Genomics of Human Fibrotic Diseases: Disordered Wound Healing Response

 , ,
, ,
Abstract
:1. Introduction
1.1. Background and Scope
1.2. Inflammation, Remodeling and Fibrosis in Wound Healing
2. Fibrosing Disorders of the Skin
2.1. Keloids and Hypertrophic Scars
2.2. Scleroderma (Systemic Sclerosis, Localized Scleroderma, and Lichen Sclerosus)
2.3. Chronic Ulcers
2.4. Exogenous Triggers
3. Hepatic Fibrosis
4. Renal Fibrosis
4.1. Chronic Kidney Disease
4.2. SLE and Lupus Nephritis
5. Lung Fibrosis
6. Cardiac Fibrosis
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | Angiotensin-converting enzyme |
| ADORA2A | Adenosine A2a receptor |
| AFF3 | AF4FMR2 family member 3 |
| AGAP2 | ArfGAP With GTPase Domain, Ankyrin Repeat And PH Domain 2 |
| AGES | Age, gene/environment susceptibility |
| ALMS1 | Alstrom syndrome 1 |
| ARB | Angiotensin II receptor blocker |
| ARHGAP31 | Rho GTPase activating protein 31 |
| ATG5 | Autophagy related 5 |
| ATXN2 | Ataxin 2 |
| BANK1 | B Cell Scaffold Protein With Ankyrin Repeats 1 |
| BCAS3 | Breast carcinoma amplified sequence 3 |
| BLCC | Bioengineered bilayered living cell construct |
| CACNA1G-AS1 | CACNA1G antisense RNA 1 |
| CAV1 | Caveolin-1 |
| CCN2 | Cellular communication network factor 2 |
| CD80 | Cluster of differentiation 80 |
| CDH1 | Cadherin 1 |
| CDH23 | Cadherin 23 |
| CDK4 | Cyclin-dependent kinase 4 |
| circRNA | circular RNA |
| CKD | Chronic kidney disease |
| COL1A1 | Collagen type I alpha 1 |
| COL4A3 | Collagen type IV alpha 3 |
| COL5A1 | Collagen type V alpha 1 |
| CORIN | Corin, atrial natriuretic peptide-converting enzyme |
| CPS1 | Carbamyl phosphate synthetase I |
| CSF1R | Colony stimulating factor 1 receptor |
| CSMD1 | CUB And Sushi Multiple Domains 1 |
| CTBP1 | C-terminal binding protein 1 |
| CTGF | Connective tissue growth factor |
| CTLA4 | Cytotoxic T-lymphocyte associated protein 4 |
| CTSS | Cathepsin S |
| CTSZ | Cathepsin Z |
| CXCL10 | C-X-C motif chemokine ligand 10 |
| CXCR5 | C-x-c motif chemokine receptor 5 |
| DAB2 | Disabled homolog 2 |
| DACH1 | Dachshund homolog 1 |
| DGKA | diacylglycerol kinase alpha |
| DPP9 | Toll-interacting protein |
| EBV | Epstein Barr virus |
| ECM | Extracellular matrix |
| EGR1 | Early Growth Response 1 |
| eMERGE | Electronic medical records and genomics |
| EMT | Epithelial-mesenchymal transition |
| eQTL | Expression of quantitative trait |
| FAN1 | Fanconi anemia associated nuclease 1 |
| FGFR | Fibroblast Growth Factor Receptor |
| FIGF | C-fos–induced growth factor |
| FKBP10 | FK506-binding protein 10 |
| FN1 | Fibronectin 1 |
| FOXL2 | Forkhead box L2 |
| GABRR1 | Gamma-aminobutyric acid type A receptor subunit rho1 |
| GALNT11 | Polypeptide N-acetylgalactosaminyltransferase 11 |
| GCKR | Glucokinase regulator |
| GRB10 | Growth factor receptor bound protein 10 |
| GWAS | Genome-wide association study |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| HGF | Hepatocyte growth factor |
| HHV | Human herpesvirus |
| HIF-1α | Hypoxia inducible factors |
| HLA | Human leukocyte antigen |
| HLA-DQB1 | Major histocompatibility complex, class II, DQ beta 1 |
| HNF1A-AS1 | HNF1A Antisense RNA 1 |
| HSC | Hepatic stellate cells |
| HSD17B13 | Hydroxysteroid 17-beta dehydrogenase 13 |
| hTERT | Human Telomerase Reverse Transcriptase |
| hTR | Human Telomerase RNA |
| ICOSLG | Inducible T-cell costimulator ligand |
| IGF, IGFBP | Insulin-like growth factor, Insulin-like growth factor binding protein |
| IIP | Idiopathic interstitial pneumonia |
| IKZF3 | IKAROS family zinc finger 3 |
| IL11 | Interleukin-11 |
| IL12A | Interleukin-12 subunit alpha |
| IL12B | Interleukin-12 subunit beta |
| IL12RB2 | Interleukin 12 receptor subunit beta 2 |
| IL17RA | Interleukin 17 receptor A |
| IL7R | Interleukin 7 receptor |
| IPF | Idiopathic pulmonary fibrosis |
| IRAK1 | Interleukin 1 receptor associated kinase 1 |
| IRF4, 5, 7 | Interferon regulatory factor |
| IRF5-TNPO3 | Interferon regulatory factor 5 - transportin 3 |
| ITGB2 | Integrin subunit beta 2 |
| LAPTM5 | Lysosomal protein transmembrane 5 |
| LASS2 | Ceramide synthase 2 |
| LINC00923 | Long intergenic non-protein coding RNA 923 |
| LIPCAR | Long non-coding cardiac associated RNA |
| lncRNA | Long non-coding RNA |
| LY75 | Lymphocyte antigen 75 |
| LYPLAL1 | Lysophospholipase like 1 |
| MAPK | mitogen-activated protein kinase |
| MIAT | Myocardial infarction associated transcript |
| miRNA | MicroRNA |
| MMP 1 | Matrix metallopeptidase 1 |
| MMP2 | Matrix metalloproteinase 2 |
| MMP9 | Matrix metallopeptidase 9 |
| MYO 1E, MYO7 | Myosin 1E, Myosin 7 |
| NAFLD | Non-alcoholic fatty liver disease |
| NCAN | Neurocan |
| NEDD4 | Neural precursor cell expressed developmentally down-regulated protein 4 |
| NET | Neutrophil extracellular trap |
| NFKB1 | Nuclear factor kappa B subunit 1 |
| NOD2/CARD15 | Nucleotide-binding oligomerization domain-containing protein 2 / Caspase Recruitment Domain-Containing Protein 15 |
| NOTCH4 | Neurogenic locus notch homolog 4 |
| OTUD6B | Ovarian tumor domain deubiquitinase 6B |
| PAI1/ PAI2 | Plasminogen Activator Inhibitor 1/ Plasminogen Activator Inhibitor 2 |
| PBC | Primary biliary cirrhosis |
| PDGF | Platelet-derived growth factor |
| PDGFR | Platelet Derived Growth Factor Receptor |
| PIP5K1B | Phosphatidylinositol-4-phosphate 5-kinase type 1 beta |
| PLEK | Pleckstrin |
| PNPLA3 | Patatin like phospholipase domain containing 3 |
| POGLUT1 | Protein O-glucosyltransferase 1 |
| POLD1 | DNA Polymerase Delta 1, Catalytic Subunit |
| POU2AF1 | POU domain class 2-associating factor 1 |
| PRKAG2 | Protein kinase AMP-activated non-catalytic subunit gamma 2 |
| PTPN5 | Protein tyrosine phosphatase non-receptor type 5 |
| PYGO1 | Pygopus family PHD finger 1 |
| RAAS | Renin-angiotensin-aldosterone system |
| RASAL1 | RAS protein activator like 1 |
| RUNX2 | Runt-related transcription factor 2 |
| RXFP1 | Relaxin family peptide receptor 1 |
| SAMM50 | Sorting and assembly machinery component 50 homolog |
| SFTPA2 | surfactant protein A2 |
| SHROOM3 | Shroom family member 3 |
| SLC22A2 | Solute carrier family 22 member 2 |
| SLC34A1 | Solute carrier family 34 member 1 |
| SLC6A13 | Solute carrier family 6 member 13 |
| SLC7A9 | Solute carrier family 7 member 9 |
| SLE | Systemic lupus erythematosus |
| α-SMA | Alpha smooth muscle actin |
| SOCS1 | Suppressor of cytokine signaling 1 |
| SOX9 | SRY-box transcription factor 9 |
| SPIB | Spi-b transcription factor |
| SPP1 | Secreted phosphoprotein 1 |
| SSc | Systemic sclerosis |
| STAT4 | Signal transducer and activator of transcription |
| STC1 | Stanniocalcin 1 |
| TFDP2 | Transcription factor DP-2 |
| TGF-β | Transforming growth factor beta |
| TIMMDC1 | Translocase of inner mitochondrial membrane domain containing 1 |
| TM6SF2 | Transmembrane 6 superfamily member 2 |
| TMEM39A | Transmembrane protein 39a |
| TMEM60 | Transmembrane protein 60 |
| TNC | Tenascin C |
| TNFRSF1A | TNF receptor superfamily member 1a |
| TNFSF15 | TNF superfamily member 15 |
| TOLLIP | Matrix Metalloproteinase |
| TRAPPC9 | Trafficking protein particle complex 9 |
| TSIX | Antisense to Xist (X-inactive specific transcript) |
| TSPYL5 | Testis-specific Y-encoded-like protein 5 |
| TXNRD2 | thioredoxin reductase 2 |
| TYROBP | TYRO protein tyrosine kinase-binding protein |
| UBE2Q2 | Ubiquitin conjugating enzyme E2 Q2 |
| UMOD | Uromodulin |
| VEGFA | Vascular endothelial growth factor A |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| VLU | Venous leg ulcer |
| WDR37 | WD repeat domain 37 |
| WDR72 | WD repeat domain 72 |
| WWP2 | WW domain-containing protein 2 |
| XRCC1 | X-Ray Repair Cross Complementing 1 |
| ZFP90 | Zinc finger protein 90 homolog |
References
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distler, J.H.W.; Gyorfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Konigshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, M.H.; Pinzani, M. Reversal of liver fibrosis. Saudi J. Gastroenterol. 2009, 15, 72–79. [Google Scholar] [CrossRef]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef] [Green Version]
