
Alisporivir Treatment Alleviates Mitochondrial Dysfunction in the Skeletal Muscles of C57BL/6NCrl Mice with High-Fat Diet/Streptozotocin-Induced Diabetes Mellitus

 ,
, 
 and
and 
Abstract
:1. Introduction
2. Results
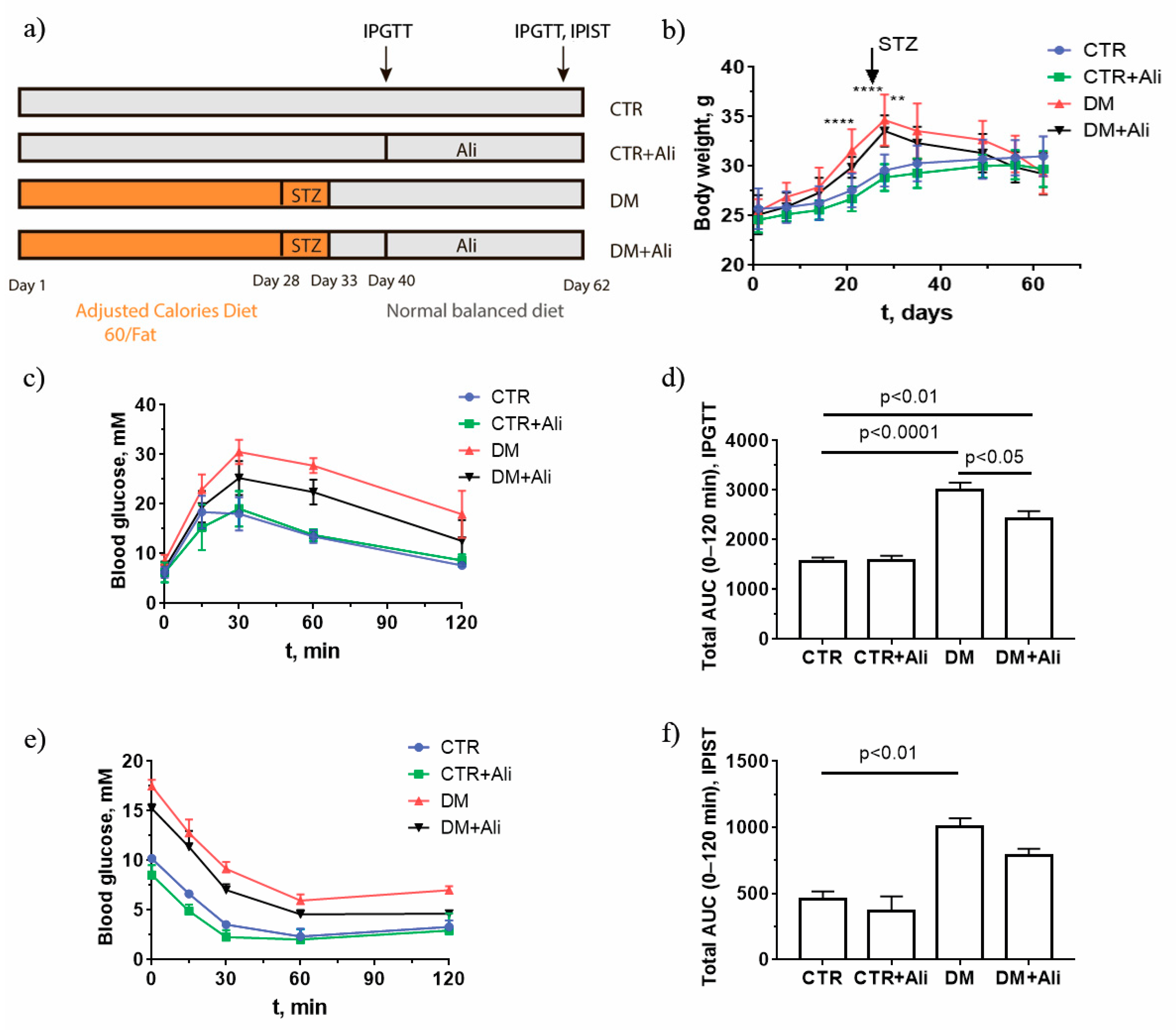
2.1. Effect of Alisporivir on Somatic and Biochemical Characteristics of Mice
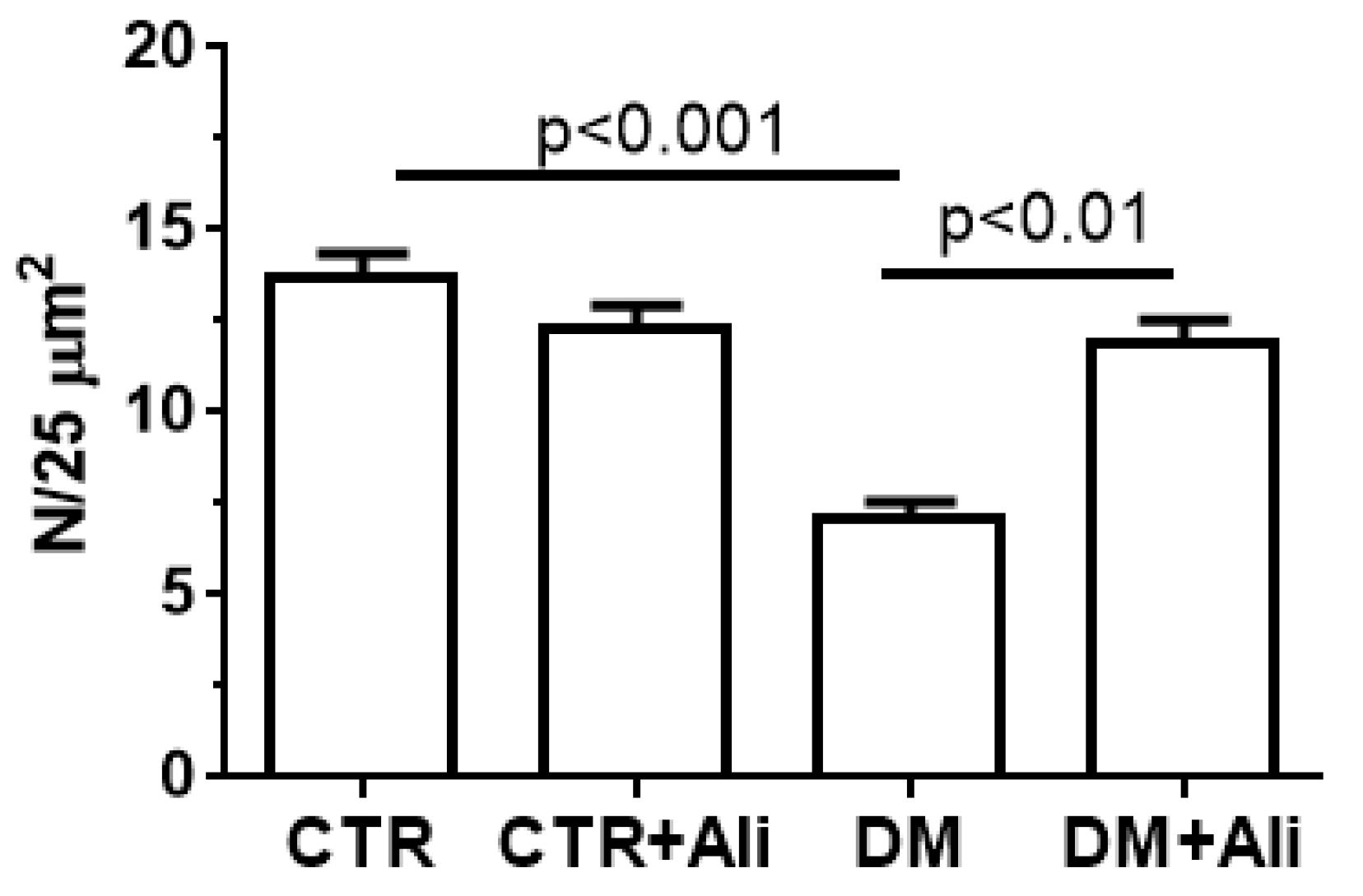
2.2. Effect of Alisporivir on the Ultrastructure of Skeletal Muscle Mitochondria in Diabetic Mice
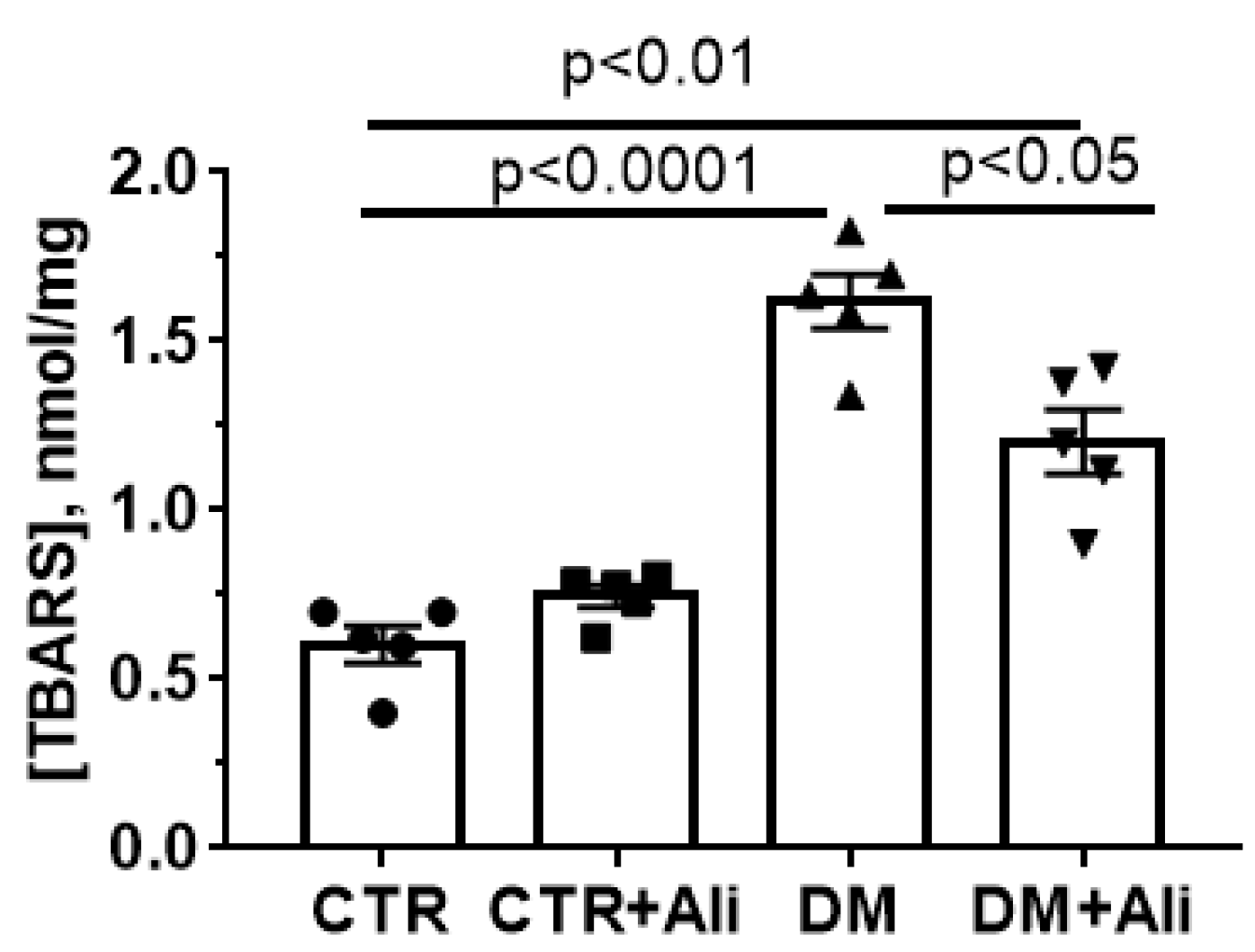
2.3. Effect of Alisporivir on DM—Induced Changes in the Functioning of Skeletal Muscle Mitochondria
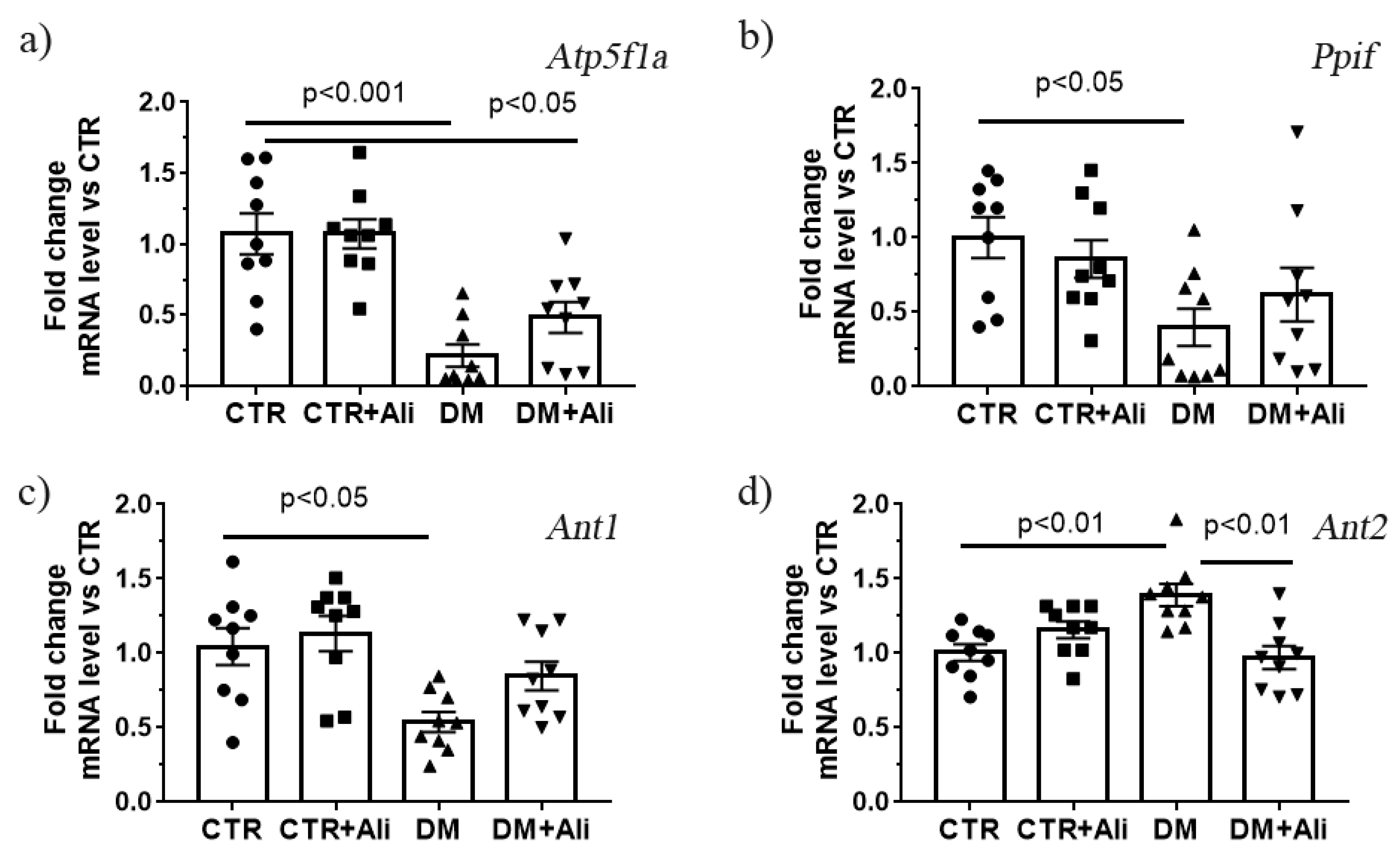
2.4. Effect of Alisporivir on Diabetes—Induced Changes in mRNA Levels of Proteins Responsible for Mitochondrial Biogenesis and Dynamics
3. Discussion
4. Materials and Methods
4.1. Animals and the Induction of Diabetes
4.2. Electron Microscopy
4.3. RNA Extraction, Reverse Transcription, and Quantitative Real-Time PCR
4.4. Isolation of Skeletal Muscle Mitochondria
4.5. Mitochondrial Respiration and Oxidative Phosphorylation
4.6. Lipid Peroxidation
4.7. Assay of the Mitochondrial Calcium Retention Capacity
4.8. Electrophoresis and Immunoblotting of Mitochondrial OXPHOS Proteins
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- American Diabetes Association. 2. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020, 43, S14–S31. [Google Scholar] [CrossRef] [Green Version]
- Winer, N.; Sowers, J.R. Epidemiology of diabetes. J. Clin. Pharmacol. 2004, 44, 397–405. [Google Scholar] [CrossRef]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, A.M.; Joanisse, D.R.; Baillot, R.G.; Hood, D.A. Mitochondrial dysregulation in the pathogenesis of diabetes: Potential for mitochondrial biogenesis-mediated interventions. Exp. Diabetes Res. 2012, 2012, 642038. [Google Scholar] [CrossRef] [PubMed]
