Computer-Aided Estimation of Biological Activity Profiles of Drug-Like Compounds Taking into Account Their Metabolism in Human Body



Abstract
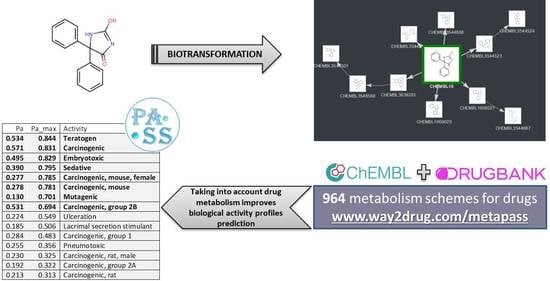
:
1. Introduction
2. Results
2.1. Information about Drug Substances and Their Metabolites
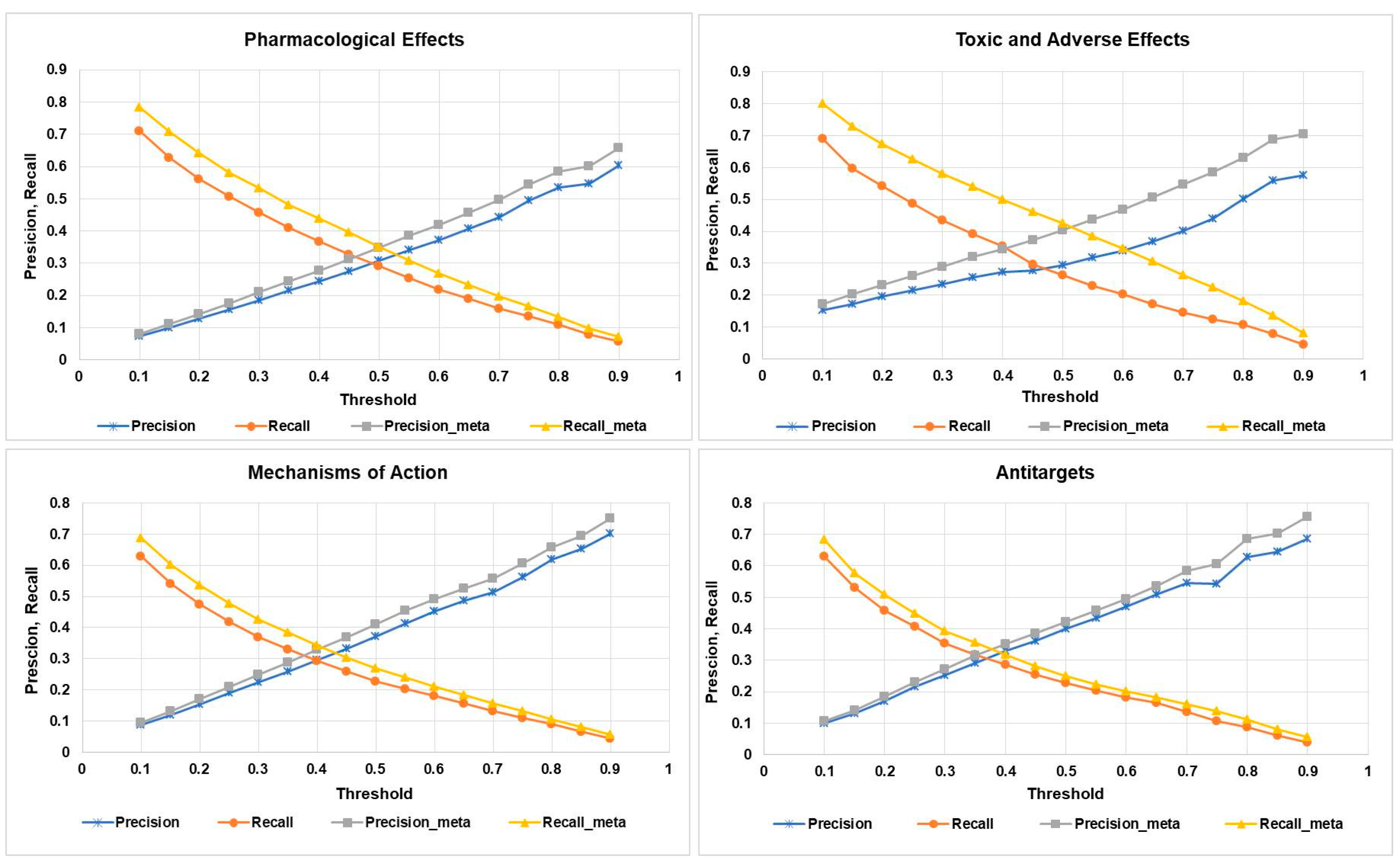
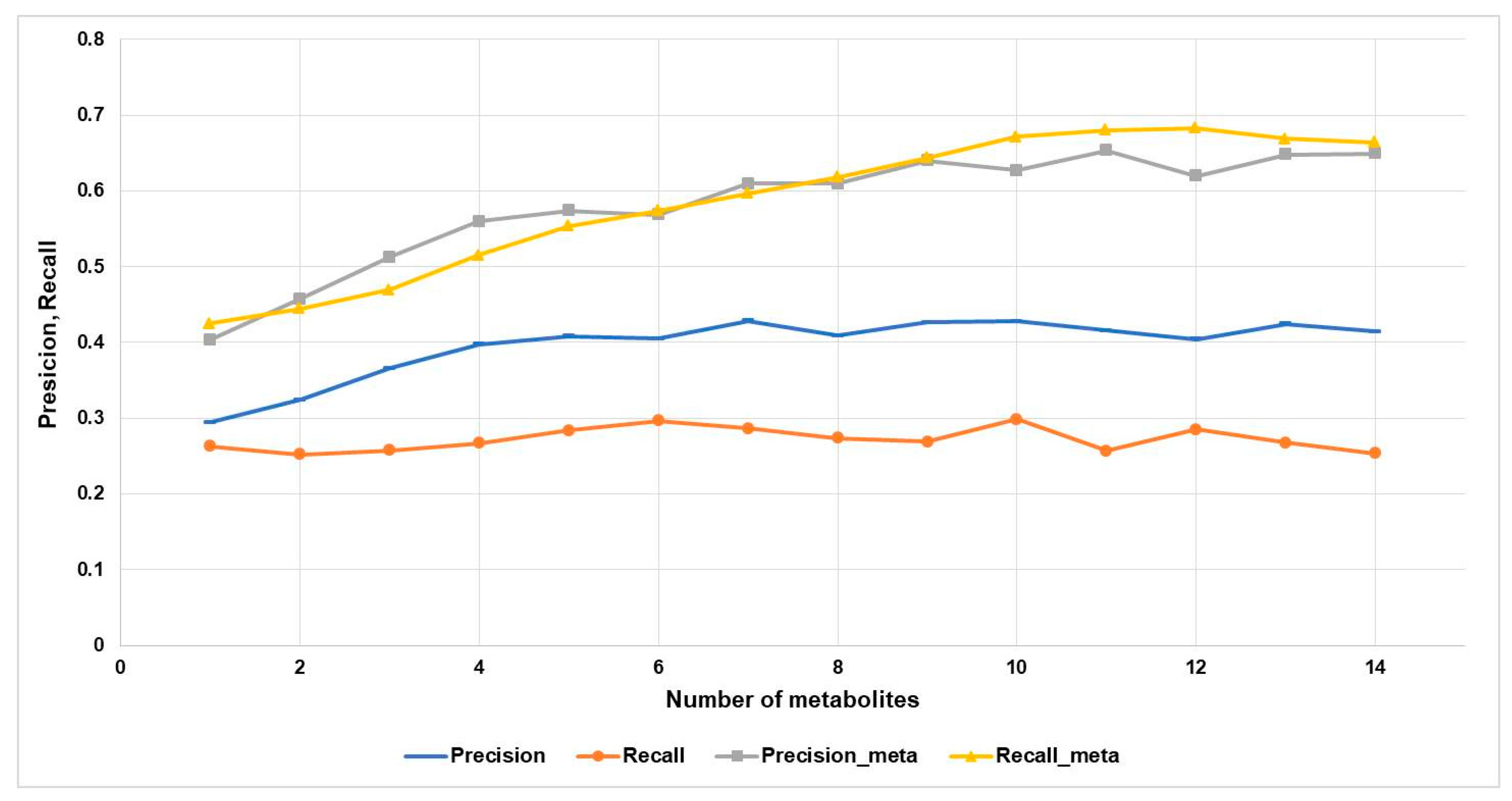
2.2. Accuracy of Prediction of Biological Activity of Compounds with and without Considering their Metabolites
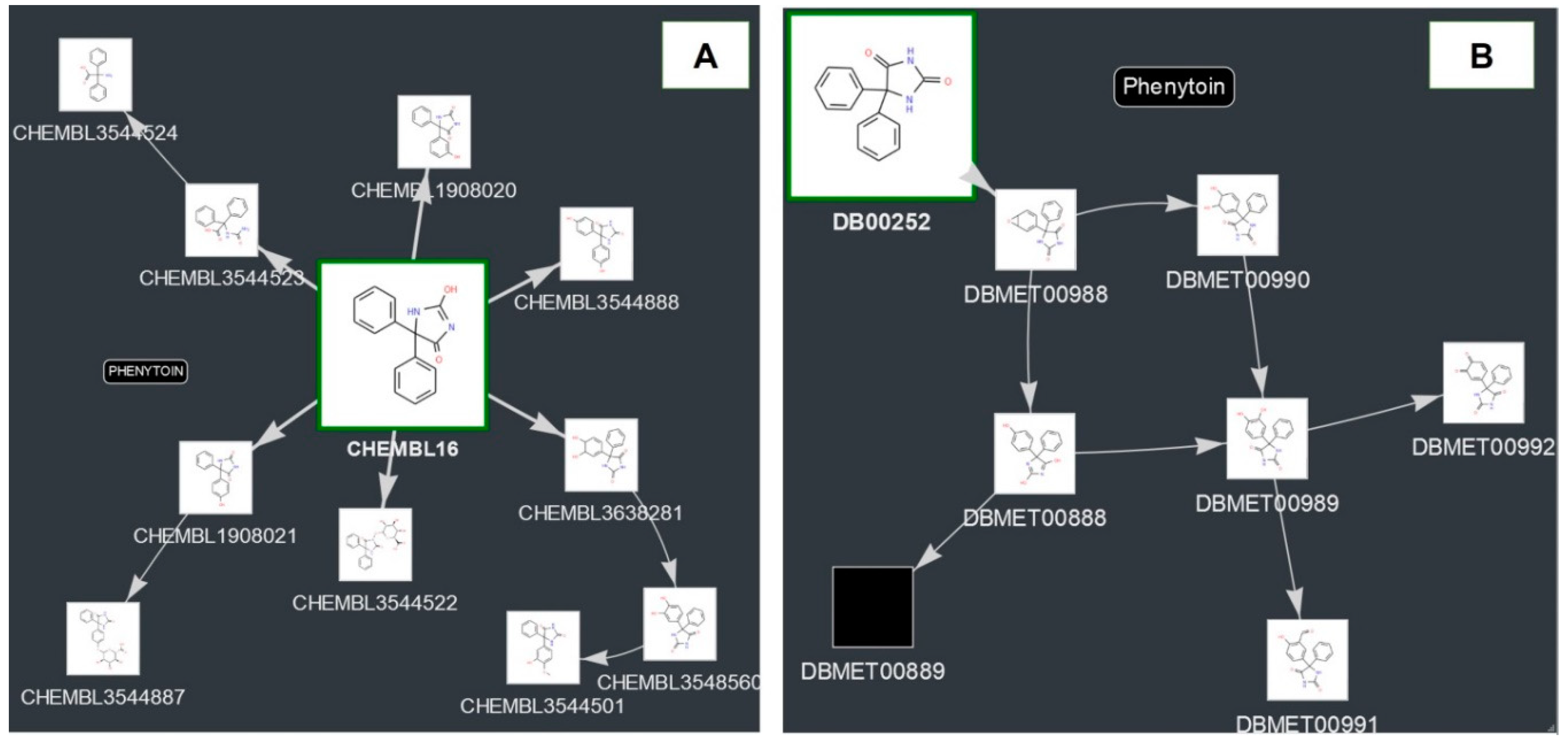
2.3. Examples of Predictions for Some Drugs
2.3.1. Toxic and Adverse Effects Predicted for Phenytoin and Its Metabolites
2.3.2. Mechanisms of Action Predicted for Clozapine and Its Metabolites
3. Discussion
4. Materials and Methods
4.1. Prediction of Activity Spectra for Substances (PASS)
4.2. Sources of Information for Creation of the Metabolic Graphs
4.3. Integral Biological Activity Spectra Assessment