- McAnulty, R.J. Fibroblasts and myofibroblasts: Their source, function and role in disease. Int. J. Biochem. Cell Biol. 2007, 39, 666–671. [Google Scholar] [CrossRef]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef]
- Kuehlmann, B.; Bonham, C.A.; Zucal, I.; Prantl, L.; Gurtner, G.C. Mechanotransduction in Wound Healing and Fibrosis. J. Clin. Med. 2020, 9, 1423. [Google Scholar] [CrossRef]
- Mack, M. Inflammation and fibrosis. Matrix Biol. 2018, 68–69, 106–121. [Google Scholar] [CrossRef]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T.; et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 2014, 233, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bartolome, R.; Uchiyama, A.; Molinolo, A.A.; Abusleme, L.; Brooks, S.R.; Callejas-Valera, J.L.; Edwards, D.; Doci, C.; Asselin-Labat, M.L.; Onaitis, M.W.; et al. Transcriptional signature primes human oral mucosa for rapid wound healing. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, R.C.; Stojadinovic, O.; Sawaya, A.P.; Glinos, G.D.; Lindley, L.E.; Pastar, I.; Badiavas, E.; Tomic-Canic, M. A bioengineered living cell construct activates metallothionein/zinc/MMP8 and inhibits TGFbeta to stimulate remodeling of fibrotic venous leg ulcers. Wound Repair Regen. 2020, 28, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, H.P.; Longaker, M.T.; Perkocha, L.A.; Jennings, R.W.; Harrison, M.R.; Adzick, N.S. Scarless wound repair: A human fetal skin model. Development 1992, 114, 253–259. [Google Scholar] [PubMed]
- Carswell, L.; Borger, J. Hypertrophic Scarring Keloids; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Ogawa, R. Keloid and Hypertrophic Scars Are the Result of Chronic Inflammation in the Reticular Dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, B.; Bayat, A. Genetics of keloid scarring. Arch. Dermatol. Res. 2010, 302, 319–339. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Chung, S.; Takahashi, A.; Kamatani, N.; Kawaguchi, T.; Tsunoda, T.; Hosono, N.; Kubo, M.; Nakamura, Y.; Zembutsu, H. A genome-wide association study identifies four susceptibility loci for keloid in the Japanese population. Nat. Genet. 2010, 42, 768–771. [Google Scholar] [CrossRef]
- Zhu, F.; Wu, B.; Li, P.; Wang, J.; Tang, H.; Liu, Y.; Zuo, X.; Cheng, H.; Ding, Y.; Wang, W.; et al. Association study confirmed susceptibility loci with keloid in the Chinese Han population. PLoS ONE 2013, 8, e62377. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, R.; Watanabe, A.; Than Naing, B.; Sasaki, M.; Fujita, A.; Akaishi, S.; Hyakusoku, H.; Shimada, T. Associations between keloid severity and single-nucleotide polymorphisms: Importance of rs8032158 as a biomarker of keloid severity. J. Investig. Dermatol. 2014, 134, 2041–2043. [Google Scholar] [CrossRef] [Green Version]
- Velez Edwards, D.R.; Tsosie, K.S.; Williams, S.M.; Edwards, T.L.; Russell, S.B. Admixture mapping identifies a locus at 15q21.2–22.3 associated with keloid formation in African Americans. Hum. Genet. 2014, 133, 1513–1523. [Google Scholar] [CrossRef] [Green Version]
- Marneros, A.G.; Norris, J.E.; Watanabe, S.; Reichenberger, E.; Olsen, B.R. Genome scans provide evidence for keloid susceptibility loci on chromosomes 2q23 and 7p11. J. Investig. Dermatol. 2004, 122, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Jumper, N.; Hodgkinson, T.; Paus, R.; Bayat, A. Site-specific gene expression profiling as a novel strategy for unravelling keloid disease pathobiology. PLoS ONE 2017, 12, e0172955. [Google Scholar] [CrossRef] [PubMed]
- Seifert, O.; Bayat, A.; Geffers, R.; Dienus, K.; Buer, J.; Löfgren, S.; Matussek, A. Identification of unique gene expression patterns within different lesional sites of keloids. Wound Repair Regen. 2008, 16, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Fu, X.; Sun, X.; Sun, T.; Zhao, Z.; Sheng, Z. Analysis of differentially expressed genes in keloids and normal skin with cDNA microarray. J. Surg. Res. 2003, 113, 208–216. [Google Scholar] [CrossRef]
- Smith, J.C.; Boone, B.E.; Opalenik, S.R.; Williams, S.M.; Russell, S.B. Gene profiling of keloid fibroblasts shows altered expression in multiple fibrosis-associated pathways. J. Investig. Dermatol. 2008, 128, 1298–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes-Duculan, J.; Bonifacio, K.M.; Suarez-Farinas, M.; Kunjravia, N.; Garcet, S.; Cruz, T.; Wang, C.Q.F.; Xu, H.; Gilleadeau, P.; Sullivan-Whalen, M.; et al. Aberrant connective tissue differentiation towards cartilage and bone underlies human keloids in African Americans. Exp. Dermatol. 2017, 26, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.K.; Lin, H.H.; Harn, H.I.; Ogawa, R.; Wang, Y.K.; Ho, Y.T.; Chen, W.R.; Lee, Y.C.; Lee, J.Y.; Shieh, S.J.; et al. Caveolin-1 Controls Hyperresponsiveness to Mechanical Stimuli and Fibrogenesis-Associated RUNX2 Activation in Keloid Fibroblasts. J. Investig. Dermatol. 2018, 138, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Naitoh, M.; Kubota, H.; Ikeda, M.; Tanaka, T.; Shirane, H.; Suzuki, S.; Nagata, K. Gene expression in human keloids is altered from dermal to chondrocytic and osteogenic lineage. Genes Cells 2005, 10, 1081–1091. [Google Scholar] [CrossRef]
- Kang, Y.; Roh, M.R.; Rajadurai, S.; Rajadurai, A.; Kumar, R.; Njauw, C.N.; Zheng, Z.; Tsao, H. Hypoxia and HIF-1alpha Regulate Collagen Production in Keloids. J. Investig. Dermatol. 2020. [Google Scholar] [CrossRef]
- Wu, X.; Li, J.; Yang, X.; Bai, X.; Shi, J.; Gao, J.; Li, Y.; Han, S.; Zhang, Y.; Han, F.; et al. miR-155 inhibits the formation of hypertrophic scar fibroblasts by targeting HIF-1alpha via PI3K/AKT pathway. J. Mol. Histol. 2018, 49, 377–387. [Google Scholar] [CrossRef]
- Liang, X.; Chai, B.; Duan, R.; Zhou, Y.; Huang, X.; Li, Q. Inhibition of FKBP10 Attenuates Hypertrophic Scarring through Suppressing Fibroblast Activity and Extracellular Matrix Deposition. J. Investig. Dermatol. 2017, 137, 2326–2335. [Google Scholar] [CrossRef] [Green Version]
- Onoufriadis, A.; Hsu, C.K.; Ainali, C.; Ung, C.Y.; Rashidghamat, E.; Yang, H.S.; Huang, H.Y.; Niazi, U.; Tziotzios, C.; Yang, J.C.; et al. Time Series Integrative Analysis of RNA Sequencing and MicroRNA Expression Data Reveals Key Biologic Wound Healing Pathways in Keloid-Prone Individuals. J. Investig. Dermatol. 2018, 138, 2690–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Guo, B.; Chang, P.; Hui, Q.; Li, W.; Tao, K. The Differential Expression of miRNAs and a Preliminary Study on the Mechanism of miR-194-3p in Keloids. Biomed. Res. Int. 2019, 2019, 8214923. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Y.; Lu, L.; Liang, J.; Guo, X.R.; Zhang, P.H.; Luo, S.J. Keloid microRNA expression analysis and the influence of miR-199a-5p on the proliferation of keloid fibroblasts. Genet. Mol. Res. 2014, 13, 2727–2738. [Google Scholar] [CrossRef] [PubMed]
- Kashiyama, K.; Mitsutake, N.; Matsuse, M.; Ogi, T.; Saenko, V.A.; Ujifuku, K.; Utani, A.; Hirano, A.; Yamashita, S. miR-196a downregulation increases the expression of type I and III collagens in keloid fibroblasts. J. Investig. Dermatol. 2012, 132, 1597–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Bian, L.; Lyu, J.; Jin, H.; Liu, Z.; Lyu, L.; Lu, D. Identification and integrated analysis of microRNA expression profiles in keloid. J. Cosmet. Dermatol. 2018, 17, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; He, Q.Y.; Luo, C.Q. Overexpression of miR-200b inhibits the cell proliferation and promotes apoptosis of human hypertrophic scar fibroblasts in vitro. J. Dermatol. 2014, 41, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Yao, S.; Chen, P.; Yang, Y.; Qian, M.; Han, Y.; Wang, N.; Zhao, Y.; He, Y.; Lyu, L.; et al. The integrative regulatory network of circRNA and microRNA in keloid scarring. Mol. Biol. Rep. 2020, 47, 201–209. [Google Scholar] [CrossRef]
- Guo, L.; Xu, K.; Yan, H.; Feng, H.; Chai, L.; Xu, G. Expression Profile of Long Noncoding RNAs in Human Earlobe Keloids: A Microarray Analysis. Biomed. Res. Int. 2016, 2016, 5893481. [Google Scholar] [CrossRef]
- Liang, X.; Ma, L.; Long, X.; Wang, X. LncRNA expression profiles and validation in keloid and normal skin tissue. Int. J. Oncol. 2015, 47, 1829–1838. [Google Scholar] [CrossRef]