- Scheele, C.; Nielsen, A.R.; Walden, T.B.; Sewell, D.A.; Fischer, C.P.; Brogan, R.J.; Petrovic, N.; Larsson, O.; Tesch, P.A.; Wennmalm, K.; et al. Altered regulation of the PINK1 locus: A link between type 2 diabetes and neurodegeneration? FASEB J. 2007, 21, 3653–3665. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, S.; Kuwabara, T. Diabetes-induced dysfunction of mitochondria and stem cells in skeletal muscle and the nervous system. Int. J. Mol. Sci. 2017, 18, 2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, M.K.; Turner, N. Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 2015, 4, R1–R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Dubinin, M.V. Diabetes Mellitus, Mitochondrial Dysfunction and Ca2+-Dependent Permeability Transition Pore. Int. J. Mol. Sci. 2020, 21, 6559. [Google Scholar] [CrossRef]
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochim. Biophys. Acta 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Carrer, A.; Tommasin, L.; Šileikytė, J.; Ciscato, F.; Filadi, R.; Urbani, A.; Forte, M.; Rasola, A.; Szabò, I.; Carraro, M.; et al. Defining the molecular mechanisms of the mitochondrial permeability transition through genetic manipulation of F-ATP synthase. Nat. Commun. 2021, 12, 4835. [Google Scholar] [CrossRef]
- Neginskaya, M.A.; Pavlov, E.V.; Sheu, S.S. Electrophysiological properties of the mitochondrial permeability transition pores: Channel diversity and disease implication. Biochim. Biophys. Acta. Bioenerg. 2021, 1862, 148357. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ transport: Mechanisms, molecular structures, and role in cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the permeability transition pore: Molecular mechanisms in human pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef]