4.4. (Q)SAR Quality Metrics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scavone, C.; di Mauro, G.; Mascolo, A.; Berrino, L.; Rossi, F.; Capuano, A. The new paradigms in clinical research: From early access programs to the novel therapeutic approaches for unmet medical needs. Front. Pharmacol. 2019, 10, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, F.; Rosich, L. The contributions of Paul Ehrlich to pharmacology: A tribute on the occasion of the centenary of his Nobel Prize. Pharmacology 2008, 82, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, A.L. Network pharmacology. Nat. Biotechnol. 2007, 25, 1110–1111. [Google Scholar] [CrossRef] [PubMed]
- Bezhentsev, V.M.; Tarasova, O.A.; Dmitriev, A.V.; Rudik, A.V.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. Computer-aided prediction of xenobiotics metabolism in the human organism. Russ. Chem. Rev. 2016, 85, 854–879. [Google Scholar] [CrossRef]
- Dixit, V.A.; Bharatam, P.V. Toxic metabolite formation from Troglitazone (TGZ): New insights from a DFT study. Chem. Res. Toxicol. 2011, 24, 1113–1122. [Google Scholar] [CrossRef]
- Martinez-Mayorga, K.; Madariaga-Mazon, A.; Medina-Franco, J.L.; Maggiora, G. The impact of chemoinformatics on drug discovery in the pharmaceutical industry. Expert Opin. Drug. Discov. 2020, 15, 293–306. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Poroikov, V.V. Probabilistic Approach in Activity Prediction. In Chemoinformatics Approaches to Virtual Screening; Varnek, A., Tropsha, A., Eds.; RSC Publishing: Cambridge, UK, 2018; pp. 182–216. [Google Scholar]
- Wang, L.; Ma, C.; Wipf, P.; Liu, H.; Su, W.; Xie, X.Q. TargetHunter: An in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. 2013, 15, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Gfeller, D.; Grosdidier, A.; Wirth, M.; Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: A web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014, 42, W32–W38. [Google Scholar] [CrossRef]
- Kringelum, J.; Kjaerulff, S.K.; Brunak, S.; Lund, O.; Oprea, T.I.; Taboureau, O. ChemProt-3.0: A global chemical biology diseases mapping. Database (Oxford) 2016, 2016, bav123. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.K. Computational modeling of P450s for toxicity prediction. Expert Opin. Drug Metab. Toxicol. 2011, 7, 1211–1231. [Google Scholar] [CrossRef]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyzack, J.D.; Kirchmair, J. Computational methods and tools to predict cytochrome P450 metabolism for drug discovery. Chem. Biol. Drug Des. 2019, 93, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Comp. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Poroikov, V.V.; Filimonov, D.A.; Gloriozova, T.A.; Lagunin, A.A.; Druzhilovskiy, D.S.; Rudik, A.V.; Stolbov, L.A.; Dmitriev, A.V.; Tarasova, O.A.; Ivanov, S.M.; et al. Computer-aided prediction of biological activity spectra for organic compounds: The possibilities and limitations. Russ. Chem. Bull. 2019, 68, 2143–2154. [Google Scholar] [CrossRef]
- DrugBank database. Available online: https://www.drugbank.ca/ (accessed on 30 August 2020).
- ChEMBL database. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 30 August 2020).
