
Mitochondriopathies as a Clue to Systemic Disorders—Analytical Tools and Mitigating Measures in Context of Predictive, Preventive, and Personalized (3P) Medicine

 ,
,  and
and
Abstract
:1. Introduction
1.1. Mitochondria as the Life-Important Energy Supplier and Potential “Troublemaker”
1.2. Liquid Biopsy Is Instrumental for the Paradigm Change from Reactive to Predictive, Preventive, and Personalized Medicine (PPPM/3PM)
1.3. Aim of Study
1.4. Source of Data
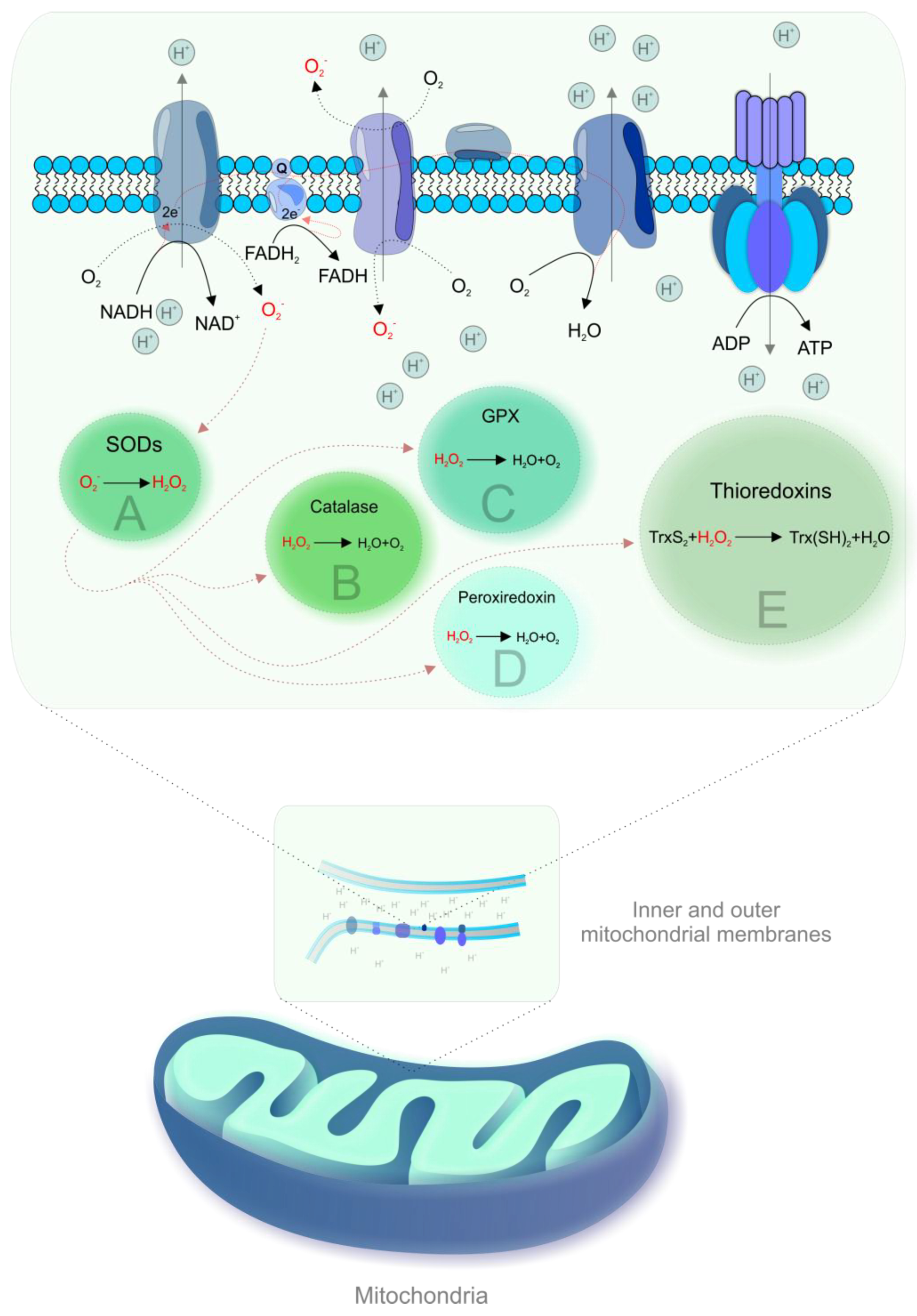
2. Mitochondria as the Powerhouse of the Cell: Energy Production and ROS Formation Insights
3. Molecular Interplay Shifted towards Excessive ROS Formation but Diminished Energy Production—A Critical “Vicious Circle” of Mitochondrial Injury
4. Mitochondriopathies
4.1. Definition and Main Characteristics
4.2. mtDNA Defects
5. Liquid Biopsy Application in MELAS Management
6. Mitochondrial Dysfunction in Neurodegenerative Disorders
6.1. Oxidative Stress and Mitochondrial Dysfunction Are Central for Neurodegeneration
6.1.1. Alzheimer’s Disease (AD)
6.1.2. Parkinson’s Disease (PD)
6.2. Mitochondria-Associated Liquid Biopsy Biomarker Panels in Neurodegenerative Disorders
6.2.1. Alzheimer’s Disease
6.2.2. Parkinson’s Disease
7. Mitochondrial Injury in Carcinogenesis: The Clue and Related Biomarker Panels
8. Antioxidant Diet Recommendations
9. Conclusions and Future Perspectives in the Context of 3P Medicine
9.1. Mitochondriopathies as an Attractive Diagnostic and Treatment Target
9.2. Mitochondriopathies in the Context of Prediction, Targeted Prevention, and Personalization of Medical Services
- A.
- Conditions
- Genotoxic environment [28]
- Inappropriate diet [31]
- Autoimmune disorders such as Sjögren’s syndrome with contributing inflammatory and vascular components [168]
- Acute infectious diseases such as COVID-19; viral infections provoke necrosis, which amplifies anti-viral immune responses, releasing damage-associated molecular patterns. Severely affected cells and tissues intrinsically secrete cell-free nucleic acids such as mtDNA. Indeed, COVID-19 patients with increased mtDNA levels are at elevated death risk and have to be intubated. Consequently, cell-free mtDNA is a potential biomarker for individualized survival status prediction in COVID-19 patients, as a model for a predictive approach under pandemic conditions [10,169,170].
- B.
- Corresponding analytical tools
- Risk assessment, predictive, and companion diagnostics [10]
- C.