- Efimov, S.V.; Dubinin, M.V.; Kobchikova, P.P.; Zgadzay, Y.O.; Khodov, I.A.; Belosludtsev, K.N.; Klochkov, V.V. Comparison of cyclosporin variants B-E based on their structural properties and activity in mitochondrial membranes. Biochem. Biophys. Res. Commun. 2020, 526, 1054–1060. [Google Scholar] [CrossRef]
- Riojas-Hernández, A.; Bernal-Ramírez, J.; Rodríguez-Mier, D.; Morales-Marroquín, F.E.; Domínguez-Barragán, E.M.; Borja-Villa, C.; Rivera-Álvarez, I.; García-Rivas, G.; Altamirano, J.; García, N. Enhanced oxidative stress sensitizes the mitochondrial permeability transition pore to opening in heart from Zucker Fa/fa rats with type 2 diabetes. Life Sci. 2015, 141, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.J.; Seiça, R.; Coxito, P.M.; Rolo, A.P.; Palmeira, C.M.; Santos, M.S.; Moreno, A.J. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003, 554, 511–514. [Google Scholar] [CrossRef] [Green Version]
- Belosludtsev, K.N.; Talanov, E.Y.; Starinets, V.S.; Agafonov, A.V.; Dubinin, M.V.; Belosludtseva, N.V. Transport of Ca2+ and Ca2+—Dependent permeability transition in rat liver mitochondria under the streptozotocin-induced type I diabetes. Cells 2019, 8, 1014. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, F.M.; Seiça, R.; Oliveira, P.J.; Coxito, P.M.; Moreno, A.J.; Palmeira, C.M.; Santos, M.S. Diabetes induces metabolic adaptations in rat liver mitochondria: Role of coenzyme Q and cardiolipin contents. Biochim. Biophys. Acta 2003, 1639, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Belosludtsev, K.N.; Starinets, V.S.; Belosludtsev, M.N.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtseva, N.V. Chronic treatment with dapagliflozin protects against mitochondrial dysfunction in the liver of C57BL/6NCrl mice with high-fat diet/streptozotocin-induced diabetes mellitus. Mitochondrion 2021, 59, 246–254. [Google Scholar] [CrossRef]