- Huang, H.; Fu, S.; Liu, D. Detection and Analysis of the Hedgehog Signaling Pathway-Related Long Non-Coding RNA (lncRNA) Expression Profiles in Keloid. Med. Sci. Monit. 2018, 24, 9032–9044. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Huang, Q.; Fu, S.; Liu, D. Aberrantly expressed long noncoding RNAs in hypertrophic scar fibroblasts in vitro: A microarray study. Int. J. Mol. Med. 2018, 41, 1917–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, L.R.; Greene, J.; Chen, K.M.; Divine, G.; Chitale, D.; Shah, V.; Datta, I.; Worsham, M.J. Biological significance of genome-wide DNA methylation profiles in keloids. Laryngoscope 2017, 127, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhu, H.; Wang, H.; Li, J.; Han, F.; Liu, Y.; Zhang, W.; He, T.; Li, N.; Zheng, Z.; et al. Methylation of secreted frizzled-related protein 1 (SFRP1) promoter downregulates Wnt/beta-catenin activity in keloids. J. Mol. Histol. 2018, 49, 185–193. [Google Scholar] [CrossRef]
- Huang, C.; Liu, L.; You, Z.; Du, Y.; Ogawa, R. Managing keloid scars: From radiation therapy to actual and potential drug deliveries. Int. Wound J. 2019, 16, 852–859. [Google Scholar] [CrossRef]
- Careta, M.F.; Romiti, R. Localized scleroderma: Clinical spectrum and therapeutic update. An. Bras. Dermatol. 2015, 90, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, J.M.; Taroni, J.; Martyanov, V.; Wood, T.A.; Greene, C.S.; Pioli, P.A.; Hinchcliff, M.E.; Whitfield, M.L. Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms. PLoS Comput. Biol. 2015, 11, e1004005. [Google Scholar] [CrossRef] [Green Version]
- Matucci-Cerinic, M.; Kahaleh, B.; Wigley, F.M. Review: Evidence that systemic sclerosis is a vascular disease. Arthritis Rheum. 2013, 65, 1953–1962. [Google Scholar] [CrossRef]
- Didier, K.; Giusti, D.; Le Jan, S.; Terryn, C.; Muller, C.; Pham, B.N.; Le Naour, R.; Antonicelli, F.D.; Servettaz, A. Neutrophil Extracellular Traps Generation Relates with Early Stage and Vascular Complications in Systemic Sclerosis. J. Clin. Med. 2020, 9, 2136. [Google Scholar] [CrossRef]
- Angiolilli, C.; Marut, W.; van der Kroef, M.; Chouri, E.; Reedquist, K.A.; Radstake, T. New insights into the genetics and epigenetics of systemic sclerosis. Nat. Rev. Rheumatol. 2018, 14, 657–673. [Google Scholar] [CrossRef]
- Chairta, P.; Nicolaou, P.; Christodoulou, K. Genomic and genetic studies of systemic sclerosis: A systematic review. Hum. Immunol. 2017, 78, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Mayes, M.D.; Bossini-Castillo, L.; Gorlova, O.; Martin, J.E.; Zhou, X.; Chen, W.V.; Assassi, S.; Ying, J.; Tan, F.K.; Arnett, F.C.; et al. Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am. J. Hum. Genet. 2014, 94, 47–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assassi, S.; Swindell, W.R.; Wu, M.; Tan, F.D.; Khanna, D.; Furst, D.E.; Tashkin, D.P.; Jahan-Tigh, R.R.; Mayes, M.D.; Gudjonsson, J.E.; et al. Dissecting the heterogeneity of skin gene expression patterns in systemic sclerosis. Arthritis Rheumatol. 2015, 67, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Hinchcliff, M.; Huang, C.C.; Wood, T.A.; Matthew Mahoney, J.; Martyanov, V.; Bhattacharyya, S.; Tamaki, Z.; Lee, J.; Carns, M.; Podlusky, S.; et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J. Investig. Dermatol. 2013, 133, 1979–1989. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.E.; Mahoney, J.M.; Taroni, J.; Sargent, J.L.; Marmarelis, E.; Wu, M.R.; Varga, J.; Hinchcliff, M.E.; Whitfield, M.L. Experimentally-derived fibroblast gene signatures identify molecular pathways associated with distinct subsets of systemic sclerosis patients in three independent cohorts. PLoS ONE 2015, 10, e0114017. [Google Scholar] [CrossRef]
- Milano, A.; Pendergrass, S.A.; Sargent, J.L.; George, L.K.; McCalmont, T.H.; Connolly, M.K.; Whitfield, M.L. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS ONE 2008, 3, e2696. [Google Scholar] [CrossRef]
- Pendergrass, S.A.; Lemaire, R.; Francis, I.P.; Mahoney, J.M.; Lafyatis, R.; Whitfield, M.L. Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. J. Investig. Dermatol. 2012, 132, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Lofgren, S.; Hinchcliff, M.; Carns, M.; Wood, T.; Aren, K.; Arroyo, E.; Cheung, P.; Kuo, A.; Valenzuela, A.; Haemel, A.; et al. Integrated, multicohort analysis of systemic sclerosis identifies robust transcriptional signature of disease severity. JCI Insight 2016, 1, e89073. [Google Scholar] [CrossRef]
- Taroni, J.N.; Martyanov, V.; Huang, C.C.; Mahoney, J.M.; Hirano, I.; Shetuni, B.; Yang, G.Y.; Brenner, D.; Jung, B.; Wood, T.A.; et al. Molecular characterization of systemic sclerosis esophageal pathology identifies inflammatory and proliferative signatures. Arthritis Res. Ther. 2015, 17, 194. [Google Scholar] [CrossRef] [Green Version]
- Maurer, B.; Stanczyk, J.; Jungel, A.; Akhmetshina, A.; Trenkmann, M.; Brock, M.; Kowal-Bielecka, O.; Gay, R.E.; Michel, B.A.; Distler, J.H.; et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010, 62, 1733–1743. [Google Scholar] [CrossRef]
- Zhu, H.; Luo, H.; Zuo, X. MicroRNAs: Their involvement in fibrosis pathogenesis and use as diagnostic biomarkers in scleroderma. Exp. Mol. Med. 2013, 45, e41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etoh, M.; Jinnin, M.; Makino, K.; Yamane, K.; Nakayama, W.; Aoi, J.; Honda, N.; Kajihara, I.; Makino, T.; Fukushima, S.; et al. microRNA-7 down-regulation mediates excessive collagen expression in localized scleroderma. Arch. Dermatol. Res. 2013, 305, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Jinnin, M.; Hirano, A.; Yamane, K.; Eto, M.; Kusano, T.; Honda, N.; Kajihara, I.; Makino, T.; Sakai, K.; et al. The downregulation of microRNA let-7a contributes to the excessive expression of type I collagen in systemic and localized scleroderma. J. Immunol. 2013, 190, 3905–3915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouri, E.; Servaas, N.H.; Bekker, C.P.J.; Affandi, A.J.; Cossu, M.; Hillen, M.R.; Angiolilli, C.; Mertens, J.S.; van den Hoogen, L.L.; Silva-Cardoso, S.; et al. Serum microRNA screening and functional studies reveal miR-483-5p as a potential driver of fibrosis in systemic sclerosis. J. Autoimmun. 2018, 89, 162–170. [Google Scholar] [CrossRef]
- Honda, N.; Jinnin, M.; Kira-Etoh, T.; Makino, K.; Kajihara, I.; Makino, T.; Fukushima, S.; Inoue, Y.; Okamoto, Y.; Hasegawa, M.; et al. miR-150 down-regulation contributes to the constitutive type I collagen overexpression in scleroderma dermal fibroblasts via the induction of integrin beta3. Am. J. Pathol. 2013, 182, 206–216. [Google Scholar] [CrossRef]
- Zhou, B.; Zhu, H.; Luo, H.; Gao, S.; Dai, X.; Li, Y.; Zuo, X. MicroRNA-202-3p regulates scleroderma fibrosis by targeting matrix metalloproteinase 1. Biomed. Pharmacother. 2017, 87, 412–418. [Google Scholar] [CrossRef]
- Wang, Z.; Jinnin, M.; Nakamura, K.; Harada, M.; Kudo, H.; Nakayama, W.; Inoue, K.; Nakashima, T.; Honda, N.; Fukushima, S.; et al. Long non-coding RNA TSIX is upregulated in scleroderma dermal fibroblasts and controls collagen mRNA stabilization. Exp. Dermatol. 2016, 25, 131–136. [Google Scholar] [CrossRef]
- Messemaker, T.C.; Chadli, L.; Cai, G.; Goelela, V.S.; Boonstra, M.; Dorjee, A.L.; Andersen, S.N.; Mikkers, H.M.M.; van ‘t Hof, P.; Mei, H.; et al. Antisense Long Non-Coding RNAs Are Deregulated in Skin Tissue of Patients with Systemic Sclerosis. J. Investig. Dermatol. 2018, 138, 826–835. [Google Scholar] [CrossRef] [Green Version]
- Rice, L.M.; Padilla, C.M.; McLaughlin, S.R.; Mathes, A.; Ziemek, J.; Goummih, S.; Nakerakanti, S.; York, M.; Farina, G.; Whitfield, M.L.; et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Investig. 2015, 125, 2795–2807. [Google Scholar] [CrossRef]
- Chakravarty, E.F.; Martyanov, V.; Fiorentino, D.; Wood, T.A.; Haddon, D.J.; Jarrell, J.A.; Utz, P.J.; Genovese, M.C.; Whitfield, M.L.; Chung, L. Gene expression changes reflect clinical response in a placebo-controlled randomized trial of abatacept in patients with diffuse cutaneous systemic sclerosis. Arthritis Res. Ther. 2015, 17, 159. [Google Scholar] [CrossRef] [Green Version]