- Comprehensive targeted prevention
- Life-style related expert recommendations based on individualized patient profiles
- Dietary habits and supplements including natural scavengers and pre- and pro-biotics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| O2− | Superoxide anion |
| OH | Hydroxyl radical |
| 8OH2′dG | 8-hydroxy-2′-deoxyguanosine |
| AD | Alzheimer’s disease |
| ADP | Adenosine diphosphate |
| ApoE4 | Apolipoprotein E4 |
| APP | Amyloid beta precursor protein |
| ATP | Adenosine triphosphate |
| Aβ | Amyloid beta |
| Ca2+ | Calcium ions |
| CPEO | Chronic progressive external ophthalmoplegia |
| CSF | Cerebrospinal fluid |
| DNA | Deoxyribonucleic acid |
| ETC | Electron transport chain |
| FGF21 | Fibroblast growth factor 21 |
| GDF-15 | Growth/differentiation factor 15 |
| GPx | Glutathione peroxidases |
| GPX | Glutathione peroxidise |
| Grx2 | Glutaredoxin-2 |
| GSH | Glutathione |
| GSSH | Oxidised glutathione |
| H2O2 | Hydrogen peroxide |
| HIF1α | Hypoxia inducible factor 1α |
| IRI | Ischemia-reperfusion injury |
| LHON | Leber hereditary optic neuropathy |
| LS | Leigh syndrome |
| MELAS | Mitochondrial encephalomyopathy lactic acidosis and stroke-like episodes |
| MERRF | Myoclonic epilepsy and ragged-red fibers |
| miRNA | MicroRNA |
| mtDNA | Mitochondrial DNA |
| NAD(+) | Nicotinamide adenine dinucleotide |
| NARP | Neurogenic muscle weakness, ataxia and retinitis pigmentosa |
| OXPHOS | Oxidative phosphorylation |
| PD | Parkinson’s disease |
| PPPM/3PM | Predictive, preventive, and personalized medicine |
| PS-1 | Presenilin-1 |
| PUFA | Polyunsaturated fatty acids |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| SOD2 | Superoxide dismutase 2 |
| Trx | Thioredoxin |
| TrxR | Thioredoxin reductase |
| α-Syn | α-synuclein |
References
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar]
- Lee, S.; Tak, E.; Lee, J.; Rashid, M.A.; Murphy, M.P.; Ha, J.; Kim, S.S. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res. 2011, 21, 817–834. [Google Scholar] [CrossRef] [Green Version]
- Gasparre, G.; Porcelli, A.M.; Lenaz, G.; Romeo, G. Relevance of Mitochondrial Genetics and Metabolism in Cancer Development. Cold Spring Harb. Perspect. Biol. 2013, 5, a011411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anglin, R. Mitochondrial Dysfunction in Psychiatric Illness. Can. J. Psychiatry 2016, 61, 444–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liskova, A.; Samec, M.; Koklesova, L.; Giordano, F.A.; Kubatka, P.; Golubnitschaja, O. Liquid Biopsy is Instrumental for 3PM Dimensional Solutions in Cancer Management. J. Clin. Med. 2020, 9, 2749. [Google Scholar] [CrossRef] [PubMed]
- Golubnitschaja, O.; Polivka, J.; Yeghiazaryan, K.; Berliner, L. Liquid biopsy and multiparametric analysis in management of liver malignancies: New concepts of the patient stratification and prognostic approach. EPMA J. 2018, 9, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Gerner, C.; Costigliola, V.; Golubnitschaja, O. Multiomic patterns in body fluids: Technological challenge with a great potential to implement the advanced paradigm of 3p medicine. Mass Spectrom. Rev. 2020, 39, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Crigna, A.T.; Samec, M.; Koklesova, L.; Liskova, A.; Giordano, F.A.; Kubatka, P.; Golubnitschaja, O. Cell-free nucleic acid patterns in disease prediction and monitoring—hype or hope? EPMA J. 2020, 11, 603–627. [Google Scholar] [CrossRef] [PubMed]
- Golubnitschaja, O.; Flammer, J. Individualised patient profile: Clinical utility of Flammer syndrome phenotype and general lessons for predictive, preventive and personalised medicine. EPMA J. 2018, 9, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubnitschaja, O. Feeling cold and other underestimated symptoms in breast cancer: Anecdotes or individual profiles for advanced patient stratification? EPMA J. 2017, 8, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Golubnitschaja, O.; Baban, B.; Boniolo, G.; Wang, W.; Bubnov, R.; Kapalla, M.; Krapfenbauer, K.; Mozaffari, M.S.; Costigliola, V. Medicine in the early twenty-first century: Paradigm and anticipation—EPMA position paper 2016. EPMA J. 2016, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Rocca, H.-P.B.-L.; Fleischhacker, L.; Golubnitschaja, O.; Heemskerk, F.; Helms, T.; Hoedemakers, T.; Allianses, S.H.; Jaarsma, T.; Kinkorova, J.; Ramaekers, J.; et al. Challenges in personalised management of chronic diseases-heart failure as prominent example to advance the care process. EPMA J. 2015, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunin, A.; Polivka, J.; Moiseeva, N.; Golubnitschaja, O. “Dry mouth” and “Flammer” syndromes—neglected risks in adolescents and new concepts by predictive, preventive and personalised approach. EPMA J. 2018, 9, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Kudela, E.; Samec, M.; Koklesova, L.; Liskova, A.; Kubatka, P.; Kozubik, E.; Rokos, T.; Pribulova, T.; Gabonova, E.; Smolar, M.; et al. miRNA Expression Profiles in Luminal A Breast Cancer—Implications in Biology, Prognosis, and Prediction of Response to Hormonal Treatment. Int. J. Mol. Sci. 2020, 21, 7691. [Google Scholar] [CrossRef]
- Tonhajzerova, I.; Olexova, L.B.; Jurko, A.; Spronck, B.; Jurko, T.; Sekaninova, N.; Visnovcova, Z.; Mestanikova, A.; Kudela, E.; Mestanik, M. Novel Biomarkers of Early Atherosclerotic Changes for Personalised Prevention of Cardiovascular Disease in Cervical Cancer and Human Papillomavirus Infection. Int. J. Mol. Sci. 2019, 20, 3720. [Google Scholar] [CrossRef] [Green Version]
- Kudela, E.; Samec, M.; Kubatka, P.; Nachajova, M.; Laucekova, Z.; Liskova, A.; Dokus, K.; Biringer, K.; Simova, D.; Gabonova, E.; et al. Breast Cancer in Young Women: Status Quo and Advanced Disease Management by a Predictive, Preventive, and Personalized Approach. Cancers 2019, 11, 1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poovathingal, S.K.; Gruber, J.; Halliwell, B.; Gunawan, R. Stochastic Drift in Mitochondrial DNA Point Mutations: A Novel Perspective Ex Silico. PLoS Comput. Biol. 2009, 5, e1000572. [Google Scholar] [CrossRef] [Green Version]
- Bergman, O.; Ben-Shachar, D. Mitochondrial Oxidative Phosphorylation System (OXPHOS) Deficits in Schizophrenia. Can. J. Psychiatry 2016, 61, 457–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Desler, C.; Lillenes, M.S.; Tønjum, T.; Rasmussen, L.J. The Role of Mitochondrial Dysfunction in the Progression of Alzheimer’s Disease. Curr. Med. Chem. 2019, 25, 5578–5587. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idelchik, M.D.P.S.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Semin. Cancer Biol. 2017, 47, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Kunin, A.; Sargheini, N.; Birkenbihl, C.; Moiseeva, N.; Fröhlich, H.; Golubnitschaja, O. Voice perturbations under the stress overload in young individuals: Phenotyping and suboptimal health as predictors for cascading pathologies. EPMA J. 2020, 11, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Sabel, B.A.; Wang, J.; Fähse, S.; Cárdenas-Morales, L.; Antal, A. Personality and stress influence vision restoration and recovery in glaucoma and optic neuropathy following alternating current stimulation: Implications for personalized neuromodulation and rehabilitation. EPMA J. 2020, 11, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Kucera, R.; Pecen, L.; Topolcan, O.; Dahal, A.R.; Costigliola, V.; Giordano, F.A.; Golubnitschaja, O. Prostate cancer management: Long-term beliefs, epidemic developments in the early twenty-first century and 3PM dimensional solutions. EPMA J. 2020, 11, 399–418. [Google Scholar] [CrossRef]
