Genetic Variants of Lipoprotein Lipase and Regulatory Factors Associated with Alzheimer’s Disease Risk


Abstract
:1. Introduction
2. LPL: Structure, Function, and Regulation
3. LPL in the Central Nervous System
4. LPL Variants and AD
5. Genetic Variants Regulating LPL Function and Processing and AD Risk
6. LPL Variants and Other CNS Disorders
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| LPL | Lipoprotein Lipase |
| Aβ | Amyloid-Beta |
| AD | Alzheimer’s Disease |
| ApoC-II | ApoproteinC-II |
| ApoC-III | ApoproteinC-III |
| ChEIs | Cholinesterase inhibitors |
| DMTs | Disease-modifying therapeutics |
| TG | Triglycerides |
| VLDL | Very-low-density lipoprotein |
| CVD | Cardiovascular Disease |
| CNS | Central Nervous system |
| OPCs | Oligodendrocytes |
| GPIHBP | Glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 |
| TRL | Triglyceride-rich lipoproteins |
| LMF1 | Lipoprotein Maturation factor 1 |
| FFAs | Free Fatty Acids |
| LRP | LDL receptor-related protein/α2-macroglobulin receptor LRP2 |
| RAP | Receptor associated protein |
| HSPGs | Heparan sulfate proteoglycans |
| ANGPTL (3,4,8) | Angiopoietin-like protein (3,4,8) |
| HDL | High-density lipoproteins |
| DOCK | Dedicator of cytokinesis |
| APOE | Apoprotein E |
| CLU | Clusterin |
| ApoJ | Apoprotein J |
| TREM2 | Triggering receptor expressed on myeloid cells |
| PUFAs | Polyunsaturated fatty acids |
| SORLA | Sortilin |
| MS | Multiple Sclerosis |
| DAMS | Disease Associated Microglia |
| PAMS | Proliferative-region-associated-microglia |
| FCS | Familial chylomicronemia syndrome |
| LOAD | Late onset AD |
| CAD | Coronary Artery Disease |
| SAD | Sporadic AD |
References
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s Disease: The Challenge of the Second Century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessesen, D.H.; Richards, C.L.; Etienne, J.; Goers, J.W.; Eckel, R.H. Spinal cord of the rat contains more lipoprotein lipase than other brain regions. J. Lipid Res. 1993, 34, 229–238. [Google Scholar] [PubMed]
- Eckel, R.H.; Robbins, R.J. Lipoprotein lipase is produced, regulated, and functional in rat brain. Proc. Natl. Acad. Sci. USA 1984, 81, 7604–7607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, I.J.; Soprano, D.R.; Wyatt, M.L.; Vanni, T.M.; Kirchgessner, T.G.; Schotz, M.C. Localization of lipoprotein lipase mRNA in selected rat tissues. J. Lipid Res. 1989, 30, 1569–1577. [Google Scholar]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Ren, X. Meta-analyses of four polymorphisms of lipoprotein lipase associated with the risk of Alzheimer’s disease. Neurosci. Lett. 2016, 619, 73–78. [Google Scholar] [CrossRef]
- Baum, L.; Wiebusch, H.; Pang, C.P. Roles for lipoprotein lipase in Alzheimer’s disease: An association study. Microsc. Res. Tech. 2000, 50, 291–296. [Google Scholar] [CrossRef]
- Gao, Y.; Vidal-Itriago, A.; Kalsbeek, M.J.; Layritz, C.; Garcia-Caceres, C.; Tom, R.Z.; Eichmann, T.O.; Vaz, F.M.; Houtkooper, R.H.; Van Der Wel, N.; et al. Lipoprotein Lipase Maintains Microglial Innate Immunity in Obesity. Cell Rep. 2017, 20, 3034–3042. [Google Scholar] [CrossRef] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.D.; Gorkhali, S.; Given, K.; Coates, A.M.; Boyle, K.E.; Macklin, W.B.; Eckel, R.H. Lipoprotein Lipase Is a Feature of Alternatively-Activated Microglia and May Facilitate Lipid Uptake in the CNS During Demyelination. Front. Mol. Neurosci. 