- Singer, A.J.; Tassiopoulos, A.; Kirsner, R.S. Evaluation and Management of Lower-Extremity Ulcers. N. Engl. J. Med. 2018, 378, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Golinko, M.S.; Joffe, R.; de Vinck, D.; Chandrasekaran, E.; Stojadinovic, O.; Barrientos, S.; Vukelic, S.; Tomic-Canic, M.; Brem, H. Surgical pathology to describe the clinical margin of debridement of chronic wounds using a wound electronic medical record. J. Am. Coll. Surg. 2009, 209, 254–260.e251. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, S.N.; Maggi, J.; Melamed, J.; Golinko, M.; Ross, F.; Chen, W. A histopathologic basis for surgical debridement to promote healing of venous ulcers. J. Am. Coll. Surg. 2012, 215, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Brem, H.; Stojadinovic, O.; Diegelmann, R.F.; Entero, H.; Lee, B.; Pastar, I.; Golinko, M.; Rosenberg, H.; Tomic-Canic, M. Molecular markers in patients with chronic wounds to guide surgical debridement. Mol. Med. 2007, 13, 30–39. [Google Scholar] [CrossRef]
- Stone, R.C.; Stojadinovic, O.; Rosa, A.M.; Ramirez, H.A.; Badiavas, E.; Blumenberg, M.; Tomic-Canic, M. A bioengineered living cell construct activates an acute wound healing response in venous leg ulcers. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Greco, J.A., 3rd; Pollins, A.C.; Boone, B.E.; Levy, S.E.; Nanney, L.B. A microarray analysis of temporal gene expression profiles in thermally injured human skin. Burns 2010, 36, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Sood, R.F.; Hocking, A.M.; Muffley, L.A.; Ga, M.; Honari, S.; Reiner, A.P.; Gibran, N.S. Genome-wide Association Study of Postburn Scarring Identifies a Novel Protective Variant. Ann. Surg. 2015, 262, 563–569. [Google Scholar] [CrossRef]
- Sood, R.F.; Arbabi, S.; Honari, S.; Gibran, N.S. Missense Variant in MAPK Inactivator PTPN5 Is Associated with Decreased Severity of Post-Burn Hypertrophic Scarring. PLoS ONE 2016, 11, e0149206. [Google Scholar] [CrossRef]
- Wallace, H.J.; Cadby, G.; Melton, P.E.; Wood, F.M.; Falder, S.; Crowe, M.M.; Martin, L.J.; Marlow, K.; Ward, S.V.; Fear, M.W. Genetic influence on scar height and pliability after burn injury in individuals of European ancestry: A prospective cohort study. Burns 2019, 45, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Citrin, D.E.; Mitchell, J.B. Mechanisms of Normal Tissue Injury From Irradiation. In Seminars in Radiation Oncology; WB Saunders: Philadelphia, PA, USA, 2017; Volume 27, pp. 316–324. [Google Scholar] [CrossRef]
- Giotopoulos, G.; Symonds, R.P.; Foweraker, K.; Griffin, M.; Peat, I.; Osman, A.; Plumb, M. The late radiotherapy normal tissue injury phenotypes of telangiectasia, fibrosis and atrophy in breast cancer patients have distinct genotype-dependent causes. Br. J. Cancer 2007, 96, 1001–1007. [Google Scholar] [CrossRef]
- Seibold, P.; Behrens, S.; Schmezer, P.; Helmbold, I.; Barnett, G.; Coles, C.; Yarnold, J.; Talbot, C.J.; Imai, T.; Azria, D.; et al. XRCC1 Polymorphism Associated With Late Toxicity After Radiation Therapy in Breast Cancer Patients. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 1084–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossberg, A.J.; Lei, X.; Xu, T.; Shaitelman, S.F.; Hoffman, K.E.; Bloom, E.S.; Stauder, M.C.; Tereffe, W.; Schlembach, P.J.; Woodward, W.A.; et al. Association of Transforming Growth Factor beta Polymorphism C-509T With Radiation-Induced Fibrosis Among Patients With Early-Stage Breast Cancer: A Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol. 2018, 4, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- Alsbeih, G.; Al-Harbi, N.; Al-Hadyan, K.; El-Sebaie, M.; Al-Rajhi, N. Association between normal tissue complications after radiotherapy and polymorphic variations in TGFB1 and XRCC1 genes. Radiat. Res. 2010, 173, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Edvardsen, H.; Landmark-Hoyvik, H.; Reinertsen, K.V.; Zhao, X.; Grenaker-Alnaes, G.I.; Nebdal, D.; Syvanen, A.C.; Rodningen, O.; Alsner, J.; Overgaard, J.; et al. SNP in TXNRD2 associated with radiation-induced fibrosis: A study of genetic variation in reactive oxygen species metabolism and signaling. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Landmark-Hoyvik, H.; Dumeaux, V.; Reinertsen, K.V.; Edvardsen, H.; Fossa, S.D.; Borresen-Dale, A.L. Blood gene expression profiling of breast cancer survivors experiencing fibrosis. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 875–883. [Google Scholar] [CrossRef]
- Weigel, C.; Veldwijk, M.R.; Oakes, C.C.; Seibold, P.; Slynko, A.; Liesenfeld, D.B.; Rabionet, M.; Hanke, S.A.; Wenz, F.; Sperk, E.; et al. Epigenetic regulation of diacylglycerol kinase alpha promotes radiation-induced fibrosis. Nat. Commun. 2016, 7, 10893. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, R.; Brunelli, S.M.; Williams, K.; Mitchell, M.D.; Feldman, H.I.; Umscheid, C.A. Gadolinium-based contrast agents and nephrogenic systemic fibrosis: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2009, 24, 856–863. [Google Scholar] [CrossRef]
- Attari, H.; Cao, Y.; Elmholdt, T.R.; Zhao, Y.; Prince, M.R. A Systematic Review of 639 Patients with Biopsy-confirmed Nephrogenic Systemic Fibrosis. Radiology 2019, 292, 376–386. [Google Scholar] [CrossRef]
- Woolen, S.A.; Shankar, P.R.; Gagnier, J.J.; MacEachern, M.P.; Singer, L.; Davenport, M.S. Risk of Nephrogenic Systemic Fibrosis in Patients With Stage 4 or 5 Chronic Kidney Disease Receiving a Group II Gadolinium-Based Contrast Agent: A Systematic Review and Meta-analysis. JAMA Intern. Med. 2020, 180, 223–230. [Google Scholar] [CrossRef]
- Jimenez, S.A.; Artlett, C.M.; Sandorfi, N.; Derk, C.; Latinis, K.; Sawaya, H.; Haddad, R.; Shanahan, J.C. Dialysis-associated systemic fibrosis (nephrogenic fibrosing dermopathy): Study of inflammatory cells and transforming growth factor beta1 expression in affected skin. Arthritis Rheum. 2004, 50, 2660–2666. [Google Scholar] [CrossRef]
- Cowper, S.E.; Su, L.D.; Bhawan, J.; Robin, H.S.; LeBoit, P.E. Nephrogenic fibrosing dermopathy. Am. J. Dermatopathol. 2001, 23, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Aydin, M.M.; Akcali, K.C. Liver fibrosis. Turk. J. Gastroenterol. 2018, 29, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotta, K.; Yoneda, M.; Hyogo, H.; Ochi, H.; Mizusawa, S.; Ueno, T.; Chayama, K.; Nakajima, A.; Nakao, K.; Sekine, A. Association of the rs738409 polymorphism in PNPLA3 with liver damage and the development of nonalcoholic fatty liver disease. BMC Med. Genet. 2010, 11, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Darlay, R.; Cockell, S.; Meroni, M.; Govaere, O.; Tiniakos, D.; Burt, A.D.; Bedossa, P.; Palmer, J.; Liu, Y.L.; et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J. Hepatol. 2020, 73, 505–515. [Google Scholar] [CrossRef]
- Namjou, B.; Lingren, T.; Huang, Y.; Parameswaran, S.; Cobb, B.L.; Stanaway, I.B.; Connolly, J.J.; Mentch, F.D.; Benoit, B.; Niu, X.; et al. GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network. BMC Med. 2019, 17, 135. [Google Scholar] [CrossRef]
- Wattacheril, J.; Lavine, J.E.; Chalasani, N.P.; Guo, X.; Kwon, S.; Schwimmer, J.; Molleston, J.P.; Loomba, R.; Brunt, E.M.; Chen, Y.I.; et al. Genome-Wide Associations Related to Hepatic Histology in Nonalcoholic Fatty Liver Disease in Hispanic Boys. J. Pediatr. 2017, 190, 100–107.e102. [Google Scholar] [CrossRef]
- Kriss, M.; Golden-Mason, L.; Kaplan, J.; Mirshahi, F.; Setiawan, V.W.; Sanyal, A.J.; Rosen, H.R. Increased hepatic and circulating chemokine and osteopontin expression occurs early in human NAFLD development. PLoS ONE 2020, 15, e0236353. [Google Scholar] [CrossRef]
- He, Y.; Hwang, S.; Cai, Y.; Kim, S.J.; Xu, M.; Yang, D.; Guillot, A.; Feng, D.; Seo, W.; Hou, X.; et al. MicroRNA-223 Ameliorates Nonalcoholic Steatohepatitis and Cancer by Targeting Multiple Inflammatory and Oncogenic Genes in Hepatocytes. Hepatology 2019, 70, 1150–1167. [Google Scholar] [CrossRef] [PubMed]