- Zhu, J.; Ying, W.; Zhang, L.; Peng, G.; Chen, W.; Anto, E.O.; Wang, X.; Lu, N.; Gao, S.; Wu, G.; et al. Psychological symptoms in Chinese nurses may be associated with predisposition to chronic disease: A cross-sectional study of suboptimal health status. EPMA J. 2020, 11, 551–563. [Google Scholar] [CrossRef]
- Goncharenko, V.; Bubnov, R.; Polivka, J.; Zubor, P.; Biringer, K.; Bielik, T.; Kuhn, W.; Golubnitschaja, O. Vaginal dryness: Individualised patient profiles, risks and mitigating measures. EPMA J. 2019, 10, 73–79. [Google Scholar] [CrossRef]
- Golubnitschaja, O. (Ed.) Flammer Syndrome: From Phenotype to Associated Pathologies, Prediction, Prevention and Personalisation; Advances in Predictive, Preventive and Personalised Medicine; Springer International Publishing: Cham, Switzerland, 2019; ISBN 978-3-030-13549-2. [Google Scholar]
- Polivka, J.; Pesta, M.; Rohan, V.; Celedova, L.; Mahajani, S.; Topolcan, O.; Golubnitschaja, O. Risks associated with the stroke predisposition at young age: Facts and hypotheses in light of individualized predictive and preventive approach. EPMA J. 2019, 10, 81–99. [Google Scholar] [CrossRef]
- Cebioglu, M.; Schild, H.H.; Golubnitschaja, O. Cancer predisposition in diabetics: Risk factors considered for predictive diagnostics and targeted preventive measures. EPMA J. 2010, 1, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, P.; Tatarkova, Z.; Sivonova, M.K.; Racay, P.; Lehotsky, J. Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems. Int. J. Mol. Sci. 2020, 21, 7698. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2008, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, M.; Fontanesi, F.; Merscher, S.; Fornoni, A. The Vicious Cycle of Renal Lipotoxicity and Mitochondrial Dysfunction. Front. Physiol. 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Horssen, J.; Van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schutt, F.; Aretz, S.; Auffarth, G.U.; Kopitz, J. Moderately Reduced ATP Levels Promote Oxidative Stress and Debilitate Autophagic and Phagocytic Capacities in Human RPE Cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5354–5361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.; Orr, A.; Perevoshchikova, I.; Quinlan, C. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br. J. Dermatol. 2013, 169, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kirkinezos, I.G.; Moraes, C.T. Reactive oxygen species and mitochondrial diseases. Semin. Cell Dev. Biol. 2001, 12, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Lawless, C.; Greaves, L.; Reeve, A.K.; Turnbull, D.M.; Vincent, A.E. The rise and rise of mitochondrial DNA mutations. Open Biol. 2020, 10, 200061. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Trifunovic, A. Origins of mtDNA mutations in ageing. Essays Biochem. 2017, 61, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Bacman, S.R.; Plastini, M.J.; Moraes, C.T. The mitochondrial DNA polymerase gamma degrades linear DNA fragments precluding the formation of deletions. Nat. Commun. 2018, 9, 2491. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Someya, S.; Kujoth, G.C.; Kim, M.-J.; Hacker, T.A.; Vermulst, M.; Weindruch, R.; Prolla, T.A. Effects of calorie restriction on the lifespan and healthspan of POLG mitochondrial mutator mice. PLoS ONE 2017, 12, e0171159. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.L.; Huang, J.; Edwards, Y.J.; Ulloa, R.H.; Dillon, L.M.; Prolla, T.A.; Vance, J.M.; Moraes, C.T.; Züchner, S. The mtDNA Mutation Spectrum of the Progeroid Polg Mutator Mouse Includes Abundant Control Region Multimers. Cell Metab. 2010, 12, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Maresca, A.; The ER-MITO Study Group; Del Dotto, V.; Romagnoli, M.; La Morgia, C.; Di Vito, L.; Capristo, M.; Valentino, M.L.; Carelli, V. Expanding and validating the biomarkers for mitochondrial diseases. J. Mol. Med. 2020, 98, 1467–1478. [Google Scholar] [CrossRef]
- Thangaraj, K.; Khan, N.A.; Govindaraj, P.; Meena, A.K. Mitochondrial disorders: Challenges in diagnosis & treatment. Indian J. Med Res. 2015, 141, 13–26. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- Szczepanowska, J.; Malińska, D.; Wieckowski, M.R.; Duszyński, J. Effect of mtDNA point mutations on cellular bioenergetics. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2012, 1817, 1740–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, G.; Cortopassi, G. Oxidative stress in inherited mitochondrial diseases. Free Radic. Biol. Med. 2015, 88, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J.; Zarrouk-Mahjoub, S.; Shoffner, J.M. MERRF Classification: Implications for Diagnosis and Clinical Trials. Pediatr. Neurol. 2018, 80, 8–23. [Google Scholar] [CrossRef]
- Finsterer, J.; Zarrouk-Mahjoub, S. Biomarkers for Detecting Mitochondrial Disorders. J. Clin. Med. 2018, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, O.; Lehmann, S.; Otto, M.; Zetterberg, H.; Lewczuk, P. Advantages and disadvantages of the use of the CSF Amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimer’s Res. Ther. 2019, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Niemantsverdriet, E.; Valckx, S.; Bjerke, M.; Engelborghs, S. Alzheimer’s disease CSF biomarkers: Clinical indications and rational use. Acta Neurol. Belg. 2017, 117, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Steele, H.E.; Horvath, R.; Lyon, J.J.; Chinnery, P.F. Monitoring clinical progression with mitochondrial disease biomarkers. Brain 2017, 140, 2530–2540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, M.; Yamadera, M.; Saito, T.; Fujimura, H.; Sakoda, S.; Koga, Y. Biomarker changes associated with clinical symptoms in MELAS patient. Neurol. Clin. Neurosci. 2019, 7, 344–346. [Google Scholar] [CrossRef]