2018, 11, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Bao, J.; Zhao, X.; Shen, H.; Lv, J.; Ma, S.; Zhang, X.; Li, Z.; Wang, S.; Wang, Q.; et al. Activated Cyclin-Dependent Kinase 5 Promotes Microglial Phagocytosis of Fibrillar β-Amyloid by Up-regulating Lipoprotein Lipase Expression. Mol. Cell. Proteom. 2013, 12, 2833–2844. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Nimonkar, A.V.; Baird, D.; Wang, C.; Chiu, C.-H.; Horton, P.A.; Hanrahan, S.; Cubbon, R.; Weldon, S.; Tschantz, W.R.; et al. Structure of lipoprotein lipase in complex with GPIHBP1. Proc. Natl. Acad. Sci. USA 2019, 116, 10360–10365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.-Z.; Xie, S.-L.; Jin, R.; Huang, Z.-M. A novel lipoprotein lipase gene missense mutation in Chinese patients with severe hypertriglyceridemia and pancreatitis. Lipids Health Dis. 2014, 13, 52. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Nakajima, T.; Inoue, I. Molecular modeling of the dimeric structure of human lipoprotein lipase and functional studies of the carboxyl-terminal domain. JBIC J. Biol. Inorg. Chem. 2002, 269, 4701–4710. [Google Scholar] [CrossRef]
- Iverius, P.H.; Ostlund-Lindqvist, A.M. Lipoprotein lipase from bovine milk. Isolation procedure, chemical characterization, and molecular weight analysis. J. Biol. Chem. 1976, 251, 7791–7795. [Google Scholar]
- Van Tilbeurgh, H.; Roussel, A.; Lalouel, J.M.; Cambillau, C. Lipoprotein lipase. Molecular model based on the pancreatic lipase x-ray structure: Consequences for heparin binding and catalysis. J. Biol. Chem. 1994, 269, 4626–4633. [Google Scholar]
- Birrane, G.; Beigneux, A.P.; Dwyer, B.; Strack-Logue, B.; Kristensen, K.K.; Francone, O.L.; Fong, L.G.; Mertens, H.D.T.; Pan, C.Q.; Ploug, M.; et al. Structure of the lipoprotein lipase–GPIHBP1 complex that mediates plasma triglyceride hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Doolittle, M.H.; Ehrhardt, N.; Péterfy, M. Lipase maturation factor 1: Structure and role in lipase folding and assembly. Curr. Opin. Lipidol. 2010, 21, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Roberts, B.S.; Babilonia-Rosa, M.A.; Broadwell, L.J.; Wu, M.J.; Neher, S.B. Lipase maturation factor 1 affects redox homeostasis in the endoplasmic reticulum. EMBO J. 2018, 37, e97379. [Google Scholar] [CrossRef]
- Ishibashi, S.; Yamada, N.; Shimano, H.; Mori, N.; Mokuno, H.; Gotohda, T.; Kawakami, M.; Murase, T.; Takaku, F. Apolipoprotein E and lipoprotein lipase secreted from human monocyte-derived macrophages modulate very low density lipoprotein uptake. J. Biol. Chem. 1990, 265, 3040–3047. [Google Scholar] [PubMed]
- Medh, J.D.; Bowen, S.L.; Fry, G.L.; Ruben, S.; Andracki, M.; Inoue, I.; Lalouel, J.-M.; Strickland, D.K.; Chappell, D.A. Lipoprotein Lipase Binds to Low Density Lipoprotein Receptors and Induces Receptor-mediated Catabolism of Very Low Density Lipoproteinsin Vitro. J. Biol. Chem. 1996, 271, 17073–17080. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.S.; Jacobsen, C.; Olivecrona, G.; Gliemann, J.; Petersen, C.M. Sortilin/Neurotensin Receptor-3 Binds and Mediates Degradation of Lipoprotein Lipase. J. Biol. Chem. 1999, 274, 8832–8836. [Google Scholar] [CrossRef] [Green Version]
- Page, S.T.; Judson, A.; Melford, K.; Bensadoun, A. Interaction of Lipoprotein Lipase and Receptor-associated Protein. J. Biol. Chem. 2006, 281, 13931–13938. [Google Scholar] [CrossRef] [Green Version]