- Gawrieh, S.; Guo, X.; Tan, J.; Lauzon, M.; Taylor, K.D.; Loomba, R.; Cummings, O.W.; Pillai, S.; Bhatnagar, P.; Kowdley, K.V.; et al. A Pilot Genome-Wide Analysis Study Identifies Loci Associated With Response to Obeticholic Acid in Patients With NASH. Hepatol. Commun. 2019, 3, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- Perakakis, N.; Stefanakis, K.; Mantzoros, C.S. The role of omics in the pathophysiology, diagnosis and treatment of non-alcoholic fatty liver disease. Metabolism 2020, 111, 154320. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Shigefuku, R.; Maeyama, S.; Suzuki, M. Cirrhosis improvement to alcoholic liver fibrosis after passive abstinence. BMJ Case Rep. 2014, 2014. [Google Scholar] [CrossRef]
- Liu, X.; Rosenthal, S.B.; Meshgin, N.; Baglieri, J.; Musallam, S.G.; Diggle, K.; Lam, K.; Wu, R.; Pan, S.Q.; Chen, Y.; et al. Primary Alcohol-Activated Human and Mouse Hepatic Stellate Cells Share Similarities in Gene-Expression Profiles. Hepatol. Commun. 2020, 4, 606–626. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Stokowski, R.P.; Kershenobich, D.; Ballinger, D.G.; Hinds, D.A. Variant in PNPLA3 is associated with alcoholic liver disease. Nat. Genet. 2010, 42, 21–23. [Google Scholar] [CrossRef]
- Trepo, E.; Gustot, T.; Degre, D.; Lemmers, A.; Verset, L.; Demetter, P.; Ouziel, R.; Quertinmont, E.; Vercruysse, V.; Amininejad, L.; et al. Common polymorphism in the PNPLA3/adiponutrin gene confers higher risk of cirrhosis and liver damage in alcoholic liver disease. J. Hepatol. 2011, 55, 906–912. [Google Scholar] [CrossRef]
- Stickel, F.; Moreno, C.; Hampe, J.; Morgan, M.Y. The genetics of alcohol dependence and alcohol-related liver disease. J. Hepatol. 2017, 66, 195–211. [Google Scholar] [CrossRef] [Green Version]
- Hitomi, Y.; Ueno, K.; Kawai, Y.; Nishida, N.; Kojima, K.; Kawashima, M.; Aiba, Y.; Nakamura, H.; Kouno, H.; Kouno, H.; et al. POGLUT1, the putative effector gene driven by rs2293370 in primary biliary cholangitis susceptibility locus chromosome 3q13.33. Sci. Rep. 2019, 9, 102. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Nishida, N.; Kawashima, M.; Aiba, Y.; Tanaka, A.; Yasunami, M.; Nakamura, H.; Komori, A.; Nakamuta, M.; Zeniya, M.; et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am. J. Hum. Genet. 2012, 91, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Aiba, Y.; Hitomi, Y.; Kawashima, M.; Kojima, K.; Kawai, Y.; Ueno, K.; Nakamura, H.; Yamashiki, N.; Tanaka, T.; et al. NELFCD and CTSZ loci are associated with jaundice-stage progression in primary biliary cholangitis in the Japanese population. Sci. Rep. 2018, 8, 8071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiba, Y.; Yamazaki, K.; Nishida, N.; Kawashima, M.; Hitomi, Y.; Nakamura, H.; Komori, A.; Fuyuno, Y.; Takahashi, A.; Kawaguchi, T.; et al. Disease susceptibility genes shared by primary biliary cirrhosis and Crohn’s disease in the Japanese population. J. Hum. Genet. 2015, 60, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Liu, X.; Xu, C.; Lu, Y.; Xie, G.; Lu, Y.; Gu, X.; Walker, E.J.; Jing, K.; Juran, B.D.; et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N. Engl. J. Med. 2009, 360, 2544–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Invernizzi, P.; Lu, Y.; Kosoy, R.; Lu, Y.; Bianchi, I.; Podda, M.; Xu, C.; Xie, G.; Macciardi, F.; et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat. Genet. 2010, 42, 658–660. [Google Scholar] [CrossRef] [Green Version]
- Mells, G.F.; Floyd, J.A.; Morley, K.I.; Cordell, H.J.; Franklin, C.S.; Shin, S.Y.; Heneghan, M.A.; Neuberger, J.M.; Donaldson, P.T.; Day, D.B.; et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 2011, 43, 329–332. [Google Scholar] [CrossRef]
- Ulveling, D.; Le Clerc, S.; Cobat, A.; Labib, T.; Noirel, J.; Laville, V.; Coulonges, C.; Carpentier, W.; Nalpas, B.; Heim, M.H.; et al. A new 3p25 locus is associated with liver fibrosis progression in human immunodeficiency virus/hepatitis C virus-coinfected patients. Hepatology 2016, 64, 1462–1472. [Google Scholar] [CrossRef] [Green Version]
- Czaja, A.J.; Carpenter, H.A. Decreased fibrosis during corticosteroid therapy of autoimmune hepatitis. J. Hepatol. 2004, 40, 646–652. [Google Scholar] [CrossRef]
- Glass, L.M.; Dickson, R.C.; Anderson, J.C.; Suriawinata, A.A.; Putra, J.; Berk, B.S.; Toor, A. Total body weight loss of >/= 10% is associated with improved hepatic fibrosis in patients with nonalcoholic steatohepatitis. Dig. Dis. Sci. 2015, 60, 1024–1030. [Google Scholar] [CrossRef]
- Sun, Y.M.; Chen, S.Y.; You, H. Regression of liver fibrosis: Evidence and challenges. Chin. Med. J. 2020, 133, 1696–1702. [Google Scholar] [CrossRef]
- Bansal, R.; Nagorniewicz, B.; Prakash, J. Clinical Advancements in the Targeted Therapies against Liver Fibrosis. Mediat. Inflamm. 2016, 2016, 7629724. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu Dayyeh, B.K.; Yang, M.; Dienstag, J.L.; Chung, R.T. The effects of angiotensin blocking agents on the progression of liver fibrosis in the HALT-C Trial cohort. Dig. Dis. Sci. 2011, 56, 564–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHutchison, J.; Goodman, Z.; Patel, K.; Makhlouf, H.; Rodriguez-Torres, M.; Shiffman, M.; Rockey, D.; Husa, P.; Chuang, W.L.; Levine, R.; et al. Farglitazar lacks antifibrotic activity in patients with chronic hepatitis C infection. Gastroenterology 2010, 138, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Zein, C.O.; Yerian, L.M.; Gogate, P.; Lopez, R.; Kirwan, J.P.; Feldstein, A.E.; McCullough, A.J. Pentoxifylline improves nonalcoholic steatohepatitis: A randomized placebo-controlled trial. Hepatology 2011, 54, 1610–1619. [Google Scholar] [CrossRef]
- Lu, Y.; Fang, Z.; Li, M.; Chen, Q.; Zeng, T.; Lu, L.; Chen, Q.; Zhang, H.; Zhou, Q.; Sun, Y.; et al. Dynamic edge-based biomarker non-invasively predicts hepatocellular carcinoma with hepatitis B virus infection for individual patients based on blood testing. J. Mol. Cell Biol. 2019, 11, 665–677. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [Green Version]
- Franceschini, N.; Haack, K.; Almasy, L.; Laston, S.; Lee, E.T.; Best, L.G.; Fabsitz, R.R.; MacCluer, J.W.; Howard, B.V.; Umans, J.G.; et al. Generalization of associations of kidney-related genetic loci to American Indians. Clin. J. Am. Soc. Nephrol. 2014, 9, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Gorski, M.; Tin, A.; Garnaas, M.; McMahon, G.M.; Chu, A.Y.; Tayo, B.O.; Pattaro, C.; Teumer, A.; Chasman, D.I.; Chalmers, J.; et al. Genome-wide association study of kidney function decline in individuals of European descent. Kidney Int. 2015, 87, 1017–1029. [Google Scholar] [CrossRef] [Green Version]
- Hishida, A.; Nakatochi, M.; Akiyama, M.; Kamatani, Y.; Nishiyama, T.; Ito, H.; Oze, I.; Nishida, Y.; Hara, M.; Takashima, N.; et al. Genome-Wide Association Study of Renal Function Traits: Results from the Japan Multi-Institutional Collaborative Cohort Study. Am. J. Nephrol. 2018, 47, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Kottgen, A.; Pattaro, C.; Boger, C.A.; Fuchsberger, C.; Olden, M.; Glazer, N.L.; Parsa, A.; Gao, X.; Yang, Q.; Smith, A.V.; et al. New loci associated with kidney function and chronic kidney disease. Nat. Genet. 2010, 42, 376–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Lee, Y.; Park, B.; Won, S.; Han, J.S.; Heo, N.J. Genome-wide association analysis identifies multiple loci associated with kidney disease-related traits in Korean populations. PLoS ONE 2018, 13, e0194044. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.M.; Nadkarni, G.N.; Tao, R.; Graff, M.; Fornage, M.; Buyske, S.; Matise, T.C.; Highland, H.M.; Wilkens, L.R.; Carlson, C.S.; et al. Genetics of Chronic Kidney Disease Stages Across Ancestries: The PAGE Study. Front. Genet. 2019, 10, 494. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.T.; Garnaas, M.K.; Tin, A.; Kottgen, A.; Franceschini, N.; Peralta, C.A.; de Boer, I.H.; Lu, X.; Atkinson, E.; Ding, J.; et al. Genetic association for renal traits among participants of African ancestry reveals new loci for renal function. PLoS Genet. 2011, 7, e1002264. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.P.; Le, T.H.; Wu, H.; Akbarov, A.; van der Most, P.J.; Hemani, G.; Smith, G.D.; Mahajan, A.; Gaulton, K.J.; Nadkarni, G.N.; et al. Trans-ethnic kidney function association study reveals putative causal genes and effects on kidney-specific disease aetiologies. Nat. Commun. 2019, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Sim, X.; Go, M.J.; Wu, J.Y.; Gu, D.; Takeuchi, F.; Takahashi, A.; Maeda, S.; Tsunoda, T.; Chen, P.; et al. Meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat. Genet. 2012, 44, 904–909. [Google Scholar] [CrossRef] [Green Version]