- Koene, S.; De Laat, P.; Van Tienoven, D.H.; Vriens, D.; Brandt, A.M.; Sweep, F.C.G.J.; Rodenburg, R.J.; Donders, A.R.T.; Janssen, M.C.; Smeitink, J.A.M. Serum FGF21 levels in adult m.3243A>G carriers: Clinical implications. Neurol. 2014, 83, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Nukui, T.; Matsui, A.; Niimi, H.; Yamamoto, M.; Mastuda, N.; Piao, J.-L.; Noguchi, K.; Kitajima, I.; Nakastuji, Y. Cerebrospinal fluid ATP as a potential biomarker in patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke like episodes (MELAS). Mitochondrion 2020, 50, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Srikun, D.; Albers, A.E.; Nam, C.I.; Iavarone, A.T.; Chang, C.J. Organelle-Targetable Fluorescent Probes for Imaging Hydrogen Peroxide in Living Cells via SNAP-Tag Protein Labeling. J. Am. Chem. Soc. 2010, 132, 4455–4465. [Google Scholar] [CrossRef] [Green Version]
- Meseguer, S.; Martínez-Zamora, A.; Garcia-Arumi, E.; Andreu, A.L.; Armengod, M.-E. The ROS-sensitive microRNA-9/9* controls the expression of mitochondrial tRNA-modifying enzymes and is involved in the molecular mechanism of MELAS syndrome. Hum. Mol. Genet. 2014, 24, 167–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxidative Med. Cell. Longev. 2016, 2016, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kausar, S.; Wang, F.; Cui, H. The Role of Mitochondria in Reactive Oxygen Species Generation and Its Implications for Neurodegenerative Diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Huang, W.-J.; Zhang, X.; Chen, W.-W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, K.; Kiko, T.; Miyazawa, T.; Sookwong, P.; Tsuduki, T.; Satoh, A.; Miyazawa, T. Amyloid β-induced erythrocytic damage and its attenuation by carotenoids. FEBS Lett. 2011, 585, 1249–1254. [Google Scholar] [CrossRef] [Green Version]
- Arrozi, A.P.; Shukri, S.N.S.; Ngah, W.Z.W.; Yusof, Y.A.M.; Damanhuri, M.H.A.; Jaafar, F.; Makpol, S. Comparative Effects of Alpha- and Gamma-Tocopherol on Mitochondrial Functions in Alzheimer’s Disease In Vitro Model. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, J.P.; De Castro, A.A.; Soares, F.V.; Da Cunha, E.F.F.; Ramalho, T.C. Future Therapeutic Perspectives into the Alzheimer’s Disease Targeting the Oxidative Stress Hypothesis. Molecules 2019, 24, 4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Monaco, M.R.L.; Bentivoglio, A.R.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial-Derived Vesicles as Candidate Biomarkers in Parkinson’s Disease: Rationale, Design and Methods of the EXosomes in PArkiNson Disease (EXPAND) Study. Int. J. Mol. Sci. 2019, 20, 2373. [Google Scholar] [CrossRef] [Green Version]
- Bougea, A. New markers in Parkinson’s disease. Adv. Clin. Chem. 2020, 96, 137–178. [Google Scholar] [PubMed]
- Marella, M.; Seo, B.B.; Yagi, T.; Matsuno-Yagi, A. Parkinson’s disease and mitochondrial complex I: A perspective on the Ndi1 therapy. J. Bioenerg. Biomembr. 2009, 41, 493–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foti, S.C.; Hargreaves, I.; Carrington, S.; Kiely, A.P.; Houlden, H.; Holton, J.L. Cerebral mitochondrial electron transport chain dysfunction in multiple system atrophy and Parkinson’s disease. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Petersen, R.C. How early can we diagnose Alzheimer disease (and is it sufficient)? Neurology 2018, 91, 395–402. [Google Scholar] [CrossRef]
- Wojsiat, J.; Laskowska-Kaszub, K.; Mietelska-Porowska, A.; Wojda, U. Search for Alzheimer’s disease biomarkers in blood cells: Hypotheses-driven approach. Biomark. Med. 2017, 11, 917–931. [Google Scholar] [CrossRef] [Green Version]
- Pinho, R.; Guedes, L.C.; Soreq, L.; Lobo, P.P.; Mestre, T.; Coelho, M.; Rosa, M.M.; Gonçalves, N.; Wales, P.; Mendes, T.; et al. Gene Expression Differences in Peripheral Blood of Parkinson’s Disease Patients with Distinct Progression Profiles. PLoS ONE 2016, 11, e0157852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gezen-Ak, D.; Alaylıoğlu, M.; Genç, G.; Şengül, B.; Keskin, E.; Sordu, P.; Güleç, Z.E.K.; Apaydın, H.; Bayram-Gürel, Ç.; Ulutin, T.; et al. Altered Transcriptional Profile of Mitochondrial DNA-Encoded OXPHOS Subunits, Mitochondria Quality Control Genes, and Intracellular ATP Levels in Blood Samples of Patients with Parkinson’s Disease. J. Alzheimer’s Dis. 2020, 74, 287–307. [Google Scholar] [CrossRef] [PubMed]
- Schmukler, E.; Solomon, S.; Simonovitch, S.; Goldshmit, Y.; Wolfson, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Altered mitochondrial dynamics and function in APOE4-expressing astrocytes. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Nielsen, M.; Carcione, T.; Li, S.; Shi, J. Apolipoprotein E regulates mitochondrial function through the PGC-1α-sirtuin 3 pathway. Aging 2019, 11, 11148–11156. [Google Scholar] [CrossRef] [PubMed]
- Llano, D.A.; Bundela, S.; Mudar, R.A.; Devanarayan, V.; (Adni), F.T.A.D.N.I. A multivariate predictive modeling approach reveals a novel CSF peptide signature for both Alzheimer’s Disease state classification and for predicting future disease progression. PLoS ONE 2017, 12, e0182098. [Google Scholar] [CrossRef] [PubMed]
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann. Neurol. 2013, 74, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Sarasija, S.; Norman, K.R. Role of Presenilin in Mitochondrial Oxidative Stress and Neurodegeneration in Caenorhabditis elegans. Antioxidants 2018, 7, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raut, S.; Patel, R.; Al-Ahmad, A.J. Presence of a mutation in PSEN1 or PSEN2 gene is associated with an impaired brain endothelial cell phenotype in vitro. Fluids Barriers CNS 2021, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Skoumalová, A.; Ivica, J.; Šantorová, P.; Topinková, E.; Wilhelm, J. The lipid peroxidation products as possible markers of Alzheimer’s disease in blood. Exp. Gerontol. 2011, 46, 38–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosenko, E.; Aliev, G.; Kaminsky, Y. Relationship between chronic disturbance of 2,3-diphosphoglycerate metabolism in erythrocytes and Alzheimer disease. CNS Neurol. Disord.-Drug Targets 2016, 15, 113–123. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Proença, M.; Santos, S.; Santana, I.; Oliveira, C.R. Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiol. Aging 2004, 25, 105–110. [Google Scholar] [CrossRef]
- Mosconi, L.; de Leon, M.; Murray, J.; Lu, J.; Javier, E.; McHugh, P.; Swerdlow, R.H. Reduced Mitochondria Cytochrome Oxidase Activity in Adult Children of Mothers with Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Baglioni, M.; Cecchetti, R.; Cai, J.; Klein, J.B.; Bastiani, P.; Ruggiero, C.; Mecocci, P.; Butterfield, D.A. Lymphocyte mitochondria: Toward identification of peripheral biomarkers in the progression of Alzheimer disease. Free Radic. Biol. Med. 2013, 65, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangialasche, F.; Baglioni, M.; Cecchetti, R.; Kivipelto, M.; Ruggiero, C.; Piobbico, D.; Kussmaul, L.; Monastero, R.; Brancorsini, S.; Mecocci, P. Lymphocytic Mitochondrial Aconitase Activity is Reduced in Alzheimer’s Disease and Mild Cognitive Impairment. J. Alzheimer’s Dis. 2015, 44, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.R.; Gu, L.; Guerrero, R.; Robinson, R.A.S. Global cPILOT analysis of the APP/PS-1 mouse liver proteome. Proteom.-Clin. Appl. 2015, 9, 872–884. [Google Scholar] [CrossRef] [PubMed]