- Mysling, S.; Kristensen, K.K.; Larsson, M.; Kovrov, O.; Bensadouen, A.; Jorgensen, T.J.D.; Olivecrona, G.; Young, S.G.; Ploug, M. The angiopoietin-like protein ANGPTL4 catalyzes unfolding of the hydrolase domain in lipoprotein lipase and the endothelial membrane protein GPIHBP1 counteracts this unfolding. eLife 2016, 5, e20958. [Google Scholar] [CrossRef]
- Mysling, S.; Kristensen, K.K.; Larsson, M.; Beigneux, A.P.; Gårdsvoll, H.; Fong, L.G.; Bensadouen, A.; Jørgensen, T.J.D.; Young, S.G.; Ploug, M. The acidic domain of the endothelial membrane protein GPIHBP1 stabilizes lipoprotein lipase activity by preventing unfolding of its catalytic domain. eLife 2016, 5, e12095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, K.K.; Leth-Espensen, K.Z.; Young, S.G.; Ploug, M. ANGPTL4 inactivates lipoprotein lipase by catalyzing the irreversible unfolding of LPL’s hydrolase domain. J. Lipid Res. 2020, 61, 1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimamura, M.; Matsuda, M.; Yasumo, H.; Okazaki, M.; Fujimoto, K.; Kono, K.; Shimizugawa, T.; Ando, Y.; Koishi, R.; Kohama, T.; et al. Angiopoietin-Like Protein3 Regulates Plasma HDL Cholesterol Through Suppression of Endothelial Lipase. Arter. Thromb. Vasc. Biol. 2007, 27, 366–372. [Google Scholar] [CrossRef] [Green Version]
- Shimizugawa, T.; Ono, M.; Shimamura, M.; Yoshida, K.; Ando, Y.; Koishi, R.; Ueda, K.; Inaba, T.; Minekura, H.; Kohama, T.; et al. ANGPTL3 Decreases Very Low Density Lipoprotein Triglyceride Clearance by Inhibition of Lipoprotein Lipase. J. Biol. Chem. 2002, 277, 33742–33748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quagliarini, F.; Wang, Y.; Kozlitina, J.; Grishin, N.V.; Hyde, R.; Boerwinkle, E.; Valenzuela, D.M.; Murphy, A.J.; Cohen, J.C.; Hobbs, H.H. Atypical angiopoietin-like protein that regulates ANGPTL3. Proc. Natl. Acad. Sci. USA 2012, 109, 19751–19756. [Google Scholar] [CrossRef] [Green Version]
- Larsson, M.; Vorrsjö, E.; Talmud, P.J.; Lookene, A.; Olivecrona, G. Apolipoproteins C-I and C-III Inhibit Lipoprotein Lipase Activity by Displacement of the Enzyme from Lipid Droplets*. J. Biol. Chem. 2013, 288, 33997–34008. [Google Scholar] [CrossRef] [Green Version]
- Rensen, P.C.; Van Berkel, T.J.C. Apolipoprotein E Effectively Inhibits Lipoprotein Lipase-mediated Lipolysis of Chylomicron-like Triglyceride-rich Lipid Emulsionsin Vitroandin Vivo. J. Biol. Chem. 1996, 271, 14791–14799. [Google Scholar] [CrossRef] [Green Version]
- Merkel, M.; Loeffler, B.; Kluger, M.; Fabig, N.; Geppert, G.; Pennacchio, L.A.; Laatsch, A.; Heeren, J. Apolipoprotein AV Accelerates Plasma Hydrolysis of Triglyceriderich Lipoproteins by Interaction with Proteoglycan-bound Lipoprotein Lipase. J. Biol. Chem. 2005, 280, 21553–21560. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, I.J.; Scheraldi, C.A.; Yacoub, L.K.; Saxena, U.; Bisgaier, C.L. Lipoprotein ApoC-II activation of lipoprotein lipase. Modulation by apolipoprotein A-IV. J. Biol. Chem. 1990, 265, 4266–4272. [Google Scholar]