- Taddeo, E.P.; Laker, R.C.; Breen, D.S.; Akhtar, Y.N.; Kenwood, B.M.; Liao, J.A.; Zhang, M.; Fazakerley, D.J.; Tomsig, J.L.; Harris, T.E.; et al. Opening of the mitochondrial permeability transition pore links mitochondrial dysfunction to insulin resistance in skeletal muscle. Mol. Metab. 2013, 3, 124–134. [Google Scholar] [CrossRef]
- Lindblom, R.; Higgins, G.C.; Nguyen, T.V.; Arnstein, M.; Henstridge, D.C.; Granata, C.; Snelson, M.; Thallas-Bonke, V.; Cooper, M.E.; Forbes, J.M.; et al. Delineating a role for the mitochondrial permeability transition pore in diabetic kidney disease by targeting cyclophilin D. Clin. Sci. 2020, 134, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.; Correia, S.; Santos, M.S.; Seiça, R.; Oliveira, C.R.; Moreira, P.I. Metformin promotes isolated rat liver mitochondria impairment. Mol. Cell Biochem. 2008, 308, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masubuchi, Y.; Kano, S.; Horie, T. Mitochondrial permeability transition as a potential determinant of hepatotoxicity of antidiabetic thiazolidinediones. Toxicology 2006, 222, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Belosludtseva, N.V.; Starinets, V.S.; Belosludtsev, K.N. Effect of dapagliflozin on the functioning of rat liver mitochondria in vitro. Bull. Exp. Biol. Med 2021, 171, 572–576. (In Russian) [Google Scholar] [CrossRef]
- Sloan, R.C.; Moukdar, F.; Frasier, C.R.; Patel, H.D.; Bostian, P.A.; Lust, R.M.; Brown, D.A. Mitochondrial permeability transition in the diabetic heart: Contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J. Mol. Cell Cardiol. 2012, 52, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farajnia, S.; Mohammadi, M.; Badalzadeh, R.; Ahmadi Asl, N.; Baradaran, B.; Amani, M. Inhibition of mitochondrial permeability transition pore restores the cardioprotection by postconditioning in diabetic hearts. J. Diabetes Metab. Disord. 2014, 13, 106. [Google Scholar] [CrossRef] [Green Version]