- González-Domínguez, R.; García-Barrera, T.; Vitorica, J.; Gómez-Ariza, J.L. Metabolomic investigation of systemic manifestations associated with Alzheimer’s disease in the APP/PS1 transgenic mouse model. Mol. BioSyst. 2015, 11, 2429–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitelpunkt, A.; Galili, T.; Kozlovski, T.; Bregman, N.; Shachar, N.; Markus-Kalish, M.; Benjamini, Y. Novel Alzheimer’s disease subtypes identified using a data and knowledge driven strategy. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fišar, Z.; Hroudová, J.; Hansíková, H.; Spáčilová, J.; Lelková, P.; Wenchich, L.; Jirák, R.; Zvěřová, M. Mitochondrial Respiration in the Platelets of Patients with Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 930–941. [Google Scholar] [CrossRef]
- Lunnon, K.; Keohane, A.; Pidsley, R.; Newhouse, S.; Riddoch-Contreras, J.; Thubron, E.B.; Devall, M.; Soininen, H.; Kłoszewska, I.; Mecocci, P.; et al. Mitochondrial genes are altered in blood early in Alzheimer’s disease. Neurobiol. Aging 2017, 53, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Dey, K.K.; Chen, P.-C.; Li, Y.; Niu, M.; Cho, J.-H.; Wang, X.; Bai, B.; Jiao, Y.; Chepyala, S.R.; et al. Integrated analysis of ultra-deep proteomes in cortex, cerebrospinal fluid and serum reveals a mitochondrial signature in Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Lee, H. Prediction of Alzheimer’s disease using blood gene expression data. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Xue, W.; Li, J.; Fu, K.; Teng, W. Differential Expression of mRNAs in Peripheral Blood Related to Prodrome and Progression of Alzheimer’s Disease. BioMed Res. Int. 2020, 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Perrotte, M.; Le Page, A.; Fournet, M.; Le Sayec, M.; Rassart, É.; Fulop, T.; Ramassamy, C. Blood-based redox-signature and their association to the cognitive scores in MCI and Alzheimer’s disease patients. Free Radic. Biol. Med. 2019, 130, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nottia, M.; Masciullo, M.; Verrigni, D.; Petrillo, S.; Modoni, A.; Rizzo, V.; Di Giuda, D.; Rizza, T.; Niceta, M.; Torraco, A.; et al. DJ-1 modulates mitochondrial response to oxidative stress: Clues from a novel diagnosis of PARK7. Clin. Genet. 2016, 92, 18–25. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy Body in Parkinson’s Disease and Related Neurodegenerative Disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zabetian, C.P.; Hancock, A.M.; Ginghina, C.; Hong, Z.; Yearout, D.; Chung, K.A.; Quinn, J.F.; Peskind, E.R.; Galasko, D.; et al. Significance and confounders of peripheral DJ-1 and alpha-synuclein in Parkinson’s disease. Neurosci. Lett. 2010, 480, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Herbert, M.K.; Eeftens, J.M.; Aerts, M.B.; Esselink, R.A.; Bloem, B.R.; Kuiperij, H.B.; Verbeek, M.M. CSF levels of DJ-1 and tau distinguish MSA patients from PD patients and controls. Park. Relat. Disord. 2014, 20, 112–115. [Google Scholar] [CrossRef]
- Garcia-Moreno, J.-M.; De Pablos, A.M.; García-Sánchez, M.-I.; Méndez-Lucena, C.; Damas-Hermoso, F.; Rus, M.; Chacón, J.; Fernández, E.; Fernández-Espejo, E.; Rus-Hidalgo, M. May Serum Levels of Advanced Oxidized Protein Products Serve as a Prognostic Marker of Disease Duration in Patients with Idiopathic Parkinson’s Disease? Antioxid. Redox Signal. 2013, 18, 1296–1302. [Google Scholar] [CrossRef]
- Luan, H.; Liu, L.-F.; Tang, Z.; Mok, V.C.; Li, M.; Cai, Z. Elevated excretion of biopyrrin as a new marker for idiopathic Parkinson’s disease. Park. Relat. Disord. 2015, 21, 1371–1372. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Depp, C.; Ryan, B.J.; Johnston, G.I.; Alegre-Abarrategui, J.; Evetts, S.; Rolinski, M.; Baig, F.; Ruffmann, C.; Simon, A.K.; et al. Mitochondrial dysfunction and increased glycolysis in prodromal and early Parkinson’s blood cells. Mov. Disord. 2018, 33, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Vida, C.; Kobayashi, H.; Garrido, A.; De Toda, I.M.; Carro, E.; Molina, J.A.; De La Fuente, M. Lymphoproliferation Impairment and Oxidative Stress in Blood Cells from Early Parkinson’s Disease Patients. Int. J. Mol. Sci. 2019, 20, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellecchia, M.T.; Savastano, R.; Moccia, M.; Picillo, M.; Siano, P.; Erro, R.; Vallelunga, A.; Amboni, M.; Vitale, C.; Santangelo, G.; et al. Lower serum uric acid is associated with mild cognitive impairment in early Parkinson’s disease: A 4-year follow-up study. J. Neural Transm. 2016, 123, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Zhou, B.; Chen, Y.-H.; Ma, Z.-L.; Gou, Y.; Zhang, C.-L.; Yu, W.-F.; Jiao, L. Serum uric acid levels in patients with Parkinson’s disease: A meta-analysis. PLoS ONE 2017, 12, e0173731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anjo, S.I.; dos Santos, P.V.; Rosado, L.; Baltazar, G.; Baldeiras, I.; Pires, D.; Gomes, A.; Januário, C.; Castelo-Branco, M.; Grãos, M.; et al. A different vision of translational research in biomarker discovery: A pilot study on circulatory mitochondrial proteins as Parkinson’s disease potential biomarkers. Transl. Neurodegener. 2020, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teves, J.M.Y.; Bhargava, V.; Kirwan, K.R.; Corenblum, M.J.; Justiniano, R.; Wondrak, G.T.; Anandhan, A.; Flores, A.J.; Schipper, D.A.; Khalpey, Z.; et al. Parkinson’s Disease Skin Fibroblasts Display Signature Alterations in Growth, Redox Homeostasis, Mitochondrial Function, and Autophagy. Front. Neurosci. 2018, 11, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.R.; et al. Mitochondrial Signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. J. Clin. Med. 2020, 9, 504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movahed, Z.G.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed. Pharmacother. 2019, 112, 108690. [Google Scholar] [CrossRef] [PubMed]
- Samec, M.; Liskova, A.; Koklesova, L.; Samuel, S.M.; Zhai, K.; Buhrmann, C.; Varghese, E.; Abotaleb, M.; Qaradakhi, T.; Zulli, A.; et al. Flavonoids against the Warburg phenotype—concepts of predictive, preventive and personalised medicine to cut the Gordian knot of cancer cell metabolism. EPMA J. 2020, 11, 377–398. [Google Scholar] [CrossRef] [PubMed]
- Samec, M.; Liskova, A.; Koklesova, L.; Mersakova, S.; Strnadel, J.; Kajo, K.; Pec, M.; Zhai, K.; Smejkal, K.; Mirzaei, S.; et al. Flavonoids Targeting HIF-1: Implications on Cancer Metabolism. Cancers 2021, 13, 130. [Google Scholar] [CrossRef] [PubMed]
