.H.; Park, C.S.; Kim, S.J. Changes in the E-cadherin-catenin complex expression in early and advanced gastric cancers. Digestion 2001, 64, 111–119. [Google Scholar] [CrossRef]
- Miyazawa, K.; Iwaya, K.; Kuroda, M.; Harada, M.; Serizawa, H.; Koyanagi, Y.; Sato, Y.; Mizokami, Y.; Matsuoka, T.; Mukai, K. Nuclear accumulation of beta-catenin in intestinal-type gastric carcinoma: Correlation with early tumor invasion. Virchows Arch. Int. J. Pathol. 2000, 437, 508–513. [Google Scholar] [CrossRef]
- Aust, G.; Eichler, W.; Laue, S.; Lehmann, I.; Heldin, N.E.; Lotz, O.; Scherbaum, W.A.; Dralle, H.; Hoang-Vu, C. CD97: A dedifferentiation marker in human thyroid carcinomas. Cancer Res. 1997, 57, 1798–1806. [Google Scholar]
- Brown, R.S.; Bellisario, R.L.; Botero, D.; Fournier, L.; Abrams, C.A.; Cowger, M.L.; David, R.; Fort, P.; Richman, R.A. Incidence of transient congenital hypothyroidism due to maternal thyrotropin receptor-blocking antibodies in over one million babies. J. Clin. Endocrinol. Metab. 1996, 81, 1147–1151. [Google Scholar] [CrossRef] [Green Version]
- Post, Y.; Clevers, H. Defining Adult Stem Cell Function at Its Simplest: The Ability to Replace Lost Cells through Mitosis. Cell Stem Cell 2019, 25, 174–183. [Google Scholar] [CrossRef]
- Lazzari, E.; Butler, J.M. The Instructive Role of the Bone Marrow Niche in Aging and Leukemia. Curr. Stem Cell Rep. 2018, 4, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Kweon, S.M.; Teo, J.L.; Yuan, Y.C.; Melstrom, L.G.; Waldron, R.T.; Lugea, A.; Urrutia, R.A.; Pandol, S.J.; Lai, K.K.Y. Targeting the CBP/beta-Catenin Interaction to Suppress Activation of Cancer-Promoting Pancreatic Stellate Cells. Cancers 2020, 12, 1476. [Google Scholar] [CrossRef]
- Giordano, M.; Cavallaro, U. Different Shades of L1CAM in the Pathophysiology of Cancer Stem Cells. J. Clin. Med. 2020, 9, 1502. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, A.I.; Trinidad, J.R.; Sandiford, O.; Etchegaray, J.P.; Rameshwar, P. Epigenetic dynamics in cancer stem cell dormancy. Cancer Metastasis Rev. 2020, 33, 413–427. [Google Scholar] [CrossRef]
- Rahmani, W.; Sinha, S.; Biernaskie, J. Immune modulation of hair follicle regeneration. NPJ Regen. Med. 2020, 5, 9. [Google Scholar] [CrossRef]
- Siveen, K.S.; Prabhu, K.; Krishnankutty, R.; Kuttikrishnan, S.; Tsakou, M.; Alali, F.Q.; Dermime, S.; Mohammad, R.M.; Uddin, S. Vascular Endothelial Growth Factor (VEGF) Signaling in Tumour Vascularization: Potential and Challenges. Curr. Vasc. Pharmacol. 2017, 15, 339–351. [Google Scholar] [CrossRef]
- Isidori, A.M.; Venneri, M.A.; Fiore, D. Angiopoietin-1 and Angiopoietin-2 in metabolic disorders: Therapeutic strategies to restore the highs and lows of angiogenesis in diabetes. J. Endocrinol. Investig. 2016, 39, 1235–1246. [Google Scholar] [CrossRef]
- Sabir, S.; Saleem, A.; Akhtar, M.F.; Saleem, M.; Raza, M. Increasing beta cell mass to treat diabetes mellitus. Adv. Clin. Exp. Med. Off. Organ Wroc. Med. Univ. 2018, 27, 1309–1315. [Google Scholar] [CrossRef]
- Li, J.N.; Li, W.; Cao, L.Q.; Liu, N.; Zhang, K. Efficacy of mesenchymal stem cells in the treatment of gastrointestinal malignancies. World J. Gastrointest. Oncol. 2020, 12, 365–382. [Google Scholar] [CrossRef]
- Jiang, Z.; White, R.A.; Wang, T.C. Adult Pancreatic Acinar Progenitor-like Populations in Regeneration and Cancer. Trends Mol. Med. 2020, 26, 758–767. [Google Scholar] [CrossRef]