- Li, P.A.; Uchino, H.; Elmér, E.; Siesjö, B.K. Amelioration by cyclosporin A of brain damage following 5 or 10 min of ischemia in rats subjected to preischemic hyperglycemia. Brain Res. 1997, 753, 133–140. [Google Scholar] [CrossRef]
- Qi, R.; Wang, D.; Xing, L.; Wu, Z. Cyclosporin A inhibits mitochondrial biogenesis in Hep G2 cells. Biochem. Biophys. Res. Commun. 2018, 496, 941–946. [Google Scholar] [CrossRef]
- Alam, M.R.; Baetz, D.; Ovize, M. Cyclophilin D and myocardial ischemia-reperfusion injury: A fresh perspective. J. Mol. Cell Cardiol. 2015, 78, 80–89. [Google Scholar] [CrossRef]
- Hansson, M.J.; Mattiasson, G.; Mansson, R.; Karlsson, J.; Keep, M.F.; Waldmeier, P.; Ruegg, U.T.; Dumont, J.M.; Besseghir, K.; Elmer, E. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J Bioenerg Biomembr. 2004, 36, 407–413. [Google Scholar] [CrossRef]
- Jezek, P.; Dlaskova, A. Dynamic of mitochondrial network, cristae, and mitochondrial nucleoids in pancreatic β-cells. Mitochondrion 2019, 49, 245–258. [Google Scholar] [CrossRef]
- Lewis, M.T.; Kasper, J.D.; Bazil, J.N.; Frisbee, J.C.; Wiseman, R.W. Quantification of mitochondrial oxidative phosphorylation in metabolic disease: Application to type 2 diabetes. Int. J. Mol. Sci. 2019, 20, 5271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharer, J.D. The adenine nucleotide translocase type 1 (ANT1): A new factor in mitochondrial disease. IUBMB Life 2005, 57, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elrod, J.W.; Wong, R.; Mishra, S.; Vagnozzi, R.J.; Sakthievel, B.; Goonasekera, S.A.; Karch, J.; Gabel, S.; Farber, J.; Force, T.; et al. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Investig. 2010, 120, 3680–3687. [Google Scholar] [CrossRef]