- Gwangwa, M.V.; Joubert, A.M.; Visagie, M.H. Crosstalk between the Warburg effect, redox regulation and autophagy induction in tumourigenesis. Cell. Mol. Biol. Lett. 2018, 23, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Shao, L.; Pan, C.; Ye, J.; Ding, Z.; Wu, J.; Du, Q.; Ren, Y.; Zhu, C. Elevated level of mitochondrial reactive oxygen species via fatty acid β-oxidation in cancer stem cells promotes cancer metastasis by inducing epithelial–mesenchymal transition. Stem Cell Res. Ther. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodic, S.; Vincent, M.D. Reactive oxygen species (ROS) are a key determinant of cancer’s metabolic phenotype. Int. J. Cancer 2018, 142, 440–448. [Google Scholar] [CrossRef]
- Boakye, D.; Jansen, L.; Schöttker, B.; Jansen, E.H.J.M.; Schneider, M.; Halama, N.; Gào, X.; Chang-Claude, J.; Hoffmeister, M.; Brenner, H. Blood markers of oxidative stress are strongly associated with poorer prognosis in colorectal cancer patients. Int. J. Cancer 2020, 147, 2373–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Soud, M.R.A.; Hewala, T.I.M. The clinical significance of serum oxidative stress biomarkers in breast cancer females. Med Res. J. 2019, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Antošová, M.; Bencova, A.; Mikolka, P.; Košútová, P.; Mokra, D.; Rozborilová, E. The markers of oxidative stress in patient with lung cancer. Eur. Respir. J. 2015, 46, PA4267. [Google Scholar] [CrossRef]
- Shukla, S.; Srivastava, J.K.; Shankar, E.; Kanwal, R.; Nawab, A.; Sharma, H.; Bhaskaran, N.; Ponsky, L.E.; Fu, P.; MacLennan, G.T.; et al. Oxidative Stress and Antioxidant Status in High-Risk Prostate Cancer Subjects. Diagnostics 2020, 10, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koklesova, L.; Liskova, A.; Samec, M.; Buhrmann, C.; Samuel, S.M.; Varghese, E.; Ashrafizadeh, M.; Najafi, M.; Shakibaei, M.; Büsselberg, D.; et al. Carotenoids in Cancer Apoptosis—The Road from Bench to Bedside and Back. Cancers 2020, 12, 2425. [Google Scholar] [CrossRef]
- Liskova, A.; Koklesova, L.; Samec, M.; Smejkal, K.; Samuel, S.M.; Varghese, E.; Abotaleb, M.; Biringer, K.; Kudela, E.; Danko, J.; et al. Flavonoids in Cancer Metastasis. Cancers 2020, 12, 1498. [Google Scholar] [CrossRef] [PubMed]
- Liskova, A.; Kubatka, P.; Samec, M.; Zubor, P.; Mlyncek, M.; Bielik, T.; Samuel, S.M.; Zulli, A.; Kwon, T.K.; Büsselberg, D. Dietary Phytochemicals Targeting Cancer Stem Cells. Molecules 2019, 24, 899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abotaleb, M.; Liskova, A.; Kubatka, P.; Büsselberg, D. Therapeutic Potential of Plant Phenolic Acids in the Treatment of Cancer. Biomolecules 2020, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapinova, A.; Kubatka, P.; Liskova, A.; Baranenko, D.; Kruzliak, P.; Matta, M.; Büsselberg, D.; Malicherova, B.; Zulli, A.; Kwon, T.K.; et al. Controlling metastatic cancer: The role of phytochemicals in cell signaling. J. Cancer Res. Clin. Oncol. 2019, 145, 1087–1109. [Google Scholar] [CrossRef] [PubMed]
- Liskova, A.; Koklesova, L.; Samec, M.; Varghese, E.; Abotaleb, M.; Samuel, S.M.; Smejkal, K.; Biringer, K.; Petras, M.; Blahutova, D.; et al. Implications of flavonoids as potential modulators of cancer neovascularity. J. Cancer Res. Clin. Oncol. 2020, 146, 3079–3096. [Google Scholar] [CrossRef] [PubMed]
- Samec, M.; Liskova, A.; Kubatka, P.; Uramova, S.; Zubor, P.; Samuel, S.M.; Zulli, A.; Pec, M.; Bielik, T.; Biringer, K.; et al. The role of dietary phytochemicals in the carcinogenesis via the modulation of miRNA expression. J. Cancer Res. Clin. Oncol. 2019, 145, 1665–1679. [Google Scholar] [CrossRef] [PubMed]
- Buhrmann, C.; Shayan, P.; Brockmueller, A.; Shakibaei, M. Resveratrol Suppresses Cross-Talk between Colorectal Cancer Cells and Stromal Cells in Multicellular Tumor Microenvironment: A Bridge between In Vitro and In Vivo Tumor Microenvironment Study. Molecules 2020, 25, 4292. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Mehrpour, O.; Buhrmann, C.; Pourbagher-Shahri, A.M.; Shakibaei, M.; Samarghandian, S. Organophosphorus Compounds and MAPK Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 4258. [Google Scholar] [CrossRef]
- Farhood, B.; Ashrafizadeh, M.; Khodamoradi, E.; Hoseini-Ghahfarokhi, M.; Afrashi, S.; Musa, A.E.; Najafi, M. Targeting of cellular redox metabolism for mitigation of radiation injury. Life Sci. 2020, 250, 117570. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Autophagy regulation using luteolin: New insight into its anti-tumor activity. Cancer Cell Int. 2020, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ashrafizadeh, M.; Ahmadi, Z.; Mohammadinejad, R.; Afshar, E.G. Tangeretin: A mechanistic review of its pharmacological and therapeutic effects. J. Basic Clin. Physiol. Pharmacol. 2020, 31. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Rafiei, H.; Mohammadinejad, R.; Afshar, E.G.; Farkhondeh, T.; Samarghandian, S. Potential therapeutic effects of curcumin mediated by JAK/STAT signaling pathway: A review. Phytother. Res. 2020, 34, 1745–1760. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Tavakol, S.; Ahmadi, Z.; Roomiani, S.; Mohammadinejad, R.; Samarghandian, S. Therapeutic effects of kaempferol affecting autophagy and endoplasmic reticulum stress. Phytother. Res. 2020, 34, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Kubatka, P.; Kapinová, A.; Kruzliak, P.; Kello, M.; Výbohová, D.; Kajo, K.; Novak, M.; Chripkova, M.; Adamkov, M.; Pec, M.; et al. Antineoplastic effects of Chlorella pyrenoidosa in the breast cancer model. Nutrition 2015, 31, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Kapinova, A.; Stefanicka, P.; Kubatka, P.; Zubor, P.; Uramova, S.; Kello, M.; Mojzis, J.; Blahutova, D.; Qaradakhi, T.; Zulli, A.; et al. Are plant-based functional foods better choice against cancer than single phytochemicals? A critical review of current breast cancer research. Biomed. Pharmacother. 2017, 96, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Kubatka, P.; Uramova, S.; Kello, M.; Kajo, K.; Kruzliak, P.; Mojzis, J.; Vybohova, D.; Adamkov, M.; Jasek, K.; Lasabova, Z.; et al. Antineoplastic effects of clove buds (Syzygium aromaticumL.) in the model of breast carcinoma. J. Cell. Mol. Med. 2017, 21, 2837–2851. [Google Scholar] [CrossRef] [PubMed]
- Kubatka, P.; Kello, M.; Kajo, K.; Kruzliak, P.; Výbohová, D.; Mojžiš, J.; Adamkov, M.; Fialová, S.; Veizerová, L.; Zulli, A.; et al. Oregano demonstrates distinct tumour-suppressive effects in the breast carcinoma model. Eur. J. Nutr. 2017, 56, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Kubatka, P.; Kello, M.; Kajo, K.; Samec, M.; Jasek, K.; Vybohova, D.; Uramova, S.; Líšková, A.; Sadlonova, V.; Koklesova, L.; et al. Chemopreventive and Therapeutic Efficacy of Cinnamomum zeylanicum L. Bark in Experimental Breast Carcinoma: Mechanistic In Vivo and In Vitro Analyses. Molecules 2020, 25, 1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubatka, P.; Kello, M.; Kajo, K.; Samec, M.; Liskova, A.; Jasek, K.; Koklesova, L.; Kuruc, T.; Adamkov, M.; Smejkal, K.; et al. Rhus coriaria L. (Sumac) Demonstrates Oncostatic Activity in the Therapeutic and Preventive Model of Breast Carcinoma. Int. J. Mol. Sci. 2020, 22, 183. [Google Scholar] [CrossRef] [PubMed]