- Pani, A.; Bragg-Gresham, J.; Masala, M.; Piras, D.; Atzeni, A.; Pilia, M.G.; Ferreli, L.; Balaci, L.; Curreli, N.; Delitala, A.; et al. Prevalence of CKD and its relationship to eGFR-related genetic loci and clinical risk factors in the SardiNIA study cohort. J. Am. Soc. Nephrol. 2014, 25, 1533–1544. [Google Scholar] [CrossRef]
- Nanayakkara, S.; Senevirathna, S.T.; Parahitiyawa, N.B.; Abeysekera, T.; Chandrajith, R.; Ratnatunga, N.; Hitomi, T.; Kobayashi, H.; Harada, K.H.; Koizumi, A. Whole-exome sequencing reveals genetic variants associated with chronic kidney disease characterized by tubulointerstitial damages in North Central Region, Sri Lanka. Environ. Health Prev. Med. 2015, 20, 354–359. [Google Scholar] [CrossRef]
- Cyrus, C.; Al-Mueilo, S.; Vatte, C.; Chathoth, S.; Li, Y.R.; Qutub, H.; Al Ali, R.; Al-Muhanna, F.; Lanktree, M.B.; Alkharsah, K.R.; et al. Assessing known chronic kidney disease associated genetic variants in Saudi Arabian populations. BMC Nephrol. 2018, 19, 88. [Google Scholar] [CrossRef] [Green Version]
- Kottgen, A.; Glazer, N.L.; Dehghan, A.; Hwang, S.J.; Katz, R.; Li, M.; Yang, Q.; Gudnason, V.; Launer, L.J.; Harris, T.B.; et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat. Genet. 2009, 41, 712–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattaro, C.; De Grandi, A.; Vitart, V.; Hayward, C.; Franke, A.; Aulchenko, Y.S.; Johansson, A.; Wild, S.H.; Melville, S.A.; Isaacs, A.; et al. A meta-analysis of genome-wide data from five European isolates reveals an association of COL22A1, SYT1, and GABRR2 with serum creatinine level. BMC Med. Genet. 2010, 11, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.K.; Katz, R.; Jotwani, V.; Garimella, P.S.; Ambrosius, W.T.; Cheung, A.K.; Gren, L.H.; Neyra, J.A.; Punzi, H.; Raphael, K.L.; et al. Distinct Dimensions of Kidney Health and Risk of Cardiovascular Disease, Heart Failure, and Mortality. Hypertension 2019, 74, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Salem, R.M.; Todd, J.N.; Sandholm, N.; Cole, J.B.; Chen, W.M.; Andrews, D.; Pezzolesi, M.G.; McKeigue, P.M.; Hiraki, L.T.; Qiu, C.; et al. Genome-Wide Association Study of Diabetic Kidney Disease Highlights Biology Involved in Glomerular Basement Membrane Collagen. J. Am. Soc. Nephrol. 2019, 30, 2000–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Zuydam, N.R.; Ahlqvist, E.; Sandholm, N.; Deshmukh, H.; Rayner, N.W.; Abdalla, M.; Ladenvall, C.; Ziemek, D.; Fauman, E.; Robertson, N.R.; et al. A Genome-Wide Association Study of Diabetic Kidney Disease in Subjects With Type 2 Diabetes. Diabetes 2018, 67, 1414–1427. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Huang, S.; Park, J.; Park, Y.; Ko, Y.A.; Seasock, M.J.; Bryer, J.S.; Xu, X.X.; Song, W.C.; Palmer, M.; et al. Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat. Med. 2018, 24, 1721–1731. [Google Scholar] [CrossRef]
- Sandholm, N.; Salem, R.M.; McKnight, A.J.; Brennan, E.P.; Forsblom, C.; Isakova, T.; McKay, G.J.; Williams, W.W.; Sadlier, D.M.; Makinen, V.P.; et al. New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet. 2012, 8, e1002921. [Google Scholar] [CrossRef] [Green Version]
- Tampe, B.; Zeisberg, M. Contribution of genetics and epigenetics to progression of kidney fibrosis. Nephrol. Dial. Transplant. 2014, 29, iv72–iv79. [Google Scholar] [CrossRef] [Green Version]
- Fragiadaki, M.; Mason, R.M. Epithelial-mesenchymal transition in renal fibrosis—Evidence for and against. Int. J. Exp. Pathol. 2011, 92, 143–150. [Google Scholar] [CrossRef]
- Zhou, W.; Otto, E.A.; Cluckey, A.; Airik, R.; Hurd, T.W.; Chaki, M.; Diaz, K.; Lach, F.P.; Bennett, G.R.; Gee, H.Y.; et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 2012, 44, 910–915. [Google Scholar] [CrossRef] [Green Version]
- Heylen, L.; Thienpont, B.; Busschaert, P.; Sprangers, B.; Kuypers, D.; Moisse, M.; Lerut, E.; Lambrechts, D.; Naesens, M. Age-related changes in DNA methylation affect renal histology and post-transplant fibrosis. Kidney Int. 2019, 96, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.; Kanetsky, P.A.; Xiao, R.; Gupta, J.; Mitra, N.; Limou, S.; Xie, D.; Xu, H.; Anderson, A.H.; Ojo, A.; et al. Genome-Wide Association of CKD Progression: The Chronic Renal Insufficiency Cohort Study. J. Am. Soc. Nephrol. 2017, 28, 923–934. [Google Scholar] [CrossRef]
- Rowland, J.; Akbarov, A.; Eales, J.; Xu, X.; Dormer, J.P.; Guo, H.; Denniff, M.; Jiang, X.; Ranjzad, P.; Nazgiewicz, A.; et al. Uncovering genetic mechanisms of kidney aging through transcriptomics, genomics, and epigenomics. Kidney Int. 2019, 95, 624–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckerman, P.; Qiu, C.; Park, J.; Ledo, N.; Ko, Y.A.; Park, A.D.; Han, S.Y.; Choi, P.; Palmer, M.; Susztak, K. Human Kidney Tubule-Specific Gene Expression Based Dissection of Chronic Kidney Disease Traits. EBioMedicine 2017, 24, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, E.P.; Morine, M.J.; Walsh, D.W.; Roxburgh, S.A.; Lindenmeyer, M.T.; Brazil, D.P.; Gaora, P.O.; Roche, H.M.; Sadlier, D.M.; Cohen, C.D.; et al. Next-generation sequencing identifies TGF-beta1-associated gene expression profiles in renal epithelial cells reiterated in human diabetic nephropathy. Biochim. Biophys. Acta 2012, 1822, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Cai, G.Y.; Duan, Z.Y.; Liu, S.W.; Wu, J.; Lv, Y.; Hou, K.; Li, Z.X.; Zhang, X.G.; Chen, X.M. Urinary sediment miRNAs reflect tubulointerstitial damage and therapeutic response in IgA nephropathy. BMC Nephrol. 2017, 18, 63. [Google Scholar] [CrossRef] [Green Version]
- Decleves, A.E.; Sharma, K. New pharmacological treatments for improving renal outcomes in diabetes. Nat. Rev. Nephrol. 2010, 6, 371–380. [Google Scholar] [CrossRef]
- Voelker, J.; Berg, P.H.; Sheetz, M.; Duffin, K.; Shen, T.; Moser, B.; Greene, T.; Blumenthal, S.S.; Rychlik, I.; Yagil, Y.; et al. Anti-TGF-beta1 Antibody Therapy in Patients with Diabetic Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Trachtman, H.; Fervenza, F.C.; Gipson, D.S.; Heering, P.; Jayne, D.R.; Peters, H.; Rota, S.; Remuzzi, G.; Rump, L.C.; Sellin, L.K.; et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 2011, 79, 1236–1243. [Google Scholar] [CrossRef] [Green Version]
- Vincenti, F.; Fervenza, F.C.; Campbell, K.N.; Diaz, M.; Gesualdo, L.; Nelson, P.; Praga, M.; Radhakrishnan, J.; Sellin, L.; Singh, A.; et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients With Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2017, 2, 800–810. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; Ix, J.H.; Mathew, A.V.; Cho, M.; Pflueger, A.; Dunn, S.R.; Francos, B.; Sharma, S.; Falkner, B.; McGowan, T.A.; et al. Pirfenidone for diabetic nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Maria, N.I.; Davidson, A. Protecting the kidney in systemic lupus erythematosus: From diagnosis to therapy. Nat. Rev. Rheumatol. 2020, 16, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.S.; Huang, J.F.; Zhu, J.; D’Agati, V.; Liu, X.; Miller, N.; Erlander, M.G.; Jackson, M.R.; Winchester, R.J. Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. J. Clin. Investig. 2004, 113, 1722–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Der, E.; Suryawanshi, H.; Morozov, P.; Kustagi, M.; Goilav, B.; Ranabothu, S.; Izmirly, P.; Clancy, R.; Belmont, H.M.; Koenigsberg, M.; et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat. Immunol. 2019, 20, 915–927. [Google Scholar] [CrossRef]
- Zhou, H.; Hasni, S.A.; Perez, P.; Tandon, M.; Jang, S.I.; Zheng, C.; Kopp, J.B.; Austin, H., 3rd; Balow, J.E.; Alevizos, I.; et al. miR-150 promotes renal fibrosis in lupus nephritis by downregulating SOCS1. J. Am. Soc. Nephrol. 2013, 24, 1073–1087. [Google Scholar] [CrossRef] [Green Version]