- Kinnunen, P.K.; Jackson, R.L.; Smith, L.C.; Gotto, A.M.; Sparrow, J.T. Activation of lipoprotein lipase by native and synthetic fragments of human plasma apolipoprotein C-II. Proc. Natl. Acad. Sci. USA 1977, 74, 4848–4851. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.-C.; The European Alzheimer’s Disease Initiative Investigators; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.O.; Combarros, O.; Zelenika, D.; Bullido, M.J.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Zandl-Lang, M.; Fanaee-Danesh, E.; Sun, Y.; Albrecher, N.M.; Gali, C.C.; Čančar, I.; Kober, A.; Tam-Amersdorfer, C.; Stracke, A.; Storck, S.M.; et al. Regulatory effects of simvastatin and apoJ on APP processing and amyloid-β clearance in blood-brain barrier endothelial cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2018, 1863, 40–60. [Google Scholar] [CrossRef]
- Mulder, S.D.; Nielsen, H.M.; Blankenstein, M.; Eikelenboom, P.; Veerhuis, R. Apolipoproteins E and J interfere with amyloid-beta uptake by primary human astrocytes and microgliain vitro. Glia 2014, 62, 493–503. [Google Scholar] [CrossRef]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons from Other Pathologies. Front. Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef] [Green Version]
- Yerbury, J.J.; Wilson, M.R. Extracellular chaperones modulate the effects of Alzheimer’s patient cerebrospinal fluid on Aβ1-42 toxicity and uptake. Cell Stress Chaperones 2009, 15, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Laperrousaz, E.; Moullé, V.S.; Denis, R.G.P.; Kassis, N.; Berland, C.; Colsch, B.; Fioramonti, X.; Philippe, E.; Lacombe, A.; Vanacker, C.; et al. Lipoprotein lipase in hypothalamus is a key regulator of body weight gain and glucose homeostasis in mice. Diabetology 2017, 60, 1314–1324. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Astarita, G.; Taussig, M.D.; Bharadwaj, K.G.; DiPatrizio, N.V.; Nave, K.-A.; Piomelli, D.; Goldberg, I.J.; Eckel, R.H. Deficiency of Lipoprotein Lipase in Neurons Modifies the Regulation of Energy Balance and Leads to Obesity. Cell Metab. 2011, 13, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Taussig, M.D.; DiPatrizio, N.V.; Astarita, G.; Piomelli, D.; Bergman, B.C.; Dell’Acqua, M.L.; Eckel, R.H.; Wang, H. Deficiency of Lipoprotein Lipase in Neurons Decreases AMPA Receptor Phosphorylation and Leads to Neurobehavioral Abnormalities in Mice. PLoS ONE 2015, 10, e0135113. [Google Scholar] [CrossRef] [Green Version]
- Bruce, K.D.; Dobrinskikh, E.; Wang, H.; Rudenko, I.; Gao, H.; Libby, A.E.; Gorkhali, S.; Yu, T.; Zsombok, A.; Eckel, R.H. Neuronal Lipoprotein Lipase Deficiency Alters Neuronal Function and Hepatic Metabolism. Metabolites 2020, 10, 385. [Google Scholar] [CrossRef]
- Klinger, S.C.; Glerup, S.; Raarup, M.K.; Mari, M.C.; Nyegaard, M.; Koster, G.; Prabakaran, T.; Nilsson, S.K.; Kjaergaard, M.M.; Bakke, O.; et al. SorLA regulates the activity of lipoprotein lipase by intracellular trafficking. J. Cell Sci. 2011, 124, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Layritz, C.; Legutko, B.; Eichmann, T.O.; Laperrousaz, E.; Moullé, V.S.; Cruciani-Guglielmacci, C.; Magnan, C.; Luquet, S.; Woods, S.C.; et al. Disruption of Lipid Uptake in Astroglia Exacerbates Diet-Induced Obesity. Diabetes 2017, 66, 2555–2563. [Google Scholar] [CrossRef] [Green Version]
- Nishitsuji, K.; Hosono, T.; Uchimura, K.; Michikawa, M. Lipoprotein Lipase Is a Novel Amyloid β (Aβ)-binding Protein That Promotes Glycosaminoglycan-dependent Cellular Uptake of Aβ in Astrocytes. J. Biol. Chem. 2010, 286, 6393–6401. [Google Scholar] [CrossRef] [Green Version]