- Jasek, K.; Kubatka, P.; Samec, M.; Liskova, A.; Smejkal, K.; Vybohova, D.; Bugos, O.; Biskupska-Bodova, K.; Bielik, T.; Zubor, P.; et al. DNA Methylation Status in Cancer Disease: Modulations by Plant-Derived Natural Compounds and Dietary Interventions. Biomolecules 2019, 9, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samec, M.; Liskova, A.; Koklesova, L.; Mestanova, V.; Franekova, M.; Kassayova, M.; Bojkova, B.; Uramova, S.; Zubor, P.; Janikova, K.; et al. Fluctuations of Histone Chemical Modifications in Breast, Prostate, and Colorectal Cancer: An Implication of Phytochemicals as Defenders of Chromatin Equilibrium. Biomolecules 2019, 9, 829. [Google Scholar] [CrossRef] [Green Version]
- Hano, C.; Tungmunnithum, D. Plant Polyphenols, More than Just Simple Natural Antioxidants: Oxidative Stress, Aging and Age-Related Diseases. Medicines 2020, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Avula, S.; Parikh, S.; Demarest, S.; Kurz, J.; Gropman, A. Treatment of Mitochondrial Disorders. Curr. Treat. Options Neurol. 2014, 16, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; The Mitochondrial Medicine Society; Saneto, R.; Falk, M.J.; Anselm, I.; Cohen, B.H.; Haas, R. A modern approach to the treatment of mitochondrial disease. Curr. Treat. Options Neurol. 2009, 11, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Gibellini, L.; Bianchini, E.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Natural Compounds Modulating Mitochondrial Functions. Evid.-Based Complement. Altern. Med. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kicinska, A.; Jarmuszkiewicz, W. Flavonoids and Mitochondria: Activation of Cytoprotective Pathways? Molecules 2020, 25, 3060. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.A.; Kim, J.E.; Sung, J.E.; Bin Yun, W.; Kim, D.S.; Lee, H.S.; Hong, J.T.; Hwang, D.Y. Asparagus cochinchinensis stimulates release of nerve growth factor and abrogates oxidative stress in the Tg2576 model for Alzheimer’s disease. BMC Complement. Altern. Med. 2018, 18, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy, J.A.; Lindsay, C.B.; Quintanilla, R.A.; Carvajal, F.J.; Cerpa, W.; Inestrosa, N.C. Quercetin Exerts Differential Neuroprotective Effects Against H2O2 and Aβ Aggregates in Hippocampal Neurons: The Role of Mitochondria. Mol. Neurobiol. 2017, 54, 7116–7128. [Google Scholar] [CrossRef] [PubMed]
- Malvajerd, S.S.; Izadi, Z.; Azadi, A.; Kurd, M.; Derakhshankhah, H.; Zadeh, M.S.; Javar, H.A.; Hamidi, M. Neuroprotective Potential of Curcumin-Loaded Nanostructured Lipid Carrier in an Animal Model of Alzheimer’s Disease: Behavioral and Biochemical Evidence. J. Alzheimer’s Dis. 2019, 69, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Moreno, J.C.; De La Riva, M.P.; Martínez-Lara, E.; Siles, E.; Cañuelo, A. Tyrosol, a simple phenol from EVOO, targets multiple pathogenic mechanisms of neurodegeneration in a C. elegans model of Parkinson’s disease. Neurobiol. Aging 2019, 82, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Baroli, B.; Loi, E.; Solari, P.; Kasture, A.; Moi, L.; Muroni, P.; Kasture, S.; Setzu, M.D.; Liscia, A.; Zavattari, P. Evaluation of oxidative stress mechanisms and the effects of phytotherapic extracts on Parkinson’s disease Drosophila PINK1 B9 model. FASEB J. 2019, 33, 11028–11034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koklesova, L.; Liskova, A.; Samec, M.; Qaradakhi, T.; Zulli, A.; Smejkal, K.; Kajo, K.; Jakubikova, J.; Behzadi, P.; Pec, M.; et al. Genoprotective activities of plant natural substances in cancer and chemopreventive strategies in the context of 3P medicine. EPMA J. 2020, 11, 261–287. [Google Scholar] [CrossRef]
- Liskova, A.; Stefanicka, P.; Samec, M.; Smejkal, K.; Zubor, P.; Bielik, T.; Biskupska-Bodova, K.; Kwon, T.K.; Danko, J.; Büsselberg, D.; et al. Dietary phytochemicals as the potential protectors against carcinogenesis and their role in cancer chemoprevention. Clin. Exp. Med. 2020, 20, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Mitochondriopathies. Eur. J. Neurol. 2004, 11, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Yeghiazaryan, K.; Flammer, J.; Golubnitschaja, O. Predictive molecular profiling in blood of healthy vasospastic individuals: Clue to targeted prevention as personalised medicine to effective costs. EPMA J. 2010, 1, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Bubnov, R.; Polivka, J.; Zubor, P.; Konieczka, K.; Golubnitschaja, O. “Pre-metastatic niches” in breast cancer: Are they created by or prior to the tumour onset? “Flammer Syndrome” relevance to address the question. EPMA J. 2017, 8, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Avishai, E.; Yeghiazaryan, K.; Golubnitschaja, O. Impaired wound healing: Facts and hypotheses for multi-professional considerations in predictive, preventive and personalised medicine. EPMA J. 2017, 8, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Qian, S.; Golubnitschaja, O.; Zhan, X. Chronic inflammation: Key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. 2019, 10, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baban, B.; Golubnitschaja, O. The potential relationship between Flammer and Sjögren syndromes: The chime of dysfunction. EPMA J. 2017, 8, 333–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scozzi, D.; Cano, M.; Ma, L.; Zhou, D.; Zhu, J.H.; O’Halloran, J.A.; Goss, C.W.; Rauseo, A.M.; Liu, Z.; Sahu, S.K.; et al. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight 2021. [Google Scholar] [CrossRef]
- Chaari, L.; Golubnitschaja, O. Covid-19 pandemic by the “real-time” monitoring: The Tunisian case and lessons for global epidemics in the context of 3PM strategies. EPMA J. 2020, 11, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Yeghiazaryan, K.; Cebioglu, M.; Braun, M.; Kuhn, W.; Schild, H.H.; Golubnitschaja, O. Noninvasive subcellular imaging in breast cancer risk assessment: Construction of diagnostic windows. Pers. Med. 2011, 8, 321–330. [Google Scholar] [CrossRef]
- Lu, M.; Zhan, X. The crucial role of multiomic approach in cancer research and clinically relevant outcomes. EPMA J. 2018, 9, 77–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, E.; Yeghiazaryan, K.; Ahmad, A.; Giordano, F.A.; Fröhlich, H.; Golubnitschaja, O. Optimal multiparametric set-up modelled for best survival outcomes in palliative treatment of liver malignancies: Unsupervised machine learning and 3 PM recommendations. EPMA J. 2020, 11, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Bubnov, R.; Babenko, L.; Lazarenko, L.; Kryvtsova, M.; Shcherbakov, O.; Zholobak, N.; Golubnitschaja, O.; Spivak, M. Can tailored nanoceria act as a prebiotic? Report on improved lipid profile and gut microbiota in obese mice. EPMA J. 2019, 10, 317–335. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Biomarker | Study Details | Results | Reference |
|---|---|---|---|