- Wang, W.; Bochtler, T.; Wuchter, P.; Manta, L.; He, H.; Eckstein, V.; Ho, A.D.; Lutz, C. Mesenchymal stromal cells contribute to quiescence of therapy-resistant leukemic cells in acute myeloid leukemia. Eur. J. Haematol. 2017, 99, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.S.; Capecchi, M.R. Lrig1 expression prospectively identifies stem cells in the ventricular-subventricular zone that are neurogenic throughout adult life. Neural Dev. 2020, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jo, Y.H.; Jang, M.; Nguyen, N.N.Y.; Yun, H.R.; Ko, S.H.; Shin, Y.; Lee, J.S.; Kang, I.; Ha, J.; et al. PAC-5 Gene Expression Signature for Predicting Prognosis of Patients with Pancreatic Adenocarcinoma. Cancers 2019, 11, 1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, F.M.; Yang, J.; Nasrallah, R.; Clarke, J.; Sadiyah, F.; Whiteside, S.K.; Imianowski, C.J.; Kuo, P.; Vardaka, P.; Todorov, T.; et al. BACH2 drives quiescence and maintenance of resting Treg cells to promote homeostasis and cancer immunosuppression. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Hossen, M.N.; Rao, G.; Dey, A.; Robertson, J.D.; Bhattacharya, R.; Mukherjee, P. Gold Nanoparticle Transforms Activated Cancer-Associated Fibroblasts to Quiescence. Acs Appl. Mater. Interfaces 2019, 11, 26060–26068. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Zhao, X.; Gupta, V.K.; Sharma, N.; Kesh, K.; Gnamlin, P.; Dudeja, V.; Vickers, S.M.; Banerjee, S.; Saluja, A. Inactivation of Cancer-Associated-Fibroblasts Disrupts Oncogenic Signaling in Pancreatic Cancer Cells and Promotes Its Regression. Cancer Res. 2018, 78, 1321–1333. [Google Scholar] [CrossRef] [Green Version]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef]
- Cai, Z.; Liang, Y.; Xing, C.; Wang, H.; Hu, P.; Li, J.; Huang, H.; Wang, W.; Jiang, C. Cancerassociated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol. Rep. 2019, 42, 2537–2549. [Google Scholar] [CrossRef] [Green Version]
- Le, Y.; Gao, H.; Richards, W.G.; Zhao, L.; Bleday, R.; Clancy, T.; Zhu, Z. VentX expression in tumor associated macrophages promotes phagocytosis and immunity against pancreatic cancers. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers 2017, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Mu, W.; Wang, Z.; Zoller, M. Ping-Pong-Tumor and Host in Pancreatic Cancer Progression. Front. Oncol. 2019, 9, 1359. [Google Scholar] [CrossRef] [PubMed]
- Saison-Ridinger, M.; DelGiorno, K.E.; Zhang, T.; Kraus, A.; French, R.; Jaquish, D.; Tsui, C.; Erikson, G.; Spike, B.T.; Shokhirev, M.N.; et al. Reprogramming pancreatic stellate cells via p53 activation: A putative target for pancreatic cancer therapy. PLoS ONE 2017, 12, e0189051. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Dalla Pozza, E.; Fanelli, G.; Di Carlo, C.; Vettori, A.; Cannino, G.; Cavallini, C.; Carmona-Carmona, C.A.; Brandi, J.; Rinalducci, S.; et al. Progressively De-Differentiated Pancreatic Cancer Cells Shift from Glycolysis to Oxidative Metabolism and Gain a Quiescent Stem State. Cells 2020, 9, 1572. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process/Changes | Activated Molecules | References |
|---|---|---|
| Activation Ligands | transforming growth factor-β (TGFβ), platelet-derived growth factors (PDGFs), epidermal growth factors (EGFs), fibroblast growth factors (FGFs), bone morphogenic proteins (BMPs), Sonic Hedgehog (Shh) | [103] |
| Contact Signals | notch signaling | [118] |
| Inflammatory Modulators | IL-1 →NFkB and IL-6 →STAT transcription factors → JAK–STAT signaling → contractile cytoskeleton and histone acetylation | [119,120] |
| Physical Changes in EC | stiffness and composition | [121,122,123] |
| Physiological Stress | heat-shock factors | [124] |
| Genomic Stress/DNA Damage—Chemo-/Radiotherapy | ROS | [106,125] |