- Cortellis Drug Discovery Intelligence database. Available online: https://www.cortellis.com/drugdiscovery/ (accessed on 30 August 2020).
- Iorga, A.; Horowitz, B.Z. Phenytoin Toxicity. In: StatPearls [Internet]. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK482444/ (accessed on 30 August 2020).
- Sasaki, E.; Yokoi, T. Role of cytochrome P450-mediated metabolism and involvement of reactive metabolite formations on antiepileptic drug-induced liver injuries. J. Toxicol. Sci. 2018, 43, 75–87. [Google Scholar] [CrossRef]
- Ivanov, S.M.; Lagunin, A.A.; Rudik, A.V.; Filimonov, D.A.; Poroikov, V.V. ADVERPred—Web service for prediction of adverse effects of drugs. J. Chem. Inf. Model. 2018, 58, 8–11. [Google Scholar] [CrossRef]
- Kondej, M.; Stępnicki, P.; Kaczor, A.A. Multi-target approach for drug discovery against schizophrenia. Int. J. Mol. Sci. 2018, 19, 3105. [Google Scholar] [CrossRef] [Green Version]
- Peach, M.L.; Zakharov, A.V.; Liu, R.; Pugliese, A.; Tawa, G.; Wallqvist, A.; Nicklaus, M.C. Computational tools and resources for metabolism-related property predictions. 1. Overview of publicly available (free and commercial) databases and software. Future Med. Chem. 2012, 4, 1907–1932. [Google Scholar] [CrossRef] [Green Version]
- Obach, R.S. Pharmacologically active drug metabolites: Impact on drug discovery and pharmacotherapy. Pharmacol. Rev. 2013, 65, 578–640. [Google Scholar] [CrossRef] [Green Version]
- Borodina, Y.V.; Filimonov, D.A.; Poroikov, V.V. Computer-aided prediction of prodrug activity using the PASS system. Pharm. Chem. J. 1996, 30, 760–763. [Google Scholar] [CrossRef]
- Ivanov, S.M.; Lagunin, A.A.; Poroikov, V.V. In silico assessment of adverse drug reactions and associated mechanisms. Drug Discov. Today 2016, 21, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 update: A web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res 2017, 45, W356–W360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forouzesh, A.; Samadi Foroushani, S.; Forouzesh, F.; Zand, E. Reliable target prediction of bioactive molecules based on chemical similarity without employing statistical methods. Front Pharmacol. 2019, 10, 835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Peng, Y.; Yu, Z.; Li, W.; Liu, G.; Tang, Y. NetInfer: A web server for prediction of targets and therapeutic and adverse effects via network-based inference methods. J. Chem. Inf. Model. 2020, 60, 3687–3691. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Leszczynski, J. Recent advances of computational modeling for predicting drug metabolism: A perspective. Curr. Drug Metab. 2017, 18, 1106–1122. [Google Scholar] [CrossRef]
- He, C.; Wan, H. Drug metabolism and metabolite safety assessment in drug discovery and development. Expert Opin. Drug Metab. Toxicol. 2018, 14, 1071–1085. [Google Scholar] [CrossRef]
- Kazmi, S.R.; Jun, R.; Yu, M.S.; Jung, C.; Na, D. In silico approaches and tools for the prediction of drug metabolism and fate: A review. Comput. Biol. Med. 2019, 106, 54–64. [Google Scholar] [CrossRef]
- Murtazalieva, K.A.; Druzhilovskii, D.S.; Goel, R.K.; Sastry, G.N.; Poroikov, V.V. How good are publicly available web services that predict bioactivity profiles for drug repurposing? SAR QSAR Environ. Res. 2017, 28, 843–862. [Google Scholar] [CrossRef]
- Rudik, A.V.; Dmitriev, A.V.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. PASS-based prediction of metabolites detection in biological systems. SAR QSAR Environ. Res. 2019, 30, 751–758. [Google Scholar] [CrossRef]
- MetaPASS web server. Available online: http://www.way2drug.com/metapass/ (accessed on 30 August 2020).