- Belosludtseva, N.V.; Starinets, V.S.; Pavlik, L.L.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtsev, K.N. The Effect of S-15176 Difumarate Salt on Ultrastructure and Functions of Liver Mitochondria of C57BL/6 Mice with Streptozotocin/High-Fat Diet-Induced Type 2 Diabetes. Biology 2020, 9, 309. [Google Scholar] [CrossRef]
- Gilbert, E.R.; Fu, Z.; Liu, D. Development of a nongenetic mouse model of type 2 diabetes. Exp. Diabetes Res. 2011, 2011, 416254. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Ghosh, S.K.; Choudhury, Y. A murine model of type 2 diabetes mellitus developed using a combination of high fat diet and multiple low doses of streptozotocin treatment mimics the metabolic characteristics of type 2 diabetes mellitus in humans. J. Pharmacol. Toxicol. Methods 2017, 84, 20–30. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, I. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Kosareva, E.A.; Talanov, E.Y.; Gudkov, S.V.; Dubinin, M.V. Itaconic acid impairs the mitochondrial function by the inhibition of complexes II and IV and induction of the permeability transition pore opening in rat liver mitochondria. Biochimie 2020, 176, 150–157. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | V Respiration, nmol O2 × min−1 × mg−1 Protein | RCR | ADP/O | |||
|---|---|---|---|---|---|---|
| State 2 | State 3 | State 4 | State 3UDNP | |||
| CTR | 18.4 ± 0.7 | 162.9 ± 9.8 | 22.3 ± 1.2 | 196.3 ± 19.7 | 7.2 ± 0.1 | 2.98 ± 0.02 |
| CTR+Ali | 19.9 ± 0.6 | 174.8 ± 10.1 | 23.3 ± 1.0 | 225.4 ± 17.0 | 7.8 ± 0.5 | 2.85 ± 0.07 |
| DM | 18.0 ± 1.2 | 125.2 ± 4.8 * | 25.1 ± 2.0 | 181.3 ± 20.5 | 5.1 ± 0.4 * | 2.57 ± 0.13 * |
| DM+Ali | 19.1 ± 1.1 | 158.7 ± 7.4 # | 24.8 ± 1.0 | 198.4 ± 20.2 | 6.6 ± 0.2 # | 2.77 ± 0.08 |