- Kamermans, A.; Rijnsburger, M.; Chakraborty, A.; Van Der Pol, S.; De Vries, H.E.; Van Horssen, J. Reduced Angiopoietin-Like 4 Expression in Multiple Sclerosis Lesions Facilitates Lipid Uptake by Phagocytes via Modulation of Lipoprotein-Lipase Activity. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.; Summers, K.M.; McColl, B.W. Microglial brain region−dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e10. [Google Scholar] [CrossRef] [Green Version]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.-N.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.-H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Chait, A.; Eckel, R.H. The Chylomicronemia Syndrome Is Most Often Multifactorial. Ann. Intern. Med. 2019, 170, 626–634. [Google Scholar] [CrossRef]
- Xie, C.; Wang, Z.C.; Liu, X.F.; Wang, L.; Yang, M.S. Association between schizophrenia and single nucleotide polymorphisms in lipoprotein lipase gene in a Han Chinese population. Psychiatr. Genet. 2011, 21, 307–314. [Google Scholar] [CrossRef]
- Nejati, M.; Atlasi, M.A.; Karimian, M.; Nikzad, H.; Tameh, A.A. Lipoprotein lipase gene polymorphisms as risk factors for stroke: A computational and meta-analysis. Iran J. Basic Med. Sci. 2018, 21, 701–708. [Google Scholar]
- Van Bockxmeer, F. Lipoprotein lipase D9N, N291S and S447X polymorphisms: Their influence on premature coronary heart disease and plasma lipids. Atherosclerosis 2001, 157, 123–129. [Google Scholar] [CrossRef]
- Hoffer, M.J.; Bredie, S.J.; Boomsma, D.I.; Reymer, P.W.; Kastelein, J.J.; De Knijff, P.; Demacker, P.N.; Stalenhoef, A.F.; Havekes, L.M.; Frants, R.R. The lipoprotein lipase (Asn291 → Ser) mutation is associated with elevated lipid levels in families with familial combined hyperlipidaemia. Atherosclerosis 1996, 119, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.M.; Mailly, F.; Peacock, R.E.; Hamsten, A.; Seed, M.; Yudkin, J.S.; Beisiegel, U.; Feussner, G.; Miller, G.; Humphries, S.E. Interaction of the lipoprotein lipase asparagine 291-->serine mutation with body mass index determines elevated plasma triacylglycerol concentrations: A study in hyperlipidemic subjects, myocardial infarction survivors, and healthy adults. J. Lipid Res. 1995, 36, 2104–2112. [Google Scholar]
- Reymer, P.W.; Groenemeyer, B.E.; Gagné, E.; Miao, L.; Appelman, E.E.; Seidel, J.C.; Kromhout, D.; Bijvoet, S.M.; Van De Oever, K.; Bruin, T.; et al. A frequently occurring mutation in the lipoprotein lipase gene (Asn291Ser) contributes to the expression of familial combined hyperlipidemia. Hum. Mol. Genet. 1995, 4, 1543–1549. [Google Scholar] [CrossRef]
- Baum, L.; Chen, L.; Masliah, E.; Chan, Y.S.; Ng, H.-K.; Pang, C.P. Lipoprotein lipase mutations and Alzheimer’s disease. Am. J. Med Genet. 1999, 88, 136–139. [Google Scholar] [CrossRef]
- Wittekoek, M.E.; Pimstone, S.N.; Reymer, P.W.A.; Feuth, L.; Botma, G.-J.; Defesche, J.C.; Prins, M.; Hayden, M.R.; Kastelein, J.J.P. A Common Mutation in the Lipoprotein Lipase Gene (N291S) Alters the Lipoprotein Phenotype and Risk for Cardiovascular Disease in Patients with Familial Hypercholesterolemia. Circulation 1998, 97, 729–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayad, A.; Noruzinia, M.; Zamani, M.; Harirchian, M.H.; Kazemnejad, A. Lipoprotein Lipase HindIII Intronic Polymorphism in a Subset of Iranian Patients with Late-Onset Alzheimer’s Disease. Cell J. 2012, 14, 67–72. [Google Scholar]
- Scacchi, R.; Gambina, G.; Broggio, E.; Moretto, G.; Ruggeri, M.; Corbo, R.M. The H+ allele of the lipoprotein lipase (LPL) HindIII intronic polymorphism and the risk for sporadic late-onset Alzheimer’s disease. Neurosci. Lett. 2004, 367, 177–180. [Google Scholar] [CrossRef]