| FGF21 and GDF-15 | Serum, CSF (case study) | Correlation between serum/CSF FGF21 and GDF-15 and MELAS vs conventional markers (lactate and pyruvate), of which the level decreased with the disease progression. FGF21 and GDF-15 were high in serum in the initial stage and greatly increased at the terminal stage of the disease | [59] |
| FGF21 | Serum (99 adult carriers of the m.3243A. G mutation) | FGF21 in adult carriers of m.3243A >G mutation (mtDNA mutation associated with MELAS) demonstrated little value in the monitoring and prediction of of the disease course | [60] |
| FGF21, GDF-15, ccf-mtDNA | Blood (cohort of 123 mitochondrial patients) | Increased FGF21, GDF-15, and ccf-mtDNA in MELAS; further increased during acute events (useful biomarkers for monitoring treatment effectiveness) vs. creatine, which only differentiated severe mitochondrial patients | [49] |
| ROS-sensitive miRNA-9/9* | MELAS cybrids | ROS-sensitive miRNA-9/9* used to control the expression of mitochondrial tRNA-modyfing enzymes and also to be involved in molecular mechanisms of MELAS in cybrid cells. Oxidative stress-mediated induction of miRNA-9/9* post-transcriptionally negatively regulates mt-tRNA-modification enzymes. Downregulation of these enzymes by miRNA-9/9* contributes to MELAS phenotype | [63] |
| ATP assay (luciferase luminious reaction) | Inverse correlation between CSF ATP with disease activity | [61] | |
| ROS quantification | Pre-clinical evaluation | SNAP-tag technique based on small molecule reporter SNAP-peroxy-green (molecular imaging for H2O2 in living cells) | [62] |
| Biomarker (Biofluid) | Study Participants (Details) | Result | Reference |
|---|---|---|---|
| ApoE (CSF) | AD and MCI patients, and normal control from the ADNI study (n = 287) | Level of ApoE → discrimination between AD and normal controls | [84] |
| mtDNA (CSF) | Selected from a cohort of 282 subjects (AD and other cognitive disorders Unit of the Hospital Clinic of Barcelona) | Low mtDNA in presymptomatic patients with PSEN1 mutation | [85] |
| Lipofuscin-like pigments (blood) | AD patients (n = 44) and age-matched controls (n = 16) | Increased lipofuscin-like pigments (productsof lipid peroxidation) in AD vs. controls | [88] |
| Oxidant and antioxidant metabolites (blood) | AD patients (n = 12), age-mached controls (n = 14), and young adult controls (n = 14) | Increased oxidative stress, hydrogen peroxide, organic hydroperoxide levels and reduced glutathione/glutathione disulfide ratio, glutathione transferase activity, and ATP in AD patients and age-matched control vs. young adult control | [89] |
| COX (mitochodnria isolated platelets) | AD and age-matched controls | Decrease in COX activity, diminished platelet ATP levels, and increased ROS in AD | [90] |
| COX | Cognitively normal (n = 36) individuals divided into 3 groups (parental history of late-onset AD) | Reduced COX activity in platelet mitochondria among cognitively normal individuals with maternal history of late-onset AD | [91] |
| Oxidative stress markers (mitochodnria isolated from lymphocytes) | Subjects with mild cognitive impairment (n = 12) and controls (n = 10) | Increased oxidative stress markers (protein carbonyls, 3-nitrotyrosine) | [92] |
| Mitochondrial aconitase (lymphocytes) | AD (n = 28), subjects with mild cognitive impairment (n = 22), older adults with normal cognition (n = 21), and younger adults with normal cognition (n = 19) | Mitochondrial aconitase reduced in AD and mild cognitive impairment | [93] |
| Antioxidants (uric acid, glutathione, homocarnosine) | In vivo model of AD (APP/PS-1 transgenic mice) | Reduced uric acid, glutathione, homocarnosine (marker of systemic oxidative stress as a hallmark of AD) | [95] |
| Biomarker | Study Participants | Result | Reference |
|---|---|---|---|
| DJ-1 (CSF) | PD patients (n = 43) and MSA patients (n = 23), and non-neurological control (n = 30) | CSF DJ-1 levels to distinguish MSA from PD | [107] |
| DJ-1 and α-Syn (blood and recently determined CSF levels) | PD patients (n = 126) and normal controls (n = 122) | Despite accessibility in CSF, DJ-1 and α-Syn are not applicable as useful plasma biomarkers for PD diagnosis | [106] |
| Advanced oxidized protein products (CSF, serum) | PD patients (n = 60) and control subjects (n = 45) | Higher advanced oxidized protein products (which originate as a result of the activity of free radicals) in PD patients vs negative controls | [108] |
| Biopyrin (urine) | PD patients (n = 234) and controls (n = 65) | Increased biopyrin (oxidative product of bilirubin) in idiopathic PD patients | [109] |
| ROS, SOD (blood) | Increased level of mitochondrial ROS in monocytes and reduced level of antioxidant SOD in blood | [110] | |
| Oxidative stress markers (blood) | PD patients (n = 45), elderly subjetcs (n = 34), and adult healthy subjects (n = 20) | Decreased glutathione peroxidase activity, increased oxidized glutathione and malondialdehyde contents | [111] |
| Uric acid (blood) | Early PD patients (n = 42) | Lower levels of serum uric acid associated with later occurrence of mild cognitive impairments | [112] |
| Cancer Type | Biofluid | Result | Reference |
|---|---|---|---|
| LC | Blood samples, LC patients (n = 40) and healthy controls (n = 40) | Higher 8OHdG in LC vs healthy control | [126] |
| BC | Blood samples (serum), BC patients (n = 35) and healthy controls (n = 35) | Higher MDA, GSSG in BC vs control. Lower GSH, TAC, GSH/GSSG ratio in BC vs control. | [125] |
| CRC | Blood samples, CRC pacients recruited into a population-based study in Germany (n = 3361) | Higher d-ROMs and lower TTL → poorer prognosis | [124] |
| PC | Blood samples, high-risk individuals (n = 20), healthy controls (n = 20) | Higher 8OHdG in high-risk subjects | [127] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liskova, A.; Samec, M.; Koklesova, L.; Kudela, E.; Kubatka, P.; Golubnitschaja, O. Mitochondriopathies as a Clue to Systemic Disorders—Analytical Tools and Mitigating Measures in Context of Predictive, Preventive, and Personalized (3P) Medicine. Int. J. Mol. Sci. 2021, 22, 2007. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042007
Liskova A, Samec M, Koklesova L, Kudela E, Kubatka P, Golubnitschaja O. Mitochondriopathies as a Clue to Systemic Disorders—Analytical Tools and Mitigating Measures in Context of Predictive, Preventive, and Personalized (3P) Medicine. International Journal of Molecular Sciences. 2021; 22(4):2007. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042007
Chicago/Turabian StyleLiskova, Alena, Marek Samec, Lenka Koklesova, Erik Kudela, Peter Kubatka, and Olga Golubnitschaja. 2021. "Mitochondriopathies as a Clue to Systemic Disorders—Analytical Tools and Mitigating Measures in Context of Predictive, Preventive, and Personalized (3P) Medicine" International Journal of Molecular Sciences 22, no. 4: 2007. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042007