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| Atp5f1a | GATCTATCCAAGCAGGCTGT | AGGCGGGAGTGTAGGTAGAA |
| Ant1 | CTATGACACTGCCAAGGGGATG | TCAAACGGATAGGACACCAGC |
| Ant2 | TCTGGACGCAAAGGAACTGA | GACCATGCGCCCTTGAAA |
| Ppif | GCAGATGTCGTGCCAAAGACTG | GCCATTGTGGTTGGTGAAGTCG |
| Drp1 | TTACAGCACACAGGAATTGT | TTGTCACGGGCAACCTTTTA |
| Mfn2 | CACGCTGATGCAGACGGAGAA | ATCCCAGCGGTTGTTCAGG |
| Opa1 | GGACCCAAGAGCAGTGTGTT | CGAGACTCCAGGTTCTTCCG |
| Ppargc1a | CTGCCATTGTTAAGACCGAG | GTGTGAGGAGGGTCATCGTT |
| Rplp2 | CGGCTCAACAAGGTCATCAGTGA | AGCAGAAACAGCCACAGCCCCAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belosludtsev, K.N.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Dubinin, M.V.; Belosludtseva, N.V. Alisporivir Treatment Alleviates Mitochondrial Dysfunction in the Skeletal Muscles of C57BL/6NCrl Mice with High-Fat Diet/Streptozotocin-Induced Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 9524. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179524
Belosludtsev KN, Starinets VS, Talanov EY, Mikheeva IB, Dubinin MV, Belosludtseva NV. Alisporivir Treatment Alleviates Mitochondrial Dysfunction in the Skeletal Muscles of C57BL/6NCrl Mice with High-Fat Diet/Streptozotocin-Induced Diabetes Mellitus. International Journal of Molecular Sciences. 2021; 22(17):9524. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179524
Chicago/Turabian StyleBelosludtsev, Konstantin N., Vlada S. Starinets, Eugeny Yu. Talanov, Irina B. Mikheeva, Mikhail V. Dubinin, and Natalia V. Belosludtseva. 2021. "Alisporivir Treatment Alleviates Mitochondrial Dysfunction in the Skeletal Muscles of C57BL/6NCrl Mice with High-Fat Diet/Streptozotocin-Induced Diabetes Mellitus" International Journal of Molecular Sciences 22, no. 17: 9524. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179524