- Cardenas-Gonzalez, M.; Srivastava, A.; Pavkovic, M.; Bijol, V.; Rennke, H.G.; Stillman, I.E.; Zhang, X.; Parikh, S.; Rovin, B.H.; Afkarian, M.; et al. Identification, Confirmation, and Replication of Novel Urinary MicroRNA Biomarkers in Lupus Nephritis and Diabetic Nephropathy. Clin. Chem. 2017, 63, 1515–1526. [Google Scholar] [CrossRef] [Green Version]
- Nakhjavani, M.; Etemadi, J.; Pourlak, T.; Mirhosaini, Z.; Zununi Vahed, S.; Abediazar, S. Plasma levels of miR-21, miR-150, miR-423 in patients with lupus nephritis. Iran. J. Kidney Dis. 2019, 13, 198–206. [Google Scholar]
- Perez-Hernandez, J.; Martinez-Arroyo, O.; Ortega, A.; Galera, M.; Solis-Salguero, M.A.; Chaves, F.J.; Redon, J.; Forner, M.J.; Cortes, R. Urinary exosomal miR-146a as a marker of albuminuria, activity changes and disease flares in lupus nephritis. J. Nephrol. 2020. [Google Scholar] [CrossRef]
- Sole, C.; Moline, T.; Vidal, M.; Ordi-Ros, J.; Cortes-Hernandez, J. An Exosomal Urinary miRNA Signature for Early Diagnosis of Renal Fibrosis in Lupus Nephritis. Cells 2019, 8, 773. [Google Scholar] [CrossRef] [Green Version]
- Sole, C.; Cortes-Hernandez, J.; Felip, M.L.; Vidal, M.; Ordi-Ros, J. miR-29c in urinary exosomes as predictor of early renal fibrosis in lupus nephritis. Nephrol. Dial. Transplant. 2015, 30, 1488–1496. [Google Scholar] [CrossRef]
- Mendez-Flores, S.; Furuzawa-Carballeda, J.; Hernandez-Molina, G.; Ramirez-Martinez, G.; Regino-Zamarripa, N.E.; Ortiz-Quintero, B.; Jimenez-Alvarez, L.; Cruz-Lagunas, A.; Zuniga, J. MicroRNA Expression in Cutaneous Lupus: A New Window to Understand Its Pathogenesis. Mediat. Inflamm. 2019, 2019, 5049245. [Google Scholar] [CrossRef] [PubMed]
- Sole, C.; Domingo, S.; Ferrer, B.; Moline, T.; Ordi-Ros, J.; Cortes-Hernandez, J. MicroRNA Expression Profiling Identifies miR-31 and miR-485-3p as Regulators in the Pathogenesis of Discoid Cutaneous Lupus. J. Investig. Dermatol. 2019, 139, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sole, C.; Gimenez-Barcons, M.; Ferrer, B.; Ordi-Ros, J.; Cortes-Hernandez, J. Microarray study reveals a transforming growth factor-beta-dependent mechanism of fibrosis in discoid lupus erythematosus. Br. J. Dermatol. 2016, 175, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Capron, F.; Grenier, P.; Cordier, J.F. Diffuse idiopathic interstitial pneumonias. International multidisciplinary consensus classification by the American Thoracic Society and the European Respiratory Society, principal clinico-pathological entities, and diagnosis. Rev. Mal. Respir. 2004, 21, 299–318. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Coward, W.R.; Saini, G.; Jenkins, G. The pathogenesis of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2010, 4, 367–388. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef]
- Hubbard, R.; Lewis, S.; Richards, K.; Johnston, I.; Britton, J. Occupational exposure to metal or wood dust and aetiology of cryptogenic fibrosing alveolitis. Lancet 1996, 347, 284–289. [Google Scholar] [CrossRef]
- Iwai, K.; Mori, T.; Yamada, N.; Yamaguchi, M.; Hosoda, Y. Idiopathic pulmonary fibrosis. Epidemiologic approaches to occupational exposure. Am. J. Respir. Crit. Care Med. 1994, 150, 670–675. [Google Scholar] [CrossRef]
- Kuwano, K.; Nomoto, Y.; Kunitake, R.; Hagimoto, N.; Matsuba, T.; Nakanishi, Y.; Hara, N. Detection of adenovirus E1A DNA in pulmonary fibrosis using nested polymerase chain reaction. Eur. Respir. J. 1997, 10, 1445–1449. [Google Scholar] [CrossRef] [Green Version]
- Wangoo, A.; Shaw, R.J.; Diss, T.C.; Farrell, P.J.; du Bois, R.M.; Nicholson, A.G. Cryptogenic fibrosing alveolitis: Lack of association with Epstein-Barr virus infection. Thorax 1997, 52, 888–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, S.; Kubo, K.; Fujimoto, K.; Honda, T.; Sekiguchi, M.; Sodeyama, T. Analysis of bronchoalveolar lavage fluid in patients with chronic hepatitis C before and after treatment with interferon alpha. Thorax 1997, 52, 33–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idilman, R.; Cetinkaya, H.; Savas, I.; Aslan, N.; Sak, S.D.; Bastemir, M.; Sarioglu, M.; Soykan, I.; Bozdayi, M.; Colantoni, A.; et al. Bronchoalveolar lavage fluid analysis in individuals with chronic hepatitis C. J. Med. Virol. 2002, 66, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Mathai, S.K.; Schwartz, D.A. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front. Med. 2017, 4, 154. [Google Scholar] [CrossRef] [PubMed]
- Allam, J.S.; Limper, A.H. Idiopathic pulmonary fibrosis: Is it a familial disease? Curr. Opin. Pulm. Med. 2006, 12, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Bitterman, P.B.; Rennard, S.I.; Keogh, B.A.; Wewers, M.D.; Adelberg, S.; Crystal, R.G. Familial idiopathic pulmonary fibrosis. Evidence of lung inflammation in unaffected family members. N. Engl. J. Med. 1986, 314, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, U.; Pulkkinen, V.; Dixon, M.; Peyrard-Janvid, M.; Rehn, M.; Lahermo, P.; Ollikainen, V.; Salmenkivi, K.; Kinnula, V.; Kere, J.; et al. ELMOD2 is a candidate gene for familial idiopathic pulmonary fibrosis. Am. J. Hum. Genet. 2006, 79, 149–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.L.; Ryu, J.H.; Wittmer, M.H.; Hartman, T.E.; Lymp, J.F.; Tazelaar, H.D.; Limper, A.H. Familial idiopathic pulmonary fibrosis: Clinical features and outcome. Chest 2005, 127, 2034–2041. [Google Scholar] [CrossRef]
- Mageto, Y.N.; Raghu, G. Genetic predisposition of idiopathic pulmonary fibrosis. Curr. Opin. Pulm. Med. 1997, 3, 336–340. [Google Scholar] [CrossRef]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A., 3rd; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Mushiroda, T.; Wattanapokayakit, S.; Takahashi, A.; Nukiwa, T.; Kudoh, S.; Ogura, T.; Taniguchi, H.; Kubo, M.; Kamatani, N.; Nakamura, Y.; et al. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J. Med. Genet. 2008, 45, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeburn, R.W.; Kendall, H.; Dobson, L.; Egan, J.; Simler, N.J.; Millar, A.B. The 3′ untranslated region of tumor necrosis factor-alpha is highly conserved in idiopathic pulmonary fibrosis (IPF). Eur. Cytokine Netw. 2001, 12, 33–38. [Google Scholar]
- Pantelidis, P.; Fanning, G.C.; Wells, A.U.; Welsh, K.I.; Du Bois, R.M. Analysis of tumor necrosis factor-alpha, lymphotoxin-alpha, tumor necrosis factor receptor II, and interleukin-6 polymorphisms in patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2001, 163, 1432–1436. [Google Scholar] [CrossRef]
- Whyte, M.; Hubbard, R.; Meliconi, R.; Whidborne, M.; Eaton, V.; Bingle, C.; Timms, J.; Duff, G.; Facchini, A.; Pacilli, A.; et al. Increased risk of fibrosing alveolitis associated with interleukin-1 receptor antagonist and tumor necrosis factor-alpha gene polymorphisms. Am. J. Respir. Crit. Care Med. 2000, 162, 755–758. [Google Scholar] [CrossRef]
- Checa, M.; Ruiz, V.; Montano, M.; Velazquez-Cruz, R.; Selman, M.; Pardo, A. MMP-1 polymorphisms and the risk of idiopathic pulmonary fibrosis. Hum. Genet. 2008, 124, 465–472. [Google Scholar] [CrossRef]
- Falfan-Valencia, R.; Camarena, A.; Juarez, A.; Becerril, C.; Montano, M.; Cisneros, J.; Mendoza, F.; Granados, J.; Pardo, A.; Selman, M. Major histocompatibility complex and alveolar epithelial apoptosis in idiopathic pulmonary fibrosis. Hum. Genet. 2005, 118, 235–244. [Google Scholar] [CrossRef]
- Zorzetto, M.; Ferrarotti, I.; Campo, I.; Trisolini, R.; Poletti, V.; Scabini, R.; Ceruti, M.; Mazzola, P.; Crippa, E.; Ottaviani, S.; et al. NOD2/CARD15 gene polymorphisms in idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2005, 22, 180–185. [Google Scholar] [PubMed]