- Blain, J.-F.; Aumont, N.; Théroux, L.; Dea, D.; Poirier, J. A polymorphism in lipoprotein lipase affects the severity of Alzheimer’s disease pathophysiology. Eur. J. Neurosci. 2006, 24, 1245–1251. [Google Scholar] [CrossRef]
- Hayne, C.K.; Lafferty, M.J.; Eglinger, B.J.; Kane, J.P.; Neher, S.B. Biochemical Analysis of the Lipoprotein Lipase Truncation Variant, LPLS447X, Reveals Increased Lipoprotein Uptake. Biochemistry 2017, 56, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Sawano, M.; Watanabe, Y.; Ohmura, H.; Shimada, K.; Daida, H.; Mokuno, H.; Yamaguchi, H. Potentially protective effects of the Ser447-Ter mutation of the lipoprotein lipase gene against the development of coronary artery disease in Japanese subjects via a beneficial lipid profile. Jpn. Circ. J. 2001, 65, 310–314. [Google Scholar] [CrossRef] [Green Version]
- Gotoda, T.; Yamada, N.; Murase, T.; Shimano, H.; Shimada, M.; Harada, K.; Kawamura, M.; Kozaki, K.; Yazaki, Y. Detection of three separate DNA polymorphisms in the human lipoprotein lipase gene by gene amplification and restriction endonuclease digestion. J. Lipid Res. 1992, 33, 1067–1072. [Google Scholar]
- Chen, Q.; Razzaghi, H.; Demirci, F.Y.; Kamboh, M.I. Functional significance of lipoprotein lipase HindIII polymorphism associated with the risk of coronary artery disease. Atherosclerosis 2008, 200, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Munshi, A.; Babu, M.S.; Kaul, S.; Rajeshwar, K.; Balakrishna, N.; Jyothy, A. Association of LPL gene variant and LDL, HDL, VLDL cholesterol and triglyceride levels with ischemic stroke and its subtypes. J. Neurol. Sci. 2012, 318, 51–54. [Google Scholar] [CrossRef]
- Cao, L.; Li, Q.; Chen, X. The HindIII and PvuII polymorphisms of lipoprotein lipase (LPL) gene reduce the risk of ischemic stroke (IS). Medicine 2018, 97, e0483. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.; Thorn, J.A.; Oka, K.; Galton, D.; Stocks, J. DNA polymorphisms at the lipoprotein lipase gene: Associations in normal and hypertriglyceridaemic subjects. Atherosclerosis 1989, 79, 85–91. [Google Scholar] [CrossRef]
- Liu, N.; Sang, Y.; Chen, S.; Liu, X. Associations of the LPL S447X and Hind III Polymorphism with Type 2 Diabetes Mellitus Risk: A Meta-Analysis. Horm. Metab. Res. 2020, 10, 1055. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.-Q.; Wang, Y.; Han, X.-Q.; Zhu, Y.; Liu, N.-F. Associations between LPL gene polymorphisms and coronary artery disease: Evidence based on an updated and cumulative meta-analysis. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozaki, K.; Gotoda, T.; Kawamura, M.; Shimano, H.; Yazaki, Y.; Ouchi, Y.; Orimo, H.; Yamada, N. Mutational analysis of human lipoprotein lipase by carboxy-terminal truncation. J. Lipid Res. 1993, 34, 1765–1772. [Google Scholar]
- Zhang, H.; Henderson, H.; Gagne, S.; Clee, S.M.; Miao, L.; Liu, G.; Hayden, M.R. Common sequence variants of lipoprotein lipase: Standardized studies of in vitro expression and catalytic function. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1996, 1302, 159–166. [Google Scholar] [CrossRef]