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef]
- Bajorath, J. Computational studies, virtual screening, and theoretical molecular models. J. Med. Chem. 2010, 53, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Hillisch, A.; Heinrich, N.; Wild, H. Computational chemistry in the pharmaceutical industry: From childhood to adolescence. ChemMedChem 2015, 10, 1958–1962. [Google Scholar] [CrossRef] [PubMed]
- ZINC database. Available online: https://zinc.docking.org/ (accessed on 30 August 2020).
- SAVI Library. Available online: https://cactus.nci.nih.gov/download/savi_download/ (accessed on 30 August 2020).
- REAL database. Available online: https://enamine.net/hit-finding/compound-collections/real-database (accessed on 30 August 2020).
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, S.; Li, F.; Zhou, Y.; Zhang, Y.; Wang, Z.; Zhang, R.; Zhu, J.; Ren, Y.; Tan, Y.; et al. Therapeutic target database 2020: Enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res 2020, 48, D1031–D1041. [Google Scholar] [CrossRef] [Green Version]
- Westerhoff, H.V.; Nakayama, S.; Mondeel, T.D.; Barberis, M. Systems Pharmacology: An opinion on how to turn the impossible into grand challenges. Drug Discov. Today Technol. 2015, 15, 23–31. [Google Scholar] [CrossRef]
- Lipinski, C.; Hopkins, A. Navigating chemical space for biology and medicine. Nature 2004, 432, 855–861. [Google Scholar] [CrossRef]
- Burov, Y.V.; Poroikov, V.V.; Korolchenko, L.V. National system for registration and biological testing of chemical compounds: Facilities for new drugs search. Bull. Natl. Cent. Biol. Act. Compd. (Rus.) 1990, 1, 4–25. [Google Scholar]
- Poroikov, V.V.; Filimonov, D.A.; Borodina, Y.V.; Lagunin, A.A.; Kos, A. Robustness of biological activity spectra predicting by computer program PASS for non-congeneric sets of chemical compounds. J. Chem. Inform. Comput. Sci. 2000, 40, 1349–1355. [Google Scholar] [CrossRef]
- Filimonov, D.; Poroikov, V.; Borodina, Y.; Gloriozova, T. Chemical similarity assessment through multilevel neighborhoods of atoms: Definition and comparison with the other descriptors. J. Chem. Inform. Comput. Sci. 1999, 39, 666–670. [Google Scholar] [CrossRef]
- Glen, R.C.; Bender, A.; Arnby, C.H.; Carlsson, L.; Boyer, S.; Smith, J. Circular fingerprints: Flexible molecular descriptors with applications from physical chemistry to ADME. IDrugs 2006, 9, 199–204. [Google Scholar]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, 1074–1082. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pa | Pa_Max | Activity |
|---|---|---|
| 0.534 | 0.844 | Teratogen |
| 0.571 | 0.831 | Carcinogenic |
| 0.495 | 0.829 | Embryotoxic |
| 0.39 | 0.795 | Sedative |
| 0.277 | 0.785 | Carcinogenic, mouse, female |
| 0.278 | 0.781 | Carcinogenic, mouse |
| 0.13 | 0.701 | Mutagenic |
| 0.531 | 0.694 | Carcinogenic, group 2B |
| 0.224 | 0.549 | Ulceration |
| 0.185 | 0.506 | Lacrimal secretion stimulant |
| 0.284 | 0.483 | Carcinogenic, group 1 |
| 0.255 | 0.356 | Pneumotoxic |
| 0.23 | 0.325 | Carcinogenic, rat, male |
| 0.192 | 0.322 | Carcinogenic, group 2A |
| 0.213 | 0.313 | Carcinogenic, rat |
| No | Activity * | No | Activity * |
|---|---|---|---|
| 1 | 5 Hydroxytryptamine 1 antagonist | 35 | Alpha 2 adrenoreceptor antagonist |
| 2 | 5 Hydroxytryptamine 1A antagonist | 36 | Alpha 2a adrenoreceptor antagonist |