- Mathai, S.K.; Newton, C.A.; Schwartz, D.A.; Garcia, C.K. Pulmonary fibrosis in the era of stratified medicine. Thorax 2016, 71, 1154–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, B.D.; Lee, J.S.; Kozlitina, J.; Noth, I.; Devine, M.S.; Glazer, C.S.; Torres, F.; Kaza, V.; Girod, C.E.; Jones, K.D.; et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: An observational cohort study with independent validation. Lancet Respir. Med. 2014, 2, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.J.; Porte, J.; Braybrooke, R.; Flores, C.; Fingerlin, T.E.; Oldham, J.M.; Guillen-Guio, B.; Ma, S.F.; Okamoto, T.; John, A.E.; et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome-wide association study. Lancet Respir. Med. 2017, 5, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, N.; Rosas, I.O. Gene expression profiling as a window into idiopathic pulmonary fibrosis pathogenesis: Can we identify the right target genes? Proc. Am. Thorac. Soc. 2006, 3, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Emblom-Callahan, M.C.; Chhina, M.K.; Shlobin, O.A.; Ahmad, S.; Reese, E.S.; Iyer, E.P.; Cox, D.N.; Brenner, R.; Burton, N.A.; Grant, G.M.; et al. Genomic phenotype of non-cultured pulmonary fibroblasts in idiopathic pulmonary fibrosis. Genomics 2010, 96, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Pandit, K.V.; Corcoran, D.; Yousef, H.; Yarlagadda, M.; Tzouvelekis, A.; Gibson, K.F.; Konishi, K.; Yousem, S.A.; Singh, M.; Handley, D.; et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lu, J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Pottier, N.; Maurin, T.; Chevalier, B.; Puissegur, M.P.; Lebrigand, K.; Robbe-Sermesant, K.; Bertero, T.; Lino Cardenas, C.L.; Courcot, E.; Rios, G.; et al. Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: Implication in epithelial-mesenchymal interactions. PLoS ONE 2009, 4, e6718. [Google Scholar] [CrossRef] [PubMed]
- Oak, S.R.; Murray, L.; Herath, A.; Sleeman, M.; Anderson, I.; Joshi, A.D.; Coelho, A.L.; Flaherty, K.R.; Toews, G.B.; Knight, D.; et al. A micro RNA processing defect in rapidly progressing idiopathic pulmonary fibrosis. PLoS ONE 2011, 6, e21253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Zhao, G.Q.; Chen, T.F.; Chang, J.X.; Wang, H.Q.; Chen, S.S.; Zhang, G.J. Serum miR-21 and miR-155 expression in idiopathic pulmonary fibrosis. J. Asthma 2013, 50, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Njock, M.S.; Guiot, J.; Henket, M.A.; Nivelles, O.; Thiry, M.; Dequiedt, F.; Corhay, J.L.; Louis, R.E.; Struman, I. Sputum exosomes: Promising biomarkers for idiopathic pulmonary fibrosis. Thorax 2019, 74, 309–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjicharalambous, M.R.; Lindsay, M.A. Idiopathic Pulmonary Fibrosis: Pathogenesis and the Emerging Role of Long Non-Coding RNAs. Int. J. Mol. Sci. 2020, 21, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Wang, J.; Li, R.; Lv, C.; Xu, P.; Wang, Y.; Song, X.; Zhang, J. Astaxanthin attenuates pulmonary fibrosis through lncITPF and mitochondria-mediated signal pathways. J. Cell. Mol. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Landi, C.; Bergantini, L.; Cameli, P.; d’Alessandro, M.; Carleo, A.; Shaba, E.; Rottoli, P.; Bini, L.; Bargagli, E. Idiopathic Pulmonary Fibrosis Serum proteomic analysis before and after nintedanib therapy. Sci. Rep. 2020, 10, 9378. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Guan, J. Antifibrotic therapies to control cardiac fibrosis. Biomater. Res. 2016, 20, 13. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Moreno-Moral, A.; Pesce, F.; Devapragash, N.; Mancini, M.; Heng, E.L.; Rotival, M.; Srivastava, P.K.; Harmston, N.; Shkura, K.; et al. WWP2 regulates pathological cardiac fibrosis by modulating SMAD2 signaling. Nat. Commun. 2019, 10, 3616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.P.; Ding, Y.; Chen, J.; Wu, G.; Kataoka, M.; Hu, Y.; Yang, J.H.; Liu, J.; Drakos, S.G.; Selzman, C.H.; et al. Long non-coding RNAs link extracellular matrix gene expression to ischemic cardiomyopathy. Cardiovasc. Res. 2016, 112, 543–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Lv, T.; Quan, J.; Zhao, W.; Song, J.; Li, Z.; Lei, H.; Huang, W.; Ran, L. Identification of target genes in cardiomyopathy with fibrosis and cardiac remodeling. J. Biomed. Sci. 2018, 25, 63. [Google Scholar] [CrossRef] [PubMed]
- Alimadadi, A.; Manandhar, I.; Aryal, S.; Munroe, P.B.; Joe, B.; Cheng, X. Machine learning based classification and diagnosis of clinical cardiomyopathies. Physiol. Genom. 2020. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Park, Y.J.; Keller, A.; Vogel, B.; Lindroth, A.M.; Weichenhan, D.; Franke, J.; Fischer, S.; Bauer, A.; et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol. Med. 2013, 5, 413–429. [Google Scholar] [CrossRef]
- Meder, B.; Haas, J.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Frese, K.; Lai, A.; Nietsch, R.; Scheiner, C.; Mester, S.; Bordalo, D.M.; et al. Epigenome-Wide Association Study Identifies Cardiac Gene Patterning and a Novel Class of Biomarkers for Heart Failure. Circulation 2017, 136, 1528–1544. [Google Scholar] [CrossRef]
- Wooten, E.C.; Hebl, V.B.; Wolf, M.J.; Greytak, S.R.; Orr, N.M.; Draper, I.; Calvino, J.E.; Kapur, N.K.; Maron, M.S.; Kullo, I.J.; et al. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Ishii, N.; Ozaki, K.; Sato, H.; Mizuno, H.; Susumu, S.; Takahashi, A.; Miyamoto, Y.; Ikegawa, S.; Kamatani, N.; Hori, M.; et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J. Hum. Genet. 2006, 51, 1087–1099. [Google Scholar] [CrossRef] [Green Version]
- Sharma, U.C.; Mosleh, W.; Chaudhari, M.R.; Katkar, R.; Weil, B.; Evelo, C.; Cimato, T.R.; Pokharel, S.; Blankesteijn, W.M.; Suzuki, G. Myocardial and Serum Galectin-3 Expression Dynamics Marks Post-Myocardial Infarction Cardiac Remodelling. Heart Lung Circ. 2017, 26, 736–745. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [Green Version]
- Kumarswamy, R.; Bauters, C.; Volkmann, I.; Maury, F.; Fetisch, J.; Holzmann, A.; Lemesle, G.; de Groote, P.; Pinet, F.; Thum, T. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ. Res. 2014, 114, 1569–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibasaki, Y.; Nishiue, T.; Masaki, H.; Tamura, K.; Matsumoto, N.; Mori, Y.; Nishikawa, M.; Matsubara, H.; Iwasaka, T. Impact of the angiotensin II receptor antagonist, losartan, on myocardial fibrosis in patients with end-stage renal disease: Assessment by ultrasonic integrated backscatter and biochemical markers. Hypertens. Res. 2005, 28, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Kanai, A.J.; Konieczko, E.M.; Bennett, R.G.; Samuel, C.S.; Royce, S.G. Relaxin and fibrosis: Emerging targets, challenges, and future directions. Mol. Cell Endocrinol. 2019, 487, 66–74. [Google Scholar] [CrossRef]
- Metra, M.; Teerlink, J.R.; Cotter, G.; Davison, B.A.; Felker, G.M.; Filippatos, G.; Greenberg, B.H.; Pang, P.S.; Ponikowski, P.; Voors, A.A.; et al. Effects of Serelaxin in Patients with Acute Heart Failure. N. Engl. J. Med. 2019, 381, 716–726. [Google Scholar] [CrossRef]


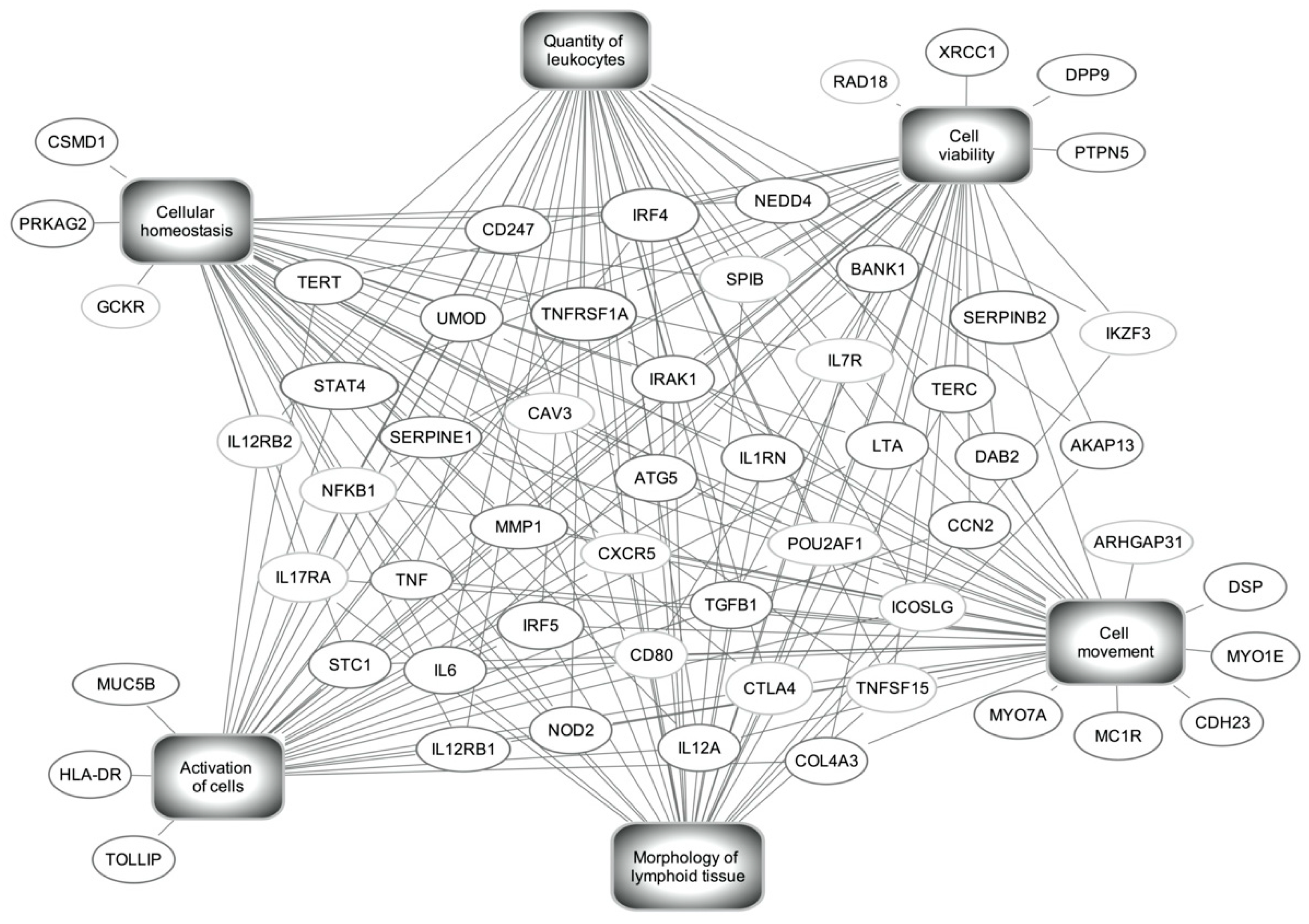{kind=link}
| Organ | Injury Trigger | Fibrotic Disease (s) |
|---|---|---|
| Skin | Breach of epidermal barrier | Keloid; hypertrophic scar; chronic ulcer |
| Vascular endothelial cell injury | Scleroderma/SSc 1; morphea; lichen sclerosus | |
| Exogenous insult (radiotherapy, heat, contrast agent) | Radiation fibrosis; burn; nephrogenic systemic fibrosis | |
| Liver | Exogenous insult (alcohol), infection, cholestasis, NASH 2 | Cirrhosis (hepatic cirrhosis; primary biliary cirrhosis) |
| Kidney | Obstruction, infection, autoimmune destruction, underlying disease (diabetes, hypertension) | Chronic kidney disease; SLE 3 |
| Lung | Alveolar epithelial damage | Idiopathic pulmonary fibrosis |
| Heart | Genetic defect, ischemia | Cardiomyopathy; heart failure |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stone, R.C.; Chen, V.; Burgess, J.; Pannu, S.; Tomic-Canic, M. Genomics of Human Fibrotic Diseases: Disordered Wound Healing Response. Int. J. Mol. Sci. 2020, 21, 8590. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228590
Stone RC, Chen V, Burgess J, Pannu S, Tomic-Canic M. Genomics of Human Fibrotic Diseases: Disordered Wound Healing Response. International Journal of Molecular Sciences. 2020; 21(22):8590. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228590
Chicago/Turabian StyleStone, Rivka C., Vivien Chen, Jamie Burgess, Sukhmani Pannu, and Marjana Tomic-Canic. 2020. "Genomics of Human Fibrotic Diseases: Disordered Wound Healing Response" International Journal of Molecular Sciences 21, no. 22: 8590. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228590