- Ranganathan, G.; Unal, R.; Pokrovskaya, I.D.; Tripathi, P.; Rotter, J.I.; Goodarzi, M.O.; Kern, P.A. The lipoprotein lipase (LPL) S447X gain of function variant involves increased mRNA translation. Atherosclerosis 2012, 221, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Martin-Rehrmann, M.D.; Cho, H.S.; Rebeck, G.W. Lack of association of two lipoprotein lipase polymorphisms with Alzheimer’s disease. Neurosci. Lett. 2002, 328, 109–112. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Y.; Wang, L.; Liao, Q.; Chang, L.; Xu, L.; Huang, Y.; Ye, H.; Xu, L.; Chen, C.; et al. Meta-Analyses of 8 Polymorphisms Associated with the Risk of the Alzheimer’s Disease. PLoS ONE 2013, 8, e73129. [Google Scholar] [CrossRef] [Green Version]
- Lamar, M.; Boots, E.A.; Arfanakis, K.; Barnes, L.L.; Schneider, J.A. Common Brain Structural Alterations Associated with Cardiovascular Disease Risk Factors and Alzheimer’s Dementia: Future Directions and Implications. Neuropsychol. Rev. 2020, 1–12. [Google Scholar] [CrossRef]
- Konttinen, H.; Cabral-Da-Silva, M.E.C.; Ohtonen, S.; Wojciechowski, S.; Shakirzyanova, A.; Caligola, S.; Giugno, R.; Ishchenko, Y.; Hernández, D.; Fazaludeen, M.F.; et al. PSEN1ΔE9, APPswe, and APOE4 Confer Disparate Phenotypes in Human iPSC-Derived Microglia. Stem Cell Rep. 2019, 13, 669–683. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Schellenberg, G.D.; Deeb, S.S.; Boehnke, M.; Bryant, E.M.; Martin, G.M.; Lampe, T.H.; Bird, T.D. Association of an apolipoprotein CII allele with familial dementia of the Alzheimer type. J. Neurogenet. 1987, 4, 97–108. [Google Scholar] [CrossRef]
- Schellenberg, G.D.; Boehnke, M.; Wijsman, E.M.; Moore, D.K.; Martin, G.M.; Bird, T.D. Genetic association and linkage analysis of the apolipoprotein CII locus and familial Alzheimer’s disease. Ann. Neurol. 1992, 31, 223–227. [Google Scholar] [CrossRef]
- Yu, C.-E.; Payami, H.; Olson, J.M.; Boehnke, M.; Wijsman, E.M.; Orr, H.T.; Kukull, W.A.; Goddard, K.A.B.; Nemens, E.; White, J.A.; et al. The Apolipoprotein E/CI/CII Gene Cluster and Late-Onset Alzheimer Disease. Am. J. Hum. Genet. 1994, 54, 631–642. [Google Scholar]
- Sun, Y.; Shi, J.; Zhang, S.; Tang, M.; Han, H.; Guo, Y.; Ma, C.; Liu, X.; Li, T. The APOC3 SstI polymorphism is weakly associated with sporadic Alzheimer’s disease in a Chinese population. Neurosci. Lett. 2005, 380, 219–222. [Google Scholar] [CrossRef]
- Bettens, K.; Brouwers, N.; Engelborghs, S.; De Deyn, P.P.; Van Broeckhoven, C.; Sleegers, K. SORL1 is genetically associated with increased risk for late-onset Alzheimer disease in the Belgian population. Hum. Mutat. 2008, 29, 769–770. [Google Scholar] [CrossRef]
- Verheijen, J.; Bossche, T.V.D.; Van Der Zee, J.; Engelborghs, S.; Sanchez-Valle, R.; Lladó, A.; Graff, C.; Thonberg, H.; Pastor, P.; Ortega-Cubero, S.; et al. A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer’s disease. Acta Neuropathol. 2016, 132, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Ba, K.L.S.; Wuu, J.; Leurgans, S.E.; Rees, H.D.; Gearing, M.; Mufson, E.J.; Levey, A.I.; Lah, J.J.; Sager, K.L.; Levey, A.I. Neuronal LR11/sorLA expression is reduced in mild cognitive impairment. Ann. Neurol. 2007, 62, 640–647. [Google Scholar] [CrossRef] [Green Version]
- Knupp, A.; Mishra, S.; Martinez, R.; Braggin, J.E.; Szabo, M.; Kinoshita, C.; Hailey, D.W.; Small, S.A.; Jayadev, S.; Young, J.E. Depletion of the AD Risk Gene SORL1 Selectively Impairs Neuronal Endosomal Traffic Independent of Amyloidogenic APP Processing. Cell Rep. 2020, 31, 107719. [Google Scholar] [CrossRef]