| 3 | 5 Hydroxytryptamine 1B antagonist | 37 | Alpha 2b adrenoreceptor antagonist |
| 4 | 5 Hydroxytryptamine 1D antagonist | 38 | Alpha 2c adrenoreceptor antagonist |
| 5 | 5 Hydroxytryptamine 2 antagonist | 39 | Alpha adrenoreceptor agonist |
| 6 | 5 Hydroxytryptamine 2A antagonist | 40 | Alpha adrenoreceptor antagonist |
| 7 | 5 Hydroxytryptamine 2B antagonist | 41 | Analgesic |
| 8 | 5 Hydroxytryptamine 2C antagonist | 42 | Antiadrenergic |
| 9 | 5 Hydroxytryptamine 3 antagonist | 42 | Antihistaminic |
| 10 | 5 Hydroxytryptamine 6 antagonist | 44 | Cholinergic antagonist |
| 11 | 5 Hydroxytryptamine 7 antagonist | 45 | Dopamine D1 antagonist |
| 12 | 5 Hydroxytryptamine agonist | 46 | Dopamine D2 antagonist |
| 13 | 5 Hydroxytryptamine antagonist | 47 | Dopamine D3 antagonist |
| 14 | Acetylcholine agonist | 48 | Dopamine D4 agonist |
| 15 | Acetylcholine antagonist | 49 | Dopamine D4 antagonist |
| 16 | Acetylcholine M1 receptor agonist | 50 | GABA A receptor antagonist |
| 17 | Acetylcholine M1 receptor antagonist | 51 | GABA receptor antagonist |
| 18 | Acetylcholine M2 receptor antagonist | 52 | Histamine agonist |
| 19 | Acetylcholine M3 receptor antagonist | 53 | Histamine antagonist |
| 20 | Acetylcholine M4 receptor antagonist | 54 | Histamine H1 receptor antagonist |
| 21 | Acetylcholine M5 receptor antagonist | 55 | Histamine H2 receptor antagonist |
| 22 | Acetylcholine muscarinic agonist | 56 | Histamine H3 receptor antagonist |
| 23 | Acetylcholine muscarinic antagonist | 57 | Histamine H4 receptor agonist |
| 24 | Adenosine A3 receptor antagonist | 58 | Histamine H4 receptor antagonist |
| 25 | Adenylate cyclase inhibitor | 59 | Immunomodulator |
| 26 | Adrenaline agonist | 60 | Opioid agonist |
| 27 | Adrenaline antagonist | 61 | Opioid delta receptor agonist |
| 28 | Adrenaline uptake inhibitor | 62 | Peptidyl-prolyl cis-trans isomerase inhibitor |
| 29 | Alpha 1 adrenoreceptor agonist | 63 | Potassium channel (Voltage-sensitive) blocker |
| 30 | Alpha 1 adrenoreceptor antagonist | 64 | Potassium channel blocker |
| 31 | Alpha 1a adrenoreceptor antagonist | 65 | Sigma receptor antagonist |
| 32 | Alpha 1b adrenoreceptor antagonist | 66 | Transcription factor inhibitor |
| 33 | Alpha 1d adrenoreceptor antagonist | 67 | Transcription factor NF kappa B inhibitor |
| 34 | Alpha 2 adrenoreceptor agonist |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filimonov, D.A.; Rudik, A.V.; Dmitriev, A.V.; Poroikov, V.V. Computer-Aided Estimation of Biological Activity Profiles of Drug-Like Compounds Taking into Account Their Metabolism in Human Body. Int. J. Mol. Sci. 2020, 21, 7492. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207492
Filimonov DA, Rudik AV, Dmitriev AV, Poroikov VV. Computer-Aided Estimation of Biological Activity Profiles of Drug-Like Compounds Taking into Account Their Metabolism in Human Body. International Journal of Molecular Sciences. 2020; 21(20):7492. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207492
Chicago/Turabian StyleFilimonov, Dmitry A., Anastassia V. Rudik, Alexander V. Dmitriev, and Vladimir V. Poroikov. 2020. "Computer-Aided Estimation of Biological Activity Profiles of Drug-Like Compounds Taking into Account Their Metabolism in Human Body" International Journal of Molecular Sciences 21, no. 20: 7492. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21207492