| Molecules Released by CAFs | Modified Processes that Orchestrate Tumor Development and Immune Evasion | References |
|---|---|---|
| VEGFs, PDGFs, HGF, IL-8, SDF-1 | enhanced angiogenesis | [140,141] |
| IL-6, IL-1 | enhanced inflammation | [142] |
| IL-6, TGFβ, SDF-1, HGF, lysophosphatidylcholines-LPCs | enhanced cell proliferation | [143,144,145] |
| TGFβ, COX-2/PGE2 | enhanced motility | [146] |
| IL-6, CXCL 12 | macrophage switch and immune evasion | [147] |
| HIF1a, lactate dehydrogenase A | altered metabolism | [148] |
| differentiation factors, activin A, FGF2 | altered cell fate | [149] |
| TGFβ, HGF, FGFs, NGF, IGF | enhanced secretion of cytokines and growth factors | [150,151] |
| Fibronectin, collagen 1, tenascin C, osteopontin—MMPs | deposition and remodeling of ECM | [141,152,153,154] |
| Cell Type | Positioning (Tumor or Associated) | Facts (or Proposed Mechanisms) | References |
|---|---|---|---|
| Stromal | “Pancreatic cancer-associated fibroblasts” | “Gold nanoparticle transforms activated cancer-associated fibroblasts to quiescence by enhancing lipid synthesis and lipid utilization”. | [227] |
| “Anticancer compound Minnelide revealed deregulation of the TGFb signalling pathway in CAF, | [228] | ||
| resulting in an apparent reversal of their activated state to a quiescent, nonproliferative state”. | [229] | ||
| “This heterogeneity explains why one type of CAF is found to support cancer invasiveness and metastases while another type does not”. | |||
| “Pancreatic cancer-associated adipocyte” | “Cancer-associated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer”, but quiescence was not examined. | [230] | |
| Pancreatic tumor-associated macrophages | “The expression of homeobox protein VentX, a master regulator of macrophage plasticity, is significantly decreased in the PDA-TAMs”. | [231] | |
| Stellate cells | “Up-regulation of Ppar-γ which is associated with quiescence”. | [214] | |
| “In the healthy pancreas, PSCs are in the quiescent state and retain vitamin A-containing lipid droplets”. | [232,233] | ||
| “PSC, quiescent in the healthy pancreas. During pancreatic injury, PSC develop a myofibroblast phenotype expressing αSMA1, actively proliferate and migrate. Activation of PSC is promoted by TGFβ, HGF, FGF, EGF, and sHH”. | [234] | ||
| “p53 activation by Nutlin-3a induces profound transcriptional changes, which reprogram activated PSCs to quiescence”. | |||
| Cancer stem cell | Quiescent stem cells are characterized by high chemo-resistance, clonogenic ability, and metastatic potential. | [235] | |
| Pancreatic progenitor-like cell | “Dclk1+ and Stmn+ cells are long-lived, largely quiescent, and lack proliferation under resting conditions”. | [222] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanase, C.; Gheorghisan-Galateanu, A.-A.; Popescu, I.D.; Mihai, S.; Codrici, E.; Albulescu, R.; Hinescu, M.E. CD36 and CD97 in Pancreatic Cancer versus Other Malignancies. Int. J. Mol. Sci. 2020, 21, 5656. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165656
Tanase C, Gheorghisan-Galateanu A-A, Popescu ID, Mihai S, Codrici E, Albulescu R, Hinescu ME. CD36 and CD97 in Pancreatic Cancer versus Other Malignancies. International Journal of Molecular Sciences. 2020; 21(16):5656. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165656
Chicago/Turabian StyleTanase, Cristiana, Ancuta-Augustina Gheorghisan-Galateanu, Ionela Daniela Popescu, Simona Mihai, Elena Codrici, Radu Albulescu, and Mihail Eugen Hinescu. 2020. "CD36 and CD97 in Pancreatic Cancer versus Other Malignancies" International Journal of Molecular Sciences 21, no. 16: 5656. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21165656