- Caglayan, S.; Takagi-Niidome, S.; Liao, F.; Carlo, A.-S.; Schmidt, V.; Burgert, T.; Kitago, Y.; Füchtbauer, E.-M.; Füchtbauer, A.; Holtzman, D.M.; et al. Lysosomal Sorting of Amyloid- by the SORLA Receptor Is Impaired by a Familial Alzheimer’s Disease Mutation. Sci. Transl. Med. 2014, 6, 223ra20. [Google Scholar] [CrossRef]
- Raghavan, N.S.; Brickman, A.M.; Andrews, H.; Manly, J.J.; Schupf, N.; Lantigua, R.; Wolock, C.J.; Kamalakaran, S.; Petrovski, S.; Tosto, G.; et al. Whole-exome sequencing in 20,197 persons for rare variants in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2018, 5, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, V.; Subkhangulova, A.; Willnow, T.E. Sorting receptor SORLA: Cellular mechanisms and implications for disease. Cell. Mol. Life Sci. 2016, 74, 1475–1483. [Google Scholar] [CrossRef] [Green Version]



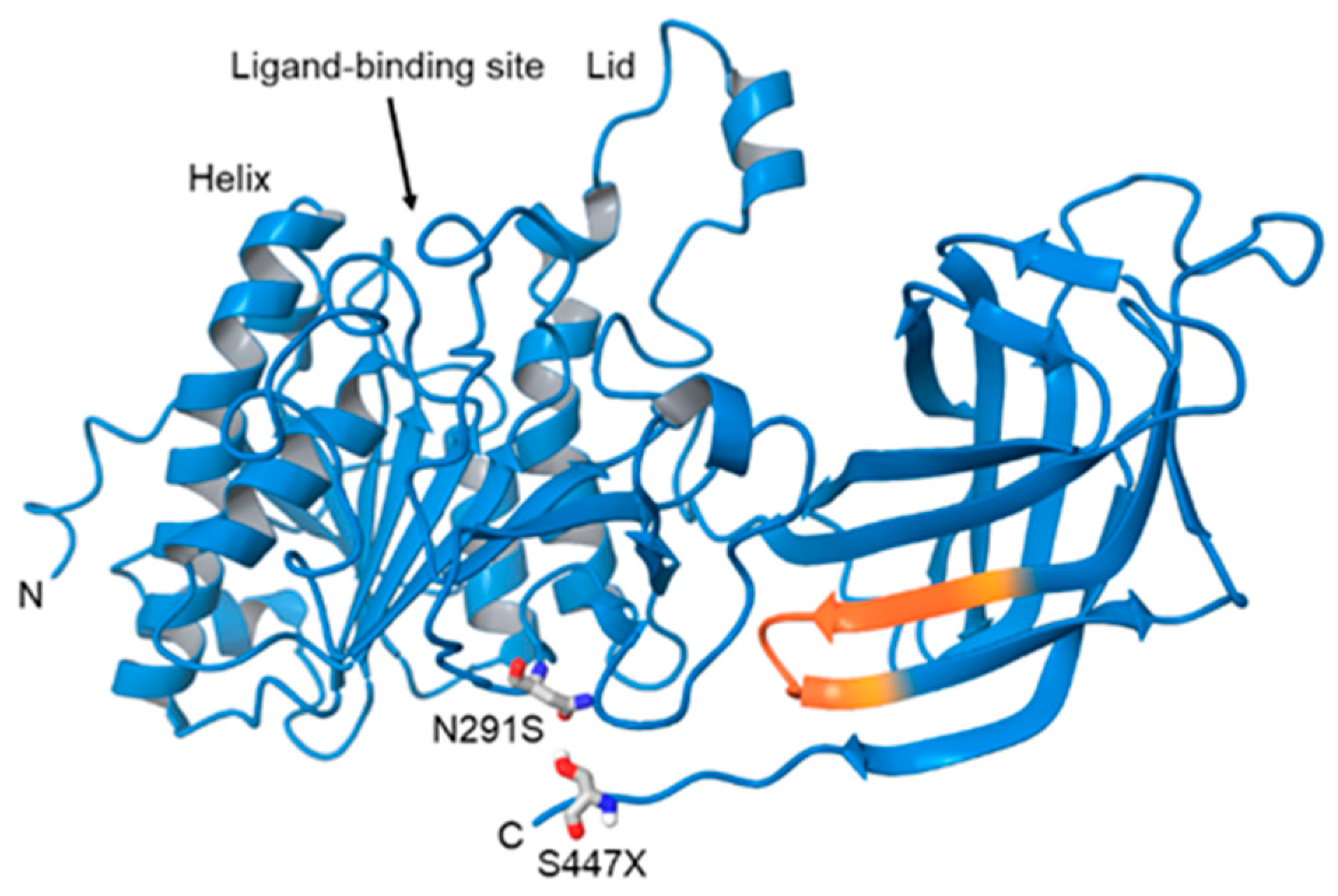{kind=link}
{kind=link}
| Mutation | Effect on LPL Structure or Function | AD Form | Prevalence (% of Population) | Clinical Characteristics |
|---|---|---|---|---|
| N291S, Asn291Ser (rs268). Also known as Asn318 | Mutation in exon 6 results in an A-G transmission [59]. Amino acid substitution occurs at base 318 changes the hydrophobicity and flexibility of residues 314-322 near the ligand bindings site [58]. | 2–5% [64] | Higher TG, lower HDL (11427211). Overrepresented in patients diagnosed with AD (5% versus 1% [63]) | |
| HindIII (rs320) | T-G substitution in intron 8, at position 481 removing HindIII restriction site | LOAD [65] | 25–39% PMID: 10830909 | Increased plasma lipid profile and susceptibility to CAD. Significant association (1.75-fold increased risk) with LOAD in an Iranian population [65]. Homozygous genotypes have an increased risk of AD, more so in females [66]. |
| PvuII (rs285) Also referred to as the P+ allele. | Mutation in intron 6 within PvuIII restriction site. Potential cis-element of unknown significance. PvuIII is associated with altered LPL mRNA [67]. | SAD [67] | 39% PMID: 10830909 | Increased risk of AD in P+ carriers. Reduced cortex cholesterol, increased NFTs and senile plaques. Combined effect of P+ and ApoE4 in the fusiform gyrus in eastern Canadian population [67]. |
| S447X (rs328). Also known as Ser447Ter. | Mutation in exon 9 leads to a C-G transversion, and loss of two final amino acids. Leads to increased receptor binding and endocytosis [68]. | 5–12% [63]. | Decreased TG, increased HDL, reduced risk of CAD and T2D [69]. Clinically underrepresented in patients with diagnosed AD. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D. Bruce, K.; Tang, M.; Reigan, P.; H. Eckel, R. Genetic Variants of Lipoprotein Lipase and Regulatory Factors Associated with Alzheimer’s Disease Risk. Int. J. Mol. Sci. 2020, 21, 8338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218338
D. Bruce K, Tang M, Reigan P, H. Eckel R. Genetic Variants of Lipoprotein Lipase and Regulatory Factors Associated with Alzheimer’s Disease Risk. International Journal of Molecular Sciences. 2020; 21(21):8338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218338
Chicago/Turabian StyleD. Bruce, Kimberley, Maoping Tang, Philip Reigan, and Robert H. Eckel. 2020. "Genetic Variants of Lipoprotein Lipase and Regulatory Factors Associated with Alzheimer’s Disease Risk" International Journal of Molecular Sciences 21, no. 21: